

Abstract
Activated partial thromboplastin time (aPTT) is associated with risk of thrombosis and coagulation disorders. We conducted a genome-wide association study for aPTT and identified significant associations with SNPs in three coagulation cascade genes, F12 (rs2731672, combined p = 2.16 × 10−30), KNG1 (rs710446, combined p = 9.52 × 10−22), and HRG (rs9898, combined p = 1.34 × 10−11). These three SNPs explain ∼18% of phenotypic variance in aPTT in the Lothian Birth Cohorts.
Main Text
Thrombotic disorders are among the most common causes of morbidity and mortality in the western world.1 Epidemiological studies show that shortening of the activated partial thromboplastin time (aPTT) is not only associated with several prothrombotic risk factors, including age, female sex, estrogen use, and obesity,2 but also increased risk of thrombosis.3 Prolonged aPTT is an indicator of coagulation disorders in patients with deficiencies of factors in the intrinsic pathway of coagulation. As a result, aPTT is considered a global test of thrombotic tendency. There is high heritability for variation in aPTT4,5 and risk of thrombosis,6 indicating the importance of genetic effects in the normal quantitative variation of aPTT.
We aimed to identify polymorphisms causing variation in aPTT in the general older population via genome-wide association methods in a total of 1477 adults from the Lothian Birth Cohorts of 1936 (LBC1936) and 1921 (LBC1921). The LBC1936 consists of 1091 relatively healthy individuals assessed on cognitive and medical traits at 70 years of age.7 They were born in 1936, most took part in the Scottish Mental Survey of 1947, and almost all lived independently in the Lothian region (Edinburgh city and surrounding area) of Scotland. The sample of 548 men and 543 women had a mean age of 69.6 years (standard deviation [SD] = 0.8). A full description of participant recruitment and testing can be found elsewhere.7,8 The LBC1921 cohort consists of 550 relatively healthy individuals, 316 females and 234 males, assessed on cognitive and medical traits at 79 years of age. They were born in 1921, most took part in the Scottish Mental Survey of 1932, and almost all lived independently in the Lothian region in Scotland. When tested, the sample had a mean age of 79.1 years (SD = 0.6). A full description of participant recruitment and testing can be found elsewhere.8 Ethics permission for the study was obtained from the Multi-Centre Research Ethics Committee for Scotland (MREC/01/0/56) and from Lothian Research Ethics Committee (LBC1936: LREC/2003/2/29 and LBC1921: LREC/1998/4/183). The research was carried out in compliance with the Helsinki Declaration. All subjects gave written, informed consent.
Blood samples were drawn from participants in both cohorts via the same protocol. Blood samples were sent for processing immediately, and the clotting tests were performed upon receipt. aPTT was measured in an ACL-Top Haemostasis testing system (Instrumentation Laboratory) with the SynthASil kit (HemosIL, Instrumentation Laboratory), the reliability of which is very high (coefficient of variation = 1.6%). Here we report the aPTT ratio for both cohorts. This is the ratio of the patient's aPTT to the aPTT reference value of the laboratory, herein referred to as aPTT. We have aPTT measurements available for LBC1936, which correlates almost perfectly (R2 = 0.95) with the aPTT ratio.
Genotyping was performed with the Illumina Human610-Quadv1 chip; 542,050 SNPs passed quality control. Of the 1091 individuals in the LBC1936, genomic DNA was isolated from 1071 by standard procedure at the Wellcome Trust Clinical Research Facility (WTCRF) Genetics Core. Twenty-nine samples failed quality control preceding the genotyping procedure. The remaining 1042 samples (all blood-extracted) were genotyped at the WTCRF Genetics Core with the Illumina Human610-Quadv1 chip. These samples were then subjected to the quality control procedures described below, and 1005 samples remained. Of these 1005 samples, 989 had aPTT measurements, and all further descriptions and analyses are based on these 989 samples. The mean age of these 989 samples was 69.53 (SD = 0.85, range = 3.69 years). Of the 550 individuals in the LBC1921, genomic DNA was isolated from whole blood from 542 samples by standard procedure at Medical Research Council Technology, Western General Hospital, Edinburgh. Sixteen samples failed quality control preceding the genotyping procedure. The remaining 526 samples were genotyped by the WTCRF Genetics Core with the Illumina Human610-Quadv1 chip. These samples were then subjected to the quality-control procedures described below, and 517 samples remained. Of these 517 samples, 488 participants had aPTT data available. The mean age of the participants with both genotype and phenotype information in LBC1921 was 79.05 (SD = 0.57, range = 2.86 years).
