

Abstract
Detection of recent identity by descent (IBD) in population samples is important for population-based linkage mapping and for highly accurate genotype imputation and haplotype-phase inference. We present a method for detection of recent IBD in population samples. Our method accounts for linkage disequilibrium between SNPs to enable full use of high-density SNP data. We find that our method can detect segments of a length of 2 cM with moderate power and negligible false discovery rate in Illumina 550K data in Northwestern Europeans. We compare our method with GERMLINE and PLINK, and we show that our method has a level of resolution that is significantly better than these existing methods, thus extending the usefulness of recent IBD in analysis of high-density SNP data. We survey four genomic regions in a sample of UK individuals of European descent and find that on average, at a given location, our method detects IBD in 2.7 per 10,000 pairs of individuals in Illumina 550K data. We also present methodology and results for detection of homozygosity by descent (HBD) and survey the whole genome in a sample of 1373 UK individuals of European descent. We detect HBD in 4.7 individuals per 10,000 on average at a given location. Our methodology is implemented in the freely available BEAGLE software package.
Introduction
Identity by descent (IBD) is fundamental to genetics. Two individuals are identical by descent (IBD) at a locus if they share identical genetic material inherited from a common ancestor. Analysis of IBD in pedigree data is important for linkage mapping. In small pedigrees, individuals are closely related and segments of IBD tend to be fairly long (> 10 cM) and are easily detected with the use of dense microsatellite or SNP marker panels. Large pedigrees from founder populations have been more difficult to analyze, partly because the segments of IBD can become quite small, but also because simultaneous analysis of all markers and all individuals is computationally intractable.
The concept of IBD is also important for population genetics, where it is approached quite differently. Whereas IBD in a pedigree is of recent origin, in a population-genetics perspective most IBD will be extremely ancient. For SNPs that have not experienced recurrent mutation, identical alleles are IBD in the population-genetics sense. Such IBD segments will tend to be extremely short, perhaps covering only a single polymorphism or, at most, a few kilobases of DNA. The population-genetics concept of IBD underlies association testing and GWAS.
It is useful to consider an intermediate definition of IBD. A pair of individuals may not know of any relationship between them, yet they may be, for example, twentieth cousins. In general, IBD segments due to sharing from a common ancestor n generations in the past (hence involving 2n meiosis) have expected length 1/(2n) Morgans (M).1 Thus, twentieth cousins, who have common ancestry 21 generations in the past, have IBD segments of average length 1/42 M, or 2.3 cM. We show that IBD segments of this length are detectable in pairs of UK individuals of European descent with dense SNP data and our proposed methodology. As an alternative to the pedigree-based and population-genetics definitions of IBD, we consider “recent IBD,” in which individuals are IBD at a locus if they share an identical haplotype at the locus and the extent of sharing is greater than would be expected by chance, given the population haplotype frequencies. Thus, unlike the population-genetics definition of IBD, the definition that we are using involves recent shared inheritance and sharing that extends beyond the background level of linkage disequilibrium (LD). We find that our method has moderate power to detect IBD segments 2 cM in length. Hence, the target range of shared ancestry with our method is up to 25 generations ago.
Because of its fundamental role in genetics, IBD has many uses. One application of recent IBD is population-based linkage analysis,2 also known as IBD mapping.3,4 IBD mapping is similar to linkage mapping, except that IBD mapping does not require pedigree information. One looks for greater levels of IBD sharing in case-case pairs than in case-control or control-control pairs. IBD mapping could prove useful in mapping disease-susceptibility genes with allelic heterogeneity (e.g., multiple rare causal variants) that are difficult to map with association analysis. It is easier and less expensive to collect population samples than to collect families for a traditional linkage analysis. Also, population samples have the advantage of representing, in essence, very extended pedigrees, which have power advantages over small pedigrees.5 Similarly, one can use homozygosity by descent (HBD) to map rare recessive mutations of strong effect.
Another application of recent IBD is genotype-imputation and haplotype-phase inference.6 Comparison of imputation and phasing accuracy in unrelated individuals and parent-offspring trios shows that the use of IBD data can substantially reduce switch-error rates and imputation-error rates.7 Recently, Kong et al.6 showed that exceptionally accurate phasing is possible when multiple individuals have inherited a genomic segment identically by descent. IBD can also be used to detect phasing errors and structural variants.8
Several methods have been proposed for detection of recent IBD and HBD. One IBD detection criterion is the length of segment over which genotypes are compatible with IBD or HBD.6,9–14 For diallelic SNP markers, genotypes are compatible with IBD unless the two individuals have discordant homozygous genotypes. Thus, long stretches of IBD compatibility are needed before recent IBD can be declared with confidence. Typically, the length threshold used is 5–10 Mb or 5–10 cM. One variant of this approach is the GERMLINE program,8 which considers identity of haplotypes rather than of genotypes. The advantage of using haplotypes is that two haplotypes are less likely than two genotypes to be consistent with IBD by chance. The disadvantage of using haplotypes is that sensitivity to detect IBD strongly depends on accurate haplotype inference because phase uncertainty is not modeled. One notable feature of the GERMLINE software is its high computational efficiency. In general, searching for IBD between all pairs of individuals in a sample has computational time that is quadratic in the number of individuals. However, the GERMLINE software is able to solve this problem in linear computing time by partitioning the genome into windows and exploiting the limited number of distinct haplotypes observed in each window. Another approach to detecting IBD is implemented in the PLINK software package.2 PLINK applies a hidden Markov model (HMM) to the IBD process, while assuming independence (linkage equilibrium) among markers. Albrechtsen et al.4 extend the PLINK approach to incorporate LD via haplotype probabilities for pairs of SNPs. Although this approach is a clear improvement on the assumption of no LD, we expect that the pairwise approach may not adequately correct for LD in some genomic regions with high levels of LD.