Quality control of the data was performed. Individuals with a disagreement between genetic and reported gender were removed (n = 12 in LBC1936; n = 1 in LBC1921). Relatedness between subjects was investigated and, for any related pair of individuals, one was removed (PI_HAT [proportion of identity by descent] > 0.25; n = 8 in LBC1936; n = 1 in LBC1921). Samples with a call rate ≤ 0.95 (n = 16 in LBC1936; n = 5 in LBC1921) and those showing evidence of non-European descent by multidimensional scaling were also removed (n = 1 in LBC1936; n = 2 in LBC1921). SNPs were included in the analyses if they met the following conditions: call rate ≥ 0.98, minor allele frequency ≥ 0.01, and Hardy-Weinberg equilibrium test with p ≥ 0.001. The final number of SNPs included in the genome-wide association study was 542,050.
The quantitative trait aPTT showed a normal distribution and is described in Table S1, available online. aPTT was adjusted for the effects of gender and age in days, because these are significant factors in aPTT.3–5 In Figure S1, the quantile-quantile plots for both the LBC1936 and LBC1921 together and separately showed no evidence of population stratification. The lambda values suggest no inflation of association signals in accordance with random expectation (joint λ = 0.9976; LBC1936 λ = 1.004; LBC1921 λ = 1.0092). Linear regression analysis covarying for cohort effect under an additive genetic model was performed with PLINK version 1.06.9 Analysis was also performed removing individuals with the following criteria: taking warfarin medication, abnormal lymphocyte count (>10 × 109/L), low platelets (<150 × 109/L), high platelets (>450 × 109/L), hemophilia, leukemia, or chronic liver disease (n = 62 in LBC1936; n = 45 in LBC1921). These exclusions included aPTT outliers (Z > ± 3.4; n = 3 in LBC1936; n = 3 in LBC1921). The results were viewed with WGAViewer.10 One SNP (rs1801020) was imputed with high confidence (R2 = 0.87 in LBC1936; R2 = 0.85 in LBC1921) with MACH.11 Associations were reported as genome-wide significant (p < 10−8). Pairwise interaction analysis was performed for the top, significantly-associated three SNPs with the additive by additive epistasis model in PLINK. To test for gender difference in the SNP associations with aPTT, we carried out a quantitative trait interaction test with PLINK. To correct for multiple testing in our gene-based search for associations based on Figure S3, we used a set-based permutation approach in PLINK.
Prediction of LBC1921 aPTT phenotype was performed with the beta weights from the best SNP in each of the genes associated with aPTT in the LBC1936 sample. They were entered as independent variables in a linear regression model to predict aPTT values in LBC1921. The resulting beta weights and constant were applied to the SNPs in LBC1921 to predict aPTT values. This analysis was performed in PASW Statistics 17.0. Pathway analysis was performed with STRING.12
Bioinformatic analyses were performed to predict whether the amino acid substitution affects protein structure and function based on sequence homology and the physical properties of amino acids with SIFT and PolyPhen.13,14
We identified highly significant associations of relatively large effect size between aPTT and SNPs in three genes: F12 (MIM ∗610619), KNG1 (MIM ∗612358) and HRG (MIM ∗142640) (Table 1; Figure 1A). The strongest association was rs2731672 (p = 2.16 × 10−30), an intergenic SNP ∼5.8 kb upstream of the gene for coagulation factor XII (F12), as illustrated in Figure 1B. The effects of rs2731672 on aPTT were additive, with each A allele increasing aPTT by 0.45 SD. The SNP explained 9.6% and 6.6% of the variation in aPTT in LBC1936 and LBC1921, respectively. The strongest KNG1 association was with rs710446 (p = 9.52 × 10−22), a nonsynonymous coding SNP (1742T>C, Ile581Thr) in exon 10 of kininogen 1 isoform 1 (high molecular weight kininogen, HMWK) (Table 1; Figure 1C). The effects of rs710446 were additive, with each G allele decreasing aPTT by 0.36 SD. The SNP explained 5.2% and 8.1% of the variance in LBC1936 and LBC1921, respectively. An association was also revealed with rs9898 (p = 1.34 × 10−11), a nonsynonymous coding SNP (610C>T, Pro204Ser) in exon 5 of the gene HRG, for histidine-rich glycoprotein (HRGP). The effects were additive, with each T allele decreasing aPTT by 0.26 SD. The SNP explained 1.8% and 0.77% of the variance in aPTT in LBC1936 and LBC1921, respectively (Table 1; Figure 1D). There was no evidence for pairwise interactions between the three significant SNPs: rs9898 and rs710446 (p = 0.044), rs9898 and rs2731672 (p = 0.14), and rs710446 and rs2731672 (p = 0.40).
Table 1.
Additive Effects of Independent, Genome-wide Significant SNPs in the Lothian Birth Cohorts on Activated Partial Thromboplastin Time
| Gene | Chr. | SNP | MA |
Joint |
LBC1936 |
LBC1921 |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | MAF | BETA | SE | R2 | P | N | MAF | BETA | SE | R2 | P | N | MAF | BETA | SE | R2 | P | ||||
| HRG | 3 | rs9898 | T | 1468 | 0.32 | −0.26 | 0.038 | 0.031 | 1.34 × 10−11 | 987 | 0.32 | −0.20 | 0.047 | 0.018 | 2.24 × 10−5 | 481 | 0.34 | −0.36 | 0.063 | 0.0077 | 1.68 × 10−8 |
| KNG1 | 3 | rs710446 | G | 1477 | 0.40 | −0.36 | 0.037 | 0.060 | 9.52 × 10−22 | 989 | 0.39 | −0.33 | 0.044 | 0.052 | 5.02 × 10−13 | 488 | 0.41 | −0.43 | 0.065 | 0.081 | 1.5 × 10−10 |
| F12 | 5 | rs2731672 | A | 1477 | 0.27 | 0.45 | 0.039 | 0.085 | 2.16 × 10−30 | 989 | 0.26 | 0.49 | 0.047 | 0.096 | 1.59 × 10−23 | 488 | 0.29 | 0.39 | 0.067 | 0.066 | 9.25 × 10−9 |
Results are also shown separately for the Lothian Birth Cohort 1936 (LBC1936) and Lothian Birth Cohort 1921 (LBC1921). The following abbreviations are used: MA, minor allele for the reported effect; MAF, minor allele frequency; BETA, regression coefficient of the trait value on the number of minor alleles; SE, standard error; R2, proportion of variance explained.
Figure 1.
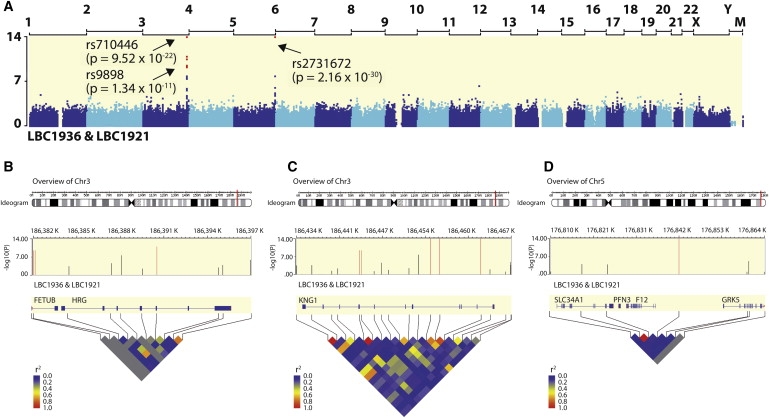
Genomic Overview of the Significant Regions Associated with Activated Partial Thromboplastin Time
(A) Associations were determined for 542,050 SNPs in two cohorts: the Lothian Birth Cohort 1936 (LBC1936; n = 989) and the Lothian Birth Cohort 1921 (LBC1921; n = 488). The x axis represents the position of each chromosome from the p terminus to the q terminus. The y axis depicts p values (-log10(P)). The y axis is truncated at 14: points above this line are placed at the top of the graph and are indicated with arrows and text. Red dots indicate SNPs that surpassed the genome-wide significance threshold (p < 10−8).
(B–D) The genotyped SNPs in the region of significance, the location of surrounding genes (HRG, KNG1, and F12, respectively), and the linkage disequilibrium in the region (HapMap CEU) are shown.