The method that we present for detecting IBD in “unrelated” individuals accounts for background levels of LD by using a comprehensive LD model incorporating data from all markers in a region. Fully accounting for LD is important, because in regions of high LD many pairs of individuals will share common haplotypes, which are not really “IBD” in the sense defined here (i.e., inherited from a recent common ancestor). Thus, including such spurious IBD would add noise when one is looking for recent IBD for the purpose of IBD mapping or improved haplotype phase inference. Incorporation of a comprehensive LD model reduces false-positive IBD detection, thus giving the ability to identify much smaller IBD segments. An alternative approach to dealing with LD is to prune markers, which is the approach taken by PLINK.2 However, we show that this approach leads to decreased power to detect short segments of IBD.
Material and Methods
Data
We analyzed genotype data from the 1958 British Birth Cohort (58BC).15 We used a prerelease version of BEAGLECALL16 to call SNP genotypes from allele signal-intensity data from the Affymetrix 500K chip and the Illumina 550K chip generated by the Wellcome Trust Case Control Consortium17 and the Wellcome Trust Sanger Centre as described previously.16 BEAGLECALL utilizes LD as well as the allele signal intensities to obtain high genotype-call accuracy. After calling genotypes, all genotypes with posterior probability < 0.985 for Illumina or < 0.975 for Affymetrix were set to missing, and all SNPs with missing data rate > 0.015 for Ilumina or > 0.025 for Affymetrix were excluded. Also, SNPs with minor allele frequency < 0.01 were excluded. After filtering, there were 399,651 autosomal SNPs in the Affymetrix data and 511,942 autosomal SNPs in the Illumina data. 1373 individuals that passed light data quality filters16,17 and that were genotyped on both platforms were included in the analyses.
As it is not yet computationally feasible to apply the proposed IBD detection method to all pairs of samples on a genome-wide scale for large sample sizes, we selected four regions to examine in detail. We chose two regions with a lower density of markers and two regions with a higher density of markers, from chromosomes 1 and 2. Each region was chosen to contain approximately 1000 SNPs in each panel, and to cover 15 cM (low-density regions, 1A and 2A) or 5 cM (high-density regions, 1B and 2B). Details of these regions are shown in Table 1. Estimates of genetic distance are taken from HapMap Phase 2.18
Table 1.
Four Genomic Regions Selected for Evaluation of IBD Detection
| Region | Chromosome | Build 35 Position (Mb) | Genetic Position (cM) | Affymetrix 500K Markers/cM | Illumina 550K Markers/cM | Combined Markers/cM |
|---|---|---|---|---|---|---|
| 1A | 1 | 5.8–14.0 | 15–30 | 54 | 94 | 136 |
| 1B | 1 | 180.7–188.1 | 200–205 | 208 | 205 | 390 |
| 2A | 2 | 5.1–11.1 | 10–25 | 76 | 91 | 153 |
| 2B | 2 | 50.4–56.3 | 75–80 | 201 | 276 | 441 |
The table shows the position of each region and the density of SNPs in each of the two panels plus the density of SNPs in the union of the two panels.
Calculation of IBD Probabilities
Our approach to calculating IBD probabilities for phased haplotypes has been outlined previously.1 Extending this to unphased genotypes is nontrivial, because it is necessary to simultaneously perform IBD probability calculation and haplotype phasing in order to account for haplotype-phase uncertainty.
In order to calculate IBD probabilities on dense genotype data, one needs to model both LD between markers and IBD between individuals. Thus, the model underlying the IBD probability calculation is a hidden Markov model (HMM) comprising two connected parts: an IBD model and an LD model.
The IBD model has two states: IBD (1) and non-IBD (0). When calculating the probability of IBD between two individuals at a locus, we ignore the possibility that one or both of the individuals may be inbred and hence HBD at the locus (this assumption can be checked in advance), and we also ignore the possibility that individuals may be bilinearly related (i.e., that each individual is related to the other through both their mother and their father, as, for example, siblings are). Although “unrelated” individuals may be distantly related through more than one pair of parents, it is unlikely that they will be IBD through more than one pair of parents at the same locus.
The IBD model is Markov. This is an approximation that has previously been used successfully in inferring relationships,19 estimating HBD,20 and estimating inbreeding coefficients.21
The prior probabilities that we use for the IBD model for a pair of individuals are: IBD at a locus with probability 0.0001, and 1 cM expected IBD tract length. Equivalently, the transition rate from IBD to non-IBD (t10) is 1 per cM, whereas the transition rate from non-IBD to IBD (t01) is 0.0001 per cM (tij is the transition rate from IBD state i to j). In our experience, these parameters are appropriate for samples of unrelated individuals of Northern European ancestry (such as the 58BC data) and roughly match the levels of IBD found in such samples (see Results).
The LD model is the localized haplotype cluster HMM implemented in BEAGLE.22,23 We combine the LD and IBD model into a single HMM as described below. Although we define the IBD probabilities for haplotypes (rather than directly for unphased genotypes) and the haplotype phases are unknown, the HMM calculations integrate over all possible phasings.
Consider one pair of individuals. When the IBD state is 0 (non-IBD), the probability of the four haplotypes is found by multiplying the probabilities of the individual haplotypes (we assume Hardy-Weinberg equilibrium). The probability of each of the haplotypes is found by multiplying the corresponding transition probabilities of the LD model. An IBD state of 1 implies one pair of IBD haplotypes (we are excluding the possibility of more complex IBD patterns). The haplotype phase of individuals is unknown, but individuals' haplotypes are stored as ordered pairs within the algorithm. Because we have no information about parental origin of the haplotypes (i.e., which haplotype is maternally inherited and which paternally inherited), there is symmetry; if the haplotypes for one individual are labeled (H1, H2), then P((H1,H2)) = P((H2,H1)). If individual 1 has ordered haplotype pair (H1, H2) and individual 2 has ordered haplotype pair (H3, H4), we define an IBD state of 1 to imply that haplotypes H1 and H3 are IBD at the given marker position. If the two individuals have data consistent with IBD, the two putatively IBD haplotypes will, during sampling from the posterior distribution, take the role of the H1 and H3 haplotypes.