Figure S2 shows that the genetic effects of the most significantly associated SNPs in the three genes—rs2731672 (F12), rs710446 (KNG1), and rs9898 (HRG)—were additive in both LBC1936 and LBC1921. There was a marginally significant effect of dominance deviation in rs2731672 (p = 0.021) and rs9898 (p = 0.024).
There was no evidence for SNP by gender interactions for the three significant SNPs (all p > 0.1). The genetic association results separated by gender are shown in Table S2. The results did not materially change after exclusion of individuals with potentially abnormal aPTT (Table S3).
Beyond association, we showed that, by using the best SNP in each of the three genes associated with aPTT in LBC1936—rs2731672 (F12), rs710446 (KNG1), and rs9898 (HRG)—and their estimated effect sizes, the aPTT phenotype in LBC1921 can be predicted (Figure 2). The Pearson correlation between the values predicted with only the information about the three SNPs and actual aPTT values was r = 0.426 (p = 1.29 × 10−22). The regression coefficient (slope) was 0.991 (standard error [SE] = 0.096), indicating that the prediction was unbiased. Furthermore, we show that F12, KNG1, and HRG form part of the same signaling pathway (Figure S3). Of the ten pathway members, F11 (MIM ∗264900) showed association with aPTT. Using a gene-based approach, we found significant evidence for three independently associated SNPs (empirical set-based p = 0.0005) out of nine F11 SNPs: an intronic SNP rs4253399 (nominal p = 1.8 × 10−5), rs3822058 (nominal p = 0.00010), and rs5971 (nominal p = 0.022).
Figure 2.
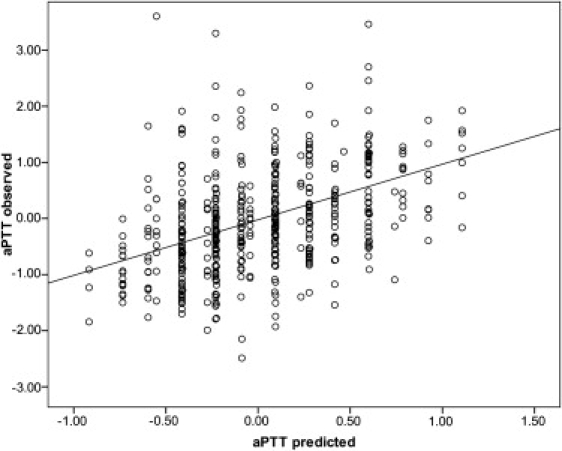
Prediction of the Lothian Birth Cohort 1921 Activated Partial Thromboplastin Time Phenotype
The estimated effect sizes from the best SNP in each of the three genes associated with aPTT in the LBC1936 sample—rs2731672 (F12), rs710446 (KNG1), and rs9898 (HRG)—were used. They were entered as independent variables in a linear regression model to predict aPTT values in LBC1921. The resulting regression coefficients were applied to the SNPs in LBC1921 to create a genetic predictor of aPTT for each person. Here, observed aPTT values are plotted against these predicted values. The observed values of aPTT correlate with their predictions (Pearson correlation = 0.426; R2 = 0.181, p = 1.3 × 10−22). The regression coefficient (slope) was 0.991 (SE = 0.096), indicating that the prediction was unbiased. The y axis was truncated at 3.5, removing three samples from the figure. aPTT values are standardized variables, having been adjusted for age and sex.
For the two nonsynonymous SNP associations in KNG1 and HRG, prediction analysis was undertaken to estimate the effect of SNP variability on protein structure and function. The nonsynonymous change in Kininogen-1 protein (HMWK) (1742T>C, Ile581Thr) was predicted to be damaging with PolyPhen analysis (score = 1.557, where score > 1.3 indicates a hydrophobicity change) but tolerated with SIFT analysis (score = 0.66). There was no predicted effect of missense mutation in HRGP (610C>T, Pro204Ser) because the residue was predicted to be benign with Polyphen (score = 0.102) and tolerated with SIFT (score = 0.73). To extend beyond bioinformatical predictive analysis, experimental work to measure the effect of the nonsynonymous changes on the protein's structure, stability, or function is necessary.
We identified genome-wide significant associations of relatively large effect size between the quantitative trait of aPTT and variants in three genes, F12, KNG1, and HRG, that all have important functional roles in the blood coagulation cascade. The F12 gene encodes the coagulation factor XII precursor. Factor XII is a plasma glycoprotein that participates in the initiation of blood coagulation, activation of the kallikrein-kinin system, fibrinolysis, and the generation of bradykinin (encoded by KNG1) and angiotensin. Factor XII is a known determinant of aPTT. Here we report association of SNPs in this genomic region with aPTT, which may reflect the influence of F12 on aPTT. Our strongest associated SNP, rs2731672, correlates highly with the F12 46C>T polymorphism (rs1801020, R2 = 0.87 CEU HapMap), which has been associated with FXII levels,15 risk of thrombosis including ischemic stroke16 (MIM #601367), and venous thrombosis.17 Analysis of our imputed data also shows a highly significant association with rs1801020 in a joint analysis (R2 = 0.077, p = 1.24 × 10−27) and separately in both cohorts, LBC1936 (R2 = 0.088, p = 2.3 × 10−21) and LBC1921 (R2 = 0.060, p = 4.7 × 10−8) (Figure S2). The A allele (corresponding to the T allele of the 46C>T polymorphism) is associated with an increase in aPTT (prolonged aPTT). This is not the expected biological direction of effect, because a decrease in aPTT (and ratio) is associated with thrombotic diseases.3 However, other prothrombotic phenotypes and coagulation deficiencies, such as lupus anticoagulants, are associated with prolongation (not shortening) of the aPTT in vitro.
The second major aPTT association was with isoform 1 of KNG1, which encodes HMWK. HMWK plays an important role in blood coagulation by positioning prekallikrein and factor XI near factor XII. Defects in KNG1 are the cause of HMWK deficiency (MIM #228960), which is an autosomal recessive coagulation defect. KNG1 gene variants are associated with essential hypertension and blood pressure in the Chinese Han population in a gender-specific manner18 and have a consistent effect on aldosterone response to antihypertensive drug therapy.19 KNG1 knockout mice demonstrate prolonged aPTT and delayed arterial thrombosis.20 Because KNG1 is also associated with blood pressure in the Chinese Han population, we attempted to replicate the findings in the LBC1921 and LBC1936 cohorts. Genotype data for the same SNPs reported in Merkulov et al.20 were available (rs1851665, rs2304456, rs4686799, and rs5030062). We found no association at the single SNP or haplotype level to sitting and standing systolic and diastolic blood pressures with an Omron 705IT monitor (all p values > 0.05) (further details available upon request).
The third major association with aPTT was with HRG, which encodes a histidine-rich glycoprotein that is located in plasma and platelets. HRG can inhibit rosette formation and interacts with heparin, thrombospondin, and plasminogen (as reviewed in 21). HRG's effect on the inhibition of fibrinolysis and the reduction of inhibition of coagulation indicates a potential prothrombotic effect. Mutations in this gene lead to thrombophilia, detected by prolonged aPTT (MIM ∗142640).
A suggestive association of F11 with aPTT was also found. F11 variants have been associated with deep vein thrombosis.22 Defects in coagulation factor XI (MIM #612416) lead to Rosenthal syndrome, a blood coagulation abnormality,23,24 and hypertension induced by pregnancy.24
We have identified single variants in three genes that account for ∼18% of the variance of aPTT. These three genes are implicated in the coagulation cascade, but their relative functional importance has yet to be established. Heritability estimates suggest that there are further contributions to aPTT's genetic variability to be discovered. The variants reported here account for a relatively substantial proportion4,5 of normal variation in aPTT in two age-homogeneous, ethnically homogeneous, and geographically homogeneous samples. The true effect sizes of the real functional variants may be even greater. Further experimental studies are required to establish the mechanisms by which these three genes prolong the aPTT in vitro and influence thrombosis in vivo.