The probability of the set of four haplotypes is the probability of the pair of IBD haplotypes (defined next) multiplied by the probabilities of the other two haplotypes (obtained from the LD model as when the IBD state is 0). If the pair of IBD haplotypes pass through the same path in the LD model (i.e., the same series of edges22,23), the probability of the pair of haplotypes is equal to the probability of a single haplotype obtained by multiplying (once) the corresponding transition probabilities from the LD model; that is, the two IBD haplotypes contribute to the overall probability as if they were only a single haplotype. When the two IBD haplotypes pass through different edges of the LD model, the minimum of the two corresponding transition probabilities is used. The reason for using the minimum of the two edge probabilities is to avoid inflating the posterior probability of IBD.
To allow for possibility of genotype error, we incorporate a genotype-error probability ɛ (we use ɛ = 0.005). If the (possibly imputed) alleles of the two IBD haplotypes are not identical at a SNP, we include a factor ɛ, whereas if they are identical, we include a factor 1− ɛ.
An HMM is defined by its state space, initial probabilities, transition probabilities, and emission probabilities (probability of observing the data given the model state). For our model of IBD and LD, the state at each marker position is two ordered pairs of haplotype states (one ordered pair for each of the two individuals for whom the IBD probabilities are being computed) plus the IBD status (i = 0/1). A haplotype state, e, is a localized haplotype cluster in the LD (BEAGLE) model. We can write the joint LD and IBD state as (e1,e2,e3,e4,i). The model runs along the chromosome, so the initial probabilities are defined at the first marker position. At the initial state, P(i = 0) = 1, and the remaining four components are with initial probabilities obtained from the LD model. We intend to change the initial IBD prior probability so that it matches the steady-state IBD prior probability (i.e., P(i = 1) = t01/(t01+t10)) in a future version of the software. The transition probabilities are:
in which a1′ and a3′ are the alleles of haplotypes H1 and H3 at the next position (corresponding to haplotype states e1′ and e3′), sij is the probability of transitioning from IBD state i at the current position to IBD state j at the next position (s11 = exp(-t10d), s00 = exp(-t01d), s10 = 1 - s11, s01 = 1 - s00, in which d is the genetic distance between the current and next positions; these expressions assume negligible probability of more than one change in IBD status between two markers and are therefore appropriate for closely spaced markers), and P(e→e′) are transition probabilities between haplotype states (from the LD model). The emission probabilities are derived from the BEAGLE LD model and are either 0 (haplotype states not consistent with genotypes) or 1 (haplotype states consistent with genotypes). This formulation is quite general and could be used with other LD models, such as the fastPHASE model.24,25
We have experimented with using a weighted mean rather than a minimum in the transition probabilities above. We found that using the mean can cause false-positive IBD because when one haplotype is very rare, the difference between the IBD and non-IBD probabilities can become very large (because the very small haplotype probability occurs in the non-IBD probability but essentially disappears into the mean in the IBD probability). As a result, in some regions several pairs of individuals were reported to have very small (< 0.1 cM) IBD segments. We found that using the minimum avoids this problem. However, the transition probabilities from a state should sum to 1. They do sum to 1 if the mean is used, but not if the minimum is used (in which case they sum to < 1). Thus, using a minimum downweights the probabilities when the two possibly IBD haplotypes are traveling through different paths of the model, which is useful. We plan to investigate this issue further in future research.
To demonstrate these probability calculations, we give a small example on four SNP markers (however, note that the method is designed for dense SNP data with thousands of markers per chromosome). Again, we assume that the haplotypes are known. However, in calculating the posterior probability of IBD, the HMM method will account for haplotype uncertainty (the full calculation of IBD probabilities for this example is not shown). The LD model for the four SNPs is taken from previous work23 and is shown in Figure 1. The transition probabilities for the model are P(eA) = 0.518, P(eB) = 0.482, P(eC) = 0.627, P(eD) = 0.373, P(eE) = 1.0, P(eF) = 0.490, P(eG) = 0.510, P(eH) = 1.0, P(eI) = 0.194, P(eJ) = 0.806, P(eK) = 1.0, P(eL) = 1.0. Individual 1 has haplotypes H1 = 1 1 1 1 and H2 = 1 2 2 1; individual 2 has haplotypes H3 = 2 1 1 1 and H4 = 2 1 2 2. We calculate the probability of the four haplotypes given that haplotypes H1 and H3 are IBD at all four marker positions.
Figure 1.
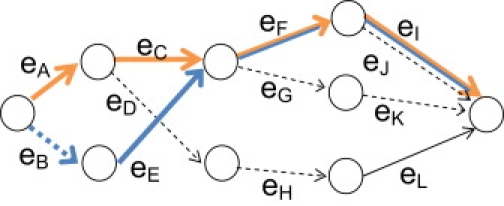
Example of an LD Model on Four SNPs
SNP 1 is represented by edges eA and eB; SNP 2 by edges eC, eD, eE; SNP 3 by edges eF, eG, eH; and SNP 4 by edges eI, eJ, eK, eL. For each SNP, allele 1 is represented by a solid line, whereas allele 2 is represented by a dashed line. Haplotype H1 (1 1 1 1) follows the orange path (eA, eC, eF, eI), and haplotye H2 (2 1 1 1) follows the blue path (eB, eE, eF, eI).
Finally,
Informally, when the probability of the data is much higher under IBD than under non-IBD (high enough to overcome the low prior probability of IBD), the posterior probability of IBD will be high.
The estimation of the IBD proceeds by first building the LD model from the unphased genotypes by using ten iterations of the model-building algorithm to obtain convergence.23 We then add the IBD model to the LD model and use the forward-backward algorithm for HMMs26,27 to obtain posterior probabilities of IBD for each pair. Our software also reports the most likely haplotype phasing given IBD, which can be useful for phasing related individuals. The procedure may be repeated several times with the use of different random number seeds, with the maximum posterior IBD probability from the multiple runs used. This avoids false negatives due to the fitted LD model converging to a local maximum that does not allow the haplotypes to follow their true IBD configuration. In this study, we use ten runs for IBD probabilities and five runs for HBD probabilities (see below).
Constructing the LD model takes the same amount of computational time as it would to phase the data set by using BEAGLE, which is relatively fast.23 However, with n individuals, there are on the order of n2 potential pairs on which to calculate IBD probabilities, thus increasing the total computation time, relative to phasing, by the order of n. Thus, it is not currently feasible to compute IBD probabilities on all pairs of individuals over the whole genome in a large data set with thousands of individuals.
Calculation of HBD probabilities involves only two haplotypes (from a single individual), but the basic principle is the same. The probability of the two haplotypes given that they are non-HBD is found by multiplying the two haplotype probabilities together. The probability of the two haplotypes given HBD is the same as the probability of two haplotypes given IBD, as described above.
For HBD, the basic unit is individuals, rather than pairs of individuals. Thus, estimating HBD probabilities for all individuals takes only slightly longer than phasing all individuals in a data set. We have estimated HBD probabilities on all individuals from several case-control cohorts from the Wellcome Trust Case Control Consortium17 with approximately 5000 individuals genotyped on 400,000 autosomal SNPs, thus demonstrating that our HBD detection method can be applied to large genome-wide association studies. Genome-wide HBD could be useful for gene mapping in diseases with rare recessive variants of strong effect.
We define as IBD or HBD any position at which the corresponding IBD or HBD probability exceeds 0.5. To define the length of an IBD or HBD region, we measure the genetic length from the first position at which the pair is IBD or the individual is HBD to the last position before the IBD or HBD probability drops below 0.5.
Comparison with Other Programs
We tested our method (implemented in BEAGLE) against GERMLINE version 1.4.08 and PLINK version 1.07,2 two existing state-of-the-art programs for IBD detection. We also attempted to include RELATE4 in our comparisons. However, we were unable to successfully run this program. We ran GERMLINE with default settings (a maximum of two mismatched homozygote markers in a slice for it to be considered a match, and slice size of 128 markers), except that we adjusted the minimum length of reported IBD segments, as described in the Results (the default is 5 cM). For PLINK, we followed the method for pruning SNPs suggested in the PLINK documentation for shared segment analysis (SNPs with > 1% missing genotypes and < 5% minor allele frequency removed, then pairwise LD-based pruning with window size 100, step size 25, and r2 threshold 0.2). The resulting numbers of SNPs remaining in each region described in Table 1 were 317 in 1A, 124 in 1B, 314 in 2A, and 154 in 2B and 7103 on chromosome 1 (reduced from 39146). We used the default parameters for segment detection (minimum 100 SNPs in a shared segment and minimum length 1000 kb), and we also ran analyses using relaxed settings, which were a minimum of 20 SNPs in a shared segment of length at least 200 kb. PLINK shared-segment analysis also requires estimates of kinship, which should usually be obtained from genome-wide data. Because we were using modified data, either with artificially inserted IBD or with composite individuals for removal of IBD, genome-wide kinship estimates were not relevant. Instead, we input as estimated kinship () the value 0.0036, which is the average value seen genome-wide in the 58BC data.
Construction of Composite Individuals for Estimation of False-Positive Rate
In order to estimate the false-positive rate for detected IBD, we created composite individuals whose genotype data is composed of a sequence of 0.2 cM segments copied from different individuals. By construction, the composite individuals will not share an IBD tract longer than 0.2 cM with any other individual in the data set. Consequently, any detected IBD sharing involving a composite individual that is substantially longer than 0.2 cM will be a false positive. We use composite individuals instead of sampled individuals to estimate the false-positive rate for IBD detection because it is not possible to be certain whether a detected IBD segment in a pair of population samples is a false or a true positive, because it is possible that the individuals sharing the segment are distantly related.
A subset of 100 58BC individuals from the Illumina 550K chip chromosome 1 data were selected and used to create ten composite individuals. The 100 individuals were divided into ten sets of ten individuals. Each set of ten samples was used to create one composite individual as follows: First, a random offset of c cM (0 ≤ c < 0.2) was selected for the composite individual. Then the samples were indexed as 1, 2, …, 10, and for K = 1,2, … the genotype data for sample ((K − 1) modulo 10) + 1 was used in the interval from (c + (K − 2)/5) ≤ x < (c + (K − 1)/5) cM. Thus, the first sample's genotype data were used in the interval 0 ≤ x < c cM, the second sample's genotype data were used in the interval c ≤ x < (c + 0.2) cM, the third sample's genotype data were used in the interval (c + 0.2) ≤ x < (c + 0.4) cM, and so on. At the eleventh segment, the sample index wraps around to 1, and the first sample's genotype data are used in the interval (c + 1.8) ≤ x < (c + 2.0) cM.
Construction of Artificial IBD and HBD for Estimation of Power
In order to calculate power of IBD or HBD detection, we constructed artificial IBD and HBD as follows. We took the 58BC data and added to them HapMap Phase 2 CEU phased parental genotypes, considering only SNPs genotyped in both sets of individuals. The HapMap Phase 2 CEU genotypes are accurately phased with the use of trio data, which allowed us to copy a haplotype from one individual into another to create artificial IBD of given segment size. To create IBD, we copied a haplotype from one parent into the other for each parent pair, whereas to create HBD, we copied a haplotype onto the other haplotype from the same individual. Although it was essential to have phased haplotypes to create realistic IBD, we wished to test our method on detecting IBD in unphased genotypes, so we randomized the genotype order to create unphased data after adding the IBD or HBD.
We added IBD or HBD segments of lengths 1, 2, 3, 4, and 5 cM. The starting position of the IBD or HBD segment within the region was random, except that we avoided placing the region within the first or last 50 markers of the region. In total, 120 IBD segments of each length were created (30 parent pairs times four regions), whereas 240 HBD segments of each length were created (60 parents times four regions). An IBD or HBD segment in a region was scored as detected if the pair of samples sharing the segment was reported to be IBD anywhere in the region.
Estimation of False Discovery Rates
The false discovery rate is the proportion of discovered IBD segments that are false positives. Consider a fixed interval of IBD sizes. Let T be the true rate of IBD segments with length in this interval in the population (all rates are per pair of individuals, per locus). Let F be the rate at which IBD segments in this length range are falsely predicted by the method under consideration in pairs of individuals with no IBD. Let P be the power to detect IBD of the specified length. Let D be the rate at which IBD segments in this length range are discovered (including false and true positives). In a data set with some IBD, the rate of false-positive discoveries will be (the rate of non-IBD multiplied by the false-positive rate) and the rate of true discoveries will be (the rate of IBD multiplied by the power). The rate of IBD discovery includes false and true discoveries, and is thus . Given estimates of F, P, and D, one can solve for an estimate of T: . The false discovery rate is estimated by . In our analyses, we estimate F and P for each method. In particular, we consider IBD segment sizes in the range x to x + 1 cM for x = 1, 2, 3, 4. For F, we use the estimated false-positive rate for segments of size x to x + 1 (obtained by subtracting the false-positive rate for segments with length ≥ (x + 1) from the false-positive rate for segments with length ≥ x). For P, we use an average of the power to detect IBD of size x and the power to detect IBD of size (x + 1). We estimate D only for the BEAGLE IBD method, using the rate of IBD detected of size between x and x+1 per locus, which is , in which the are the lengths of the detected segments that are within the size range x to x + 1 cM, “npairs” is the number of pairs interrogated, and “total length” is the length in cM of the interrogated region. T should not depend on the method, and because we have estimates of D for BEAGLE only, we use these to estimate T.
Results
Estimation of IBD False-Positive Rate
We combined chromosome 1 genotype data from the Illumina 550K chip for ten composite samples from the 58BC (see Material and Methods) with1323 individuals from the 58BC cohort. The 1323 58BC samples did not include any of the 100 individuals used to construct the ten composite samples. We then compared three IBD-detection methods (BEAGLE, PLINK, and GERMLINE) for this combined sample. Results are shown in Table 2. BEAGLE's false-positive rate is uniformly lower than that of GERMLINE and PLINK. Although these false-positive rates are very small so that differences might be thought to be of no consequence, they are in fact very important. The rate of true-positive IBD signals in an outbred population sample will also be extremely small, and thus the false-positive rate has a large impact on the false discovery rate (proportion of reported signals that are false), as we demonstrate below.
Table 2.
False-Positive Rates for BEAGLE, GERMLINE, and PLINK for Illumina 550K European Data
| Sizeb |
BEAGLE |
GERMLINE |
PLINK (Default) |
PLINK (Relaxed)a |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of Segments | No. of IBD SNPsc | FP Rated | No. of Segments | No. of IBD SNPsc | FP Rated | No. of Segments | No. of IBD SNPsc | FP Rated | No. of Segments | No. of IBD SNPsc | FP Rated | |
| ≥ 0.5 cM | 9 | 1009 | 1.9 × 10−6 | 2202 | 581010 | 1.1 × 10−3 | 19 | 2118 | 2.2 × 10−5 | 100 | 7173 | 7.6 × 10−5 |
| ≥ 1 cM | 1 | 170 | 3.3× 10−7 | 1202 | 365730 | 7.0 × 10−4 | 19 | 2118 | 2.2 × 10−5 | 97 | 7079 | 7.5 × 10−5 |
| ≥ 2 cM | 0 | 0 | 0 | 118 | 43786 | 8.4 × 10−5 | 19 | 2118 | 2.2 × 10−5 | 76 | 6077 | 6.4 × 10−5 |
| ≥ 3 cM | 0 | 0 | 0 | 14 | 3839 | 7.4 × 10−6 | 18 | 2016 | 2.1 × 10−5 | 41 | 3754 | 4.0 × 10−5 |
| ≥ 4 cM | 0 | 0 | 0 | 8 | 1398 | 2.7 × 10−6 | 11 | 1273 | 1.4 × 10−5 | 20 | 1874 | 2.0 × 10−5 |
| ≥ 5 cM | 0 | 0 | 0 | 8 | 1398 | 2.7 × 10−6 | 4 | 439 | 4.7 × 10−6 | 9 | 794 | 8.4 × 10−6 |
Detected IBD on chromosome 1 in 13,275 pairs constructed so as to have no IBD.
See Material and Methods for parameters used in the relaxed PLINK run.
Reported size of detected segments.
Sum of numbers of SNPs covered by detected segments within this size category.
False-positive rate is (no. of IBD SNPs)/(nSNPs × npairs), where “nSNPs” is 39,146 for BEAGLE and GERMLINE and 7103 for PLINK (SNPs on chromosome 1) and where “npairs” is 13,275 (number of pairs tested).
IBD and HBD Power
We created artificial IBD as described in Material and Methods, and we estimated the power to detect this IBD by using BEAGLE, GERMLINE, and PLINK. For IBD power with BEAGLE, we investigated four regions of the genome (see Table 1) and three panels of SNPs (Affymetrix 500K, Illumina 550K, and the union of these two panels). From Figure 2, we can see that power is fairly constant over the four regions, with the low density regions (1A and 2A) having lower power for the Affymetrix 500K data but not for the other two SNP panels. In the 58BC data, power to detect IBD is high for segments of size 3 cM and larger (using the Illumina 550K panel). Power to detect segments of size 2 cM is 50%, whereas power to detect segments of size 1 cM is only 10%–20%. Adding the Affymetrix data to the Illumina data does not increase power substantially over the use of only the Illumina data. It seems that the Illumina 550K chip already has good coverage of all common variants. Most SNP discovery has been performed in small samples, biasing it toward common variants, and common variants are also favored on the SNP arrays because they tend to have higher power in association studies; thus, the SNP arrays have a frequency spectrum that is biased toward common variants. Consequently, one would expect that it would be possible to increase resolution and power to detect IBD by including genotype data for a large number of rare variants.
Figure 2.
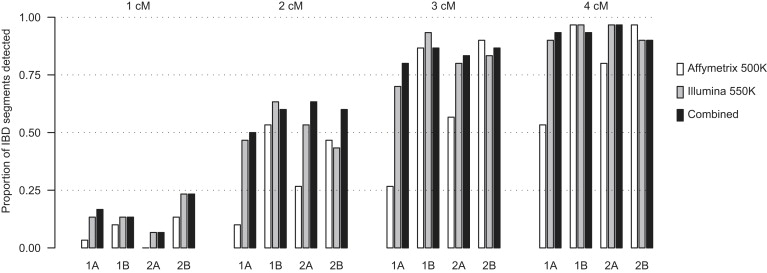
Power to Detect IBD with BEAGLE
Four sizes of IBD segments are considered, and these are labeled at the top of the plot. Four different regions of the genome are interrogated, and these are labeled at the bottom of the plot. Two SNP arrays plus their union (“Combined”) are considered for each segment size and region. Each bar is the proportion detected out of 30 artificial IBD segments.
We compared power of BEAGLE, GERMLINE, and PLINK on the Illumina data, aggregating results over the four regions. Table 3 shows the results. To ensure that GERMLINE would have the opportunity to detect regions as short as 1 cM, we ran GERMLINE with a minimum length threshold equal to 0.2 cM smaller than the size of the inserted IBD (e.g., 0.8 cM minimum when detecting the 1 cM regions, 1.8 cM when detecting the 2 cM regions). This will tend to overestimate GERMLINE's power for the small segments, because one would normally run this program with a much higher threshold, such as the default 5 cM. With low thresholds on segment size in GERMLINE, the false-positive rates are quite high (Table 2), and hence the false discovery rate will also be high (false discovery rates are given below). We see that, except for the short regions (1 cM) with GERMLINE, the power of BEAGLE is significantly higher than that of GERMLINE or PLINK, even for the larger 4 cM regions that are relatively easy to detect, and despite BEAGLE's much lower false-positive rate.
Table 3.
Comparison of Power across BEAGLE, GERMLINE, and PLINK for Illumina 550K European Data
| Size of Artificial IBD | BEAGLE | GERMLINE | PLINK (Default) | PLINK (Relaxed)a |
|---|---|---|---|---|
| 1 cM | 14 | 28 | 0 | 1 |
| 2 cM | 52 | 47 | 0 | 13 |
| 3 cM | 82 | 60 | 7 | 46 |
| 4 cM | 95 | 67 | 46 | 80 |
| 5 cM | 98 | 63 | 71 | 90 |
Reported power is the percentage of artificial IBD segments detected.
See Material and Methods for parameters used in the relaxed PLINK run.
Figure 3 shows HBD power with BEAGLE. For HBD power, we investigated the same regions and panels. Comparing Figure 3 to Figure 2, we see that HBD power is somewhat higher than IBD power. This is not surprising, given that there is no haplotype-phase uncertainty in the HBD segments. In Figure 2 and Figure 3, we can see several instances where the estimated detection power decreases with the use of the combined data-set versus only one of the panels. Stochastic differences in the population haplotype-frequency model between the different marker sets could explain the occasional lower estimated detection power in the combined data.
Figure 3.
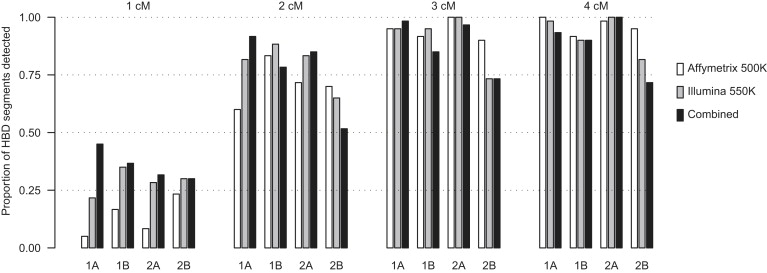
Power to Detect HBD with BEAGLE
Four sizes of HBD segments are considered, and these are labeled at the top of the plot. Four different regions of the genome are interrogated, and these are labeled at the bottom of the plot. Two SNP arrays plus their union (“Combined”) are considered for each segment size and region. Each bar is the proportion detected out of 60 artificial HBD segments.
With no phase uncertainty, Browning1 reported approximately 80% power to detect IBD segments of size 1 cM. In contrast, we find IBD power of approximately 15% and HBD power of approximately 25% for this size region. Phase uncertainty reduces IBD-detection power. However, it should not affect HBD-detection power. The main explanation for this difference is the difference in prior IBD probabilities between the two studies, although several other factors may have contributed to the difference. Browning1 used a larger prior IBD probability, of 0.001, as compared to the current study (0.0001). To investigate this factor, we reran HBD detection, using this higher prior HBD probability for HBD segments of size 1 cM on Illumina data in region 1A. This change to the prior increased the rate of detection of 1 cM HBD segments to 50% from 22%. Thus, our conservative choice of prior in this work explains a large part of the difference in results between this work and the earlier work.1 Clearly, the choice of prior plays an important role in the trade-off between power and false-positive IBD detection. The choice of marker panel and the accuracy of the genetic map could also be contributing factors. Browning1 used simulated data and data from the Affymetrix 500K panel over chromosomes 1 and 22. Differences in marker density and other characteristics of the marker panel certainly affect power to detect IBD. However, Browning1 found power > 70% for 1 cM segments over a range of simulated marker densities and over the two chromosomes.
As well as being able to detect the presence of IBD segments, we wish to estimate their endpoints. Figure 4 shows the amount of over- or underestimation of the endpoints for detected IBD segments with BEAGLE. Here, the definition of the boundary of the estimated IBD segment is given as the first position for which the posterior IBD probability exceeds 0.5 through to the last such contiguous position. The estimated IBD segment tends to be smaller than the actual IBD segment. Where the estimated IBD segment extends into the surrounding non-IBD background, the length that it does so is typically fairly short. If one changes the definition of the estimated IBD region to include positions on either side until the posterior IBD probability first drops below 0.1, the picture reverses (data not shown), with amounts of missed IBD tending to be significantly smaller than amounts of overestimated IBD. With this alternative definition, for detected IBD segments of size 1 cM, the amount of missed IBD has median 0.008 cM and maximum 0.13 cM. Whereas BEAGLE tends to underestimate the lengths of the IBD regions, PLINK and GERMLINE tend to overestimate, by an average of 0.2 to 0.6 cM for PLINK (depending on region size and default or relaxed settings) and 0.6 to 0.8 cM for GERMLINE. It is not surprising that methods with a relatively high false-positive rate will tend to overestimate IBD-segment size.
Figure 4.
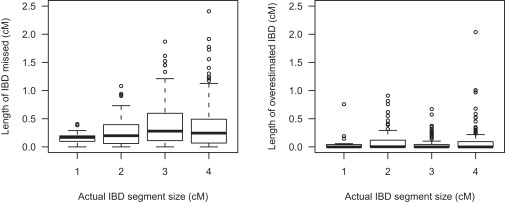
Under- and Overestimation of IBD Segments Detected in Illumina 550K Data with BEAGLE
For detected IBD segments of given size (x axis), the left plot shows the amount of the IBD segment with posterior IBD probability < 0.5, whereas the right plot shows the distance over which the posterior IBD probability remained > 0.5 beyond the boundaries of the IBD segment. The plots are box plots: the thick black line gives the median, the box gives the upper and lower quartiles, the “whiskers” extend to the furthest data point that is no more than 1.5 times the interquartile range from the box, and outlying points beyond the whiskers are individually plotted.
IBD and HBD Discovery in the 1958 British Birth Cohort
We tested 100,000 randomly selected pairs of individuals from the 58BC data for IBD in the four regions with BEAGLE. Figure 5 shows the lengths of the detected segments. For this histogram, we define estimated IBD length as the length over which the posterior IBD probability remains above 0.5. This will tend to slightly underestimate the length of the actual IBD segment (see Figure 4). All lengths greater than 5cM (from regions 1A and 2A only) are truncated to 5cM in the histogram, for comparability with results from regions 1B and 2B, which have total length 5cM.
Figure 5.
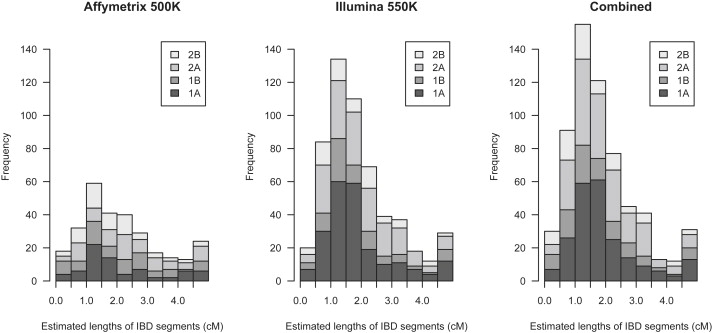
Estimated Lengths of IBD Segments Detected in 58BC Data with BEAGLE
The rightmost bar in each plot includes all estimated segment lengths > 4.5 cM. IBD segments were detected in the four regions described in Table 1. The left panel shows lengths of segments detected with the use of Affymetrix 500K data, the center panel shows lengths of segments detected with Illumina 550K data, and the right panel shows lengths of segments detected with the union of the two SNP chips. A total of 100,000 randomly selected pairs of individuals were analyzed.
The mean length of IBD segments detected with the Affymetrix data is 2.26 cM, and there are 287 detected segments (total length 647 cM); the mean length of IBD segments detected with the Illumina data is 1.99 cM, and there are 552 detected segments (total length 1098 cM); the mean length of IBD segments detected with the combined data is 1.93 cM, and there are 616 detected segments (total length 1188 cM). Thus, detection of recent IBD is significantly (approximately 70%) better with Illumina 550K data than with Affymetrix 500K data for the four regions examined, and detection of recent IBD is slightly (approximately 8%) better with the combination of the two sets of data compared with Illumina 550K data only.
Figure 6 directly compares lengths of IBD segments found by using the Affymetrix 500K and Illumina 550K platforms. In the left panel, we see that where an IBD segment is found by using both platforms, the estimated lengths are in good agreement (the correlation is 0.94), although the estimated lengths from Illumina 550K tend to be slightly higher than those from the Affymetrix 500K data for these segments (mean 2.82 cM for Illumina 550K and 2.70 cM for Affymetrix 500K). In the center and right panels, we see that the IBD segments not found by using one platform tend to be short, although even some IBD segments as large as 5 cM are found by using one platform but not the other. The mean length of IBD segments found by using Affymetrix data but not Illumina data is 1.4 cM, whereas the mean length of IBD segments found by using Illumina data but not Affymetrix data is 1.6 cM.
Figure 6.
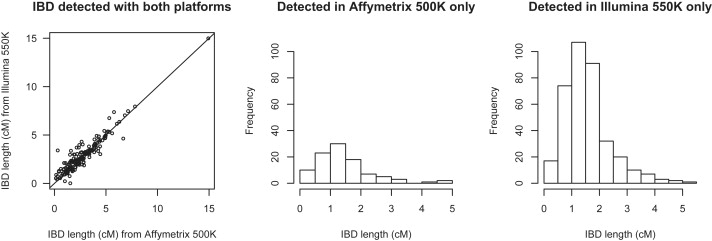
Comparison of Lengths of IBD Segments Detected with BEAGLE with Data from Both Platforms or with Data from One Platform Only
The results are based on IBD detected in 100,000 random pairs of individuals from the 58BC data in four regions (see Table 1). The left panel shows detected lengths on both platforms of IBD segments detected with the use of both the Affymetrix 500K and the Illumina 550K data (there are 188 such segments). The center and right panels show distributions of detected lengths of IBD segments found with the use of Affymetrix 500K data but not with Illumina 550K data (center panel; 99 segments) or with Illumina 550K data but not with Affymetrix 500K data (right panel; 364 segments).
Overall, we tested 100,000 pairs over 40 cM (the combined length of the four regions), and we found 1188 cM IBD with the combined data, which corresponds to an average rate of detected IBD of 3.0 per 10,000 (2.7 per 10,000 with the Illumina data). This estimate is based on only 40 cM selected from the genome, so these rates are imprecise estimates of genomic rates of IBD detectability. We used a prior IBD probability of 1 per 10,000, which is thus seen to be conservative for these data. Figure 7 shows IBD detection over the four regions. The detection rate is lower at the ends of the regions because there is less information at the ends. For the Illumina data, the IBD-detection rate stays mostly between 2.0 and 4.5 per 10,000.
Figure 7.
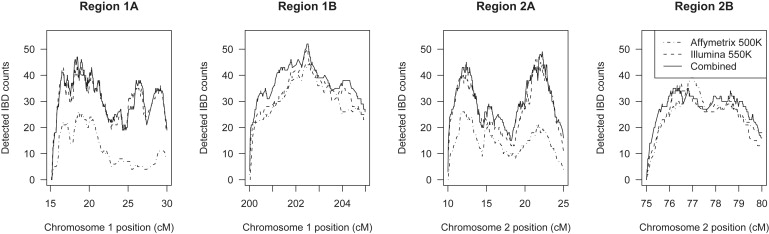
Total IBD Detected with BEAGLE across Each of the Four Regions
A total of 100,000 pairs of individuals were tested.
The increased resolution of our method is important for finding increased amounts of IBD. Although we found 1188 cM IBD in total by using the combined data, only 230 cM of that was from IBD segments of length 4 cM or greater, whereas 483 cM of it was from IBD segments of length 2 cM or less.
We tested all individuals in the 58BC data with genotypes on both platforms (1373 individuals) for HBD with BEAGLE over all autosomal chromosomes. Whereas it is not computationally feasible to test all pairs of individuals for IBD over all autosomal chromosomes for these data (due to the quadratic nature of testing all pairs), it is feasible to apply HBD detection on this scale. The total amount of detected HBD (adding all estimated segment lengths) was 2263 cM for Affymetrix (comprising 566 HBD segments) versus 2351 cM for Illumina (comprising 657 HBD segments). Although the total amounts of HBD are fairly close, they represent a significant number of smaller segments (< 2 cM) detected by Illumina but not by Affymetrix, as seen by examination of the histograms in Figure 8.
Figure 8.
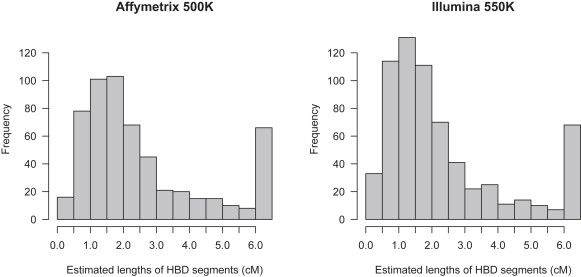
Estimated Lengths of HBD Segments Detected in 58BC Data with BEAGLE
The rightmost bar in each plot includes all estimated segment lengths > 6 cM. The left panel shows lengths of segments detected with the use of Affymetrix 500K data, whereas the right panel shows lengths of segments detected with Illumina 550K data. The data are from 1373 individuals with genotypes on both platforms.
We found five instances (two in the Affymetrix data and three in the Illumina data) for which two HBD segments in an individual were separated by a non-HBD gap of < 1 cM. Because such gaps are improbable under our HBD prior model, we investigated these to determine the cause. In particular, we looked at the number of heterozygous genotypes within the gap region on both platforms and whether the gap remained after increasing the number of runs to 10 (from 5). Table 4 shows the results. One of the gaps (gap 5) appeared to be due to an insufficient number of runs. The other gaps involved heterozygous genotypes, and thus reflected properties of the data. Small clusters of heterozygous genotypes located within an HBD region (whether resulting in a non-HBD gap or not) could be due to correlated genotype errors (gaps 1–3) or to a structural event, such as a double crossover that occurred in one of the historical meioses linking the two shared haplotypes (gaps 1–4). Table 4 suggests that when the number of heterozygous genotypes is low (1–2), the genotype error modeling allows for the HBD region to extend across the region, whereas if the number is larger (3 or more) the method inserts a non-HBD gap. We did not find any similar gaps in IBD in the IBD discovery study, but this may be because the examined regions