Acknowledgments
We thank the LBC1936 and LBC1921 participants. We thank Janie Corley, Caroline Brett, Caroline Cameron, Michelle Taylor, and Alison Pattie for data collection and data entry. We thank the study secretary Paula Davies. We thank the nurses and staff at the Wellcome Trust Clinical Research Facility, where subjects were tested and the genotyping was performed. We thank Caroline Hayward and Veronique Vitart for advice and Paul Redmond for technical assistance. We also thank the staff at the Department of Haematology, Western General Hospital for the aPTT measurements. We thank the staff at the Lothian Health Board and the staff at the Scottish Council for Research in Education Centre, University of Glasgow. This whole-genome association study was funded by the Biotechnology and Biological Sciences Research Council (BBSRC). The LBC1936 research was supported by a program grant from Research into Ageing and continues with program grants from Help the Aged and Research into Ageing (Disconnected Mind). The LBC1921 data collection was funded by the BBSRC. The study was conducted within the University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology, supported by the BBSRC, Engineering and Physical Sciences Research Council, Economic and Social Research Council, and Medical Research Council, as part of the cross-council Lifelong Health and Wellbeing Initiative. M.L. is a Royal Society of Edinburgh and Lloyds TSB Foundation for Scotland Personal Research Fellow. P.M.V. acknowledges support from the Australian National Health and Medical Research Council.
Contributor Information
Lorna M. Houlihan, Email: lorna.houlihan@ed.ac.uk.
Ian J. Deary, Email: ian.deary@ed.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
MACH 1.0: Markov Chain Haplotyping, http://www.sph.umich.edu/csg/abecasis/MACH/index.html
NCBI Sincle Nucleotide Polymorphism Database, http://www.ncbi.nlm.nih.gov/SNP/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
PLINK Whole Genome Association Analysis Toolset, http://pngu.mgh.harvard.edu/∼purcell/plink
PolyPhen: Prediction of Functional Effect of Human nsSNPs, http://genetics.bwh.harvard.edu/pph/
SIFT: Predicting Amino Acid Changes that Affect Protein Function, http://sift.jcvi.org/
STRING: Functional Protein Association Networks, http://string.embl.de/
References
- 1.Centers for Disease Control and Prevention (CDC) Mortality from coronary heart disease and acute myocardial infarction—United States, 1998. MMWR Morb. Mortal. Wkly. Rep. 2001;50:90–93. [PubMed] [Google Scholar]
- 2.Lowe G.D., Rumley A., Woodward M., Reid E., Rumley J. Activated protein C resistance and the FV:R506Q mutation in a random population sample—associations with cardiovascular risk factors and coagulation variables. Thromb. Haemost. 1999;81:918–924. [PubMed] [Google Scholar]
- 3.Lowe G.D., Haverkate F., Thompson S.G., Turner R.M., Bertina R.M., Turpie A.G., Mannucci P.M. Prediction of deep vein thrombosis after elective hip replacement surgery by preoperative clinical and haemostatic variables: The ECAT DVT Study. European Concerted Action on Thrombosis. Thromb. Haemost. 1999;81:879–886. [PubMed] [Google Scholar]
- 4.Warren D.M., Soria J.M., Souto J.C., Comuzzie A., Fontcuberta J., Blangero J., MacCluer J.W., Almasy L. Heritability of hemostasis phenotypes and their correlation with type 2 diabetes status in Mexican Americans. Hum. Biol. 2005;77:1–15. doi: 10.1353/hub.2005.0034. [DOI] [PubMed] [Google Scholar]
- 5.Souto J.C., Almasy L., Borrell M., Garí M., Martínez E., Mateo J., Stone W.H., Blangero J., Fontcuberta J. Genetic determinants of hemostasis phenotypes in Spanish families. Circulation. 2000;101:1546–1551. doi: 10.1161/01.cir.101.13.1546. [DOI] [PubMed] [Google Scholar]
- 6.Souto J.C., Almasy L., Borrell M., Blanco-Vaca F., Mateo J., Soria J.M., Coll I., Felices R., Stone W., Fontcuberta J., Blangero J. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: The GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am. J. Hum. Genet. 2000;67:1452–1459. doi: 10.1086/316903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deary I.J., Gow A.J., Taylor M.D., Corley J., Brett C., Wilson V., Campbell H., Whalley L.J., Visscher P.M., Porteous D.J., Starr J.M. The Lothian Birth Cohort 1936: A study to examine influences on cognitive ageing from age 11 to age 70 and beyond. BMC Geriatr. 2007;7:28. doi: 10.1186/1471-2318-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deary I.J., Whiteman M.C., Starr J.M., Whalley L.J., Fox H.C. The impact of childhood intelligence on later life: Following up the Scottish mental surveys of 1932 and 1947. J. Pers. Soc. Psychol. 2004;86:130–147. doi: 10.1037/0022-3514.86.1.130. [DOI] [PubMed] [Google Scholar]
- 9.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge D., Zhang K., Need A.C., Martin O., Fellay J., Urban T.J., Telenti A., Goldstein D.B. WGAViewer: Software for genomic annotation of whole genome association studies. Genome Res. 2008;18:640–643. doi: 10.1101/gr.071571.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y., Abecasis G.R. Mach 1.0: Rapid haplotype reconstruction and missing genotype inference. Am. J. Hum. Genet. 2006;S79:2290. [Google Scholar]
- 12.Jensen L.J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 14.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calafell F., Almasy L., Sabater-Lleal M., Buil A., Mordillo C., Ramírez-Soriano A., Sikora M., Souto J.C., Blangero J., Fontcuberta J., Soria J.M. Sequence variation and genetic evolution at the human F12 locus: Mapping quantitative trait nucleotides that influence FXII plasma levels. Hum. Mol. Genet. 2010;19:517–525. doi: 10.1093/hmg/ddp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santamaría A., Mateo J., Tirado I., Oliver A., Belvís R., Martí-Fábregas J., Felices R., Soria J.M., Souto J.C., Fontcuberta J. Homozygosity of the T allele of the 46 C>T polymorphism in the F12 gene is a risk factor for ischemic stroke in the Spanish population. Stroke. 2004;35:1795–1799. doi: 10.1161/01.STR.0000133127.68041.a3. [DOI] [PubMed] [Google Scholar]
- 17.Tirado I., Soria J.M., Mateo J., Oliver A., Souto J.C., Santamaria A., Felices R., Borrell M., Fontcuberta J. Association after linkage analysis indicates that homozygosity for the 46C>T polymorphism in the F12 gene is a genetic risk factor for venous thrombosis. Thromb. Haemost. 2004;91:899–904. doi: 10.1160/TH03-10-0620. [DOI] [PubMed] [Google Scholar]
- 18.Zhao W., Wang Y., Wang L., Lu X., Yang W., Huang J., Chen S., Gu D. Gender-specific association between the kininogen 1 gene variants and essential hypertension in Chinese Han population. J. Hypertens. 2009;27:484–490. doi: 10.1097/hjh.0b013e32831e19f9. [DOI] [PubMed] [Google Scholar]
- 19.Barbalic M., Schwartz G.L., Chapman A.B., Turner S.T., Boerwinkle E. Kininogen (KNG) gene variation has a consistent effect on aldosterone response to antihypertensive drug therapy: The GERA study. Physiol Genomics. 2009;39:56–60. doi: 10.1152/physiolgenomics.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merkulov S., Zhang W.M., Komar A.A., Schmaier A.H., Barnes E., Zhou Y., Lu X., Iwaki T., Castellino F.J., Luo G., McCrae K.R. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111:1274–1281. doi: 10.1182/blood-2007-06-092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones A.L., Hulett M.D., Parish C.R. Histidine-rich glycoprotein: A novel adaptor protein in plasma that modulates the immune, vascular and coagulation systems. Immunol. Cell Biol. 2005;83:106–118. doi: 10.1111/j.1440-1711.2005.01320.x. [DOI] [PubMed] [Google Scholar]
- 22.Li Y., Bezemer I.D., Rowland C.M., Tong C.H., Arellano A.R., Catanese J.J., Devlin J.J., Reitsma P.H., Bare L.A., Rosendaal F.R. Genetic variants associated with deep vein thrombosis: The F11 locus. J. Thromb. Haemost. 2009;7:1802–1808. doi: 10.1111/j.1538-7836.2009.03544.x. [DOI] [PubMed] [Google Scholar]
- 23.Quélin F., Trossaërt M., Sigaud M., Mazancourt P.D., Fressinaud E. Molecular basis of severe factor XI deficiency in seven families from the west of France. Seven novel mutations, including an ancient Q88X mutation. J. Thromb. Haemost. 2004;2:71–76. doi: 10.1111/j.1538-7836.2004.00554.x. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell M., Cutler J., Thompson S., Moore G., Jenkins Ap Rees E., Smith M., Savidge G., Alhaq A. Heterozygous factor XI deficiency associated with three novel mutations. Br. J. Haematol. 1999;107:763–765. doi: 10.1046/j.1365-2141.1999.01769.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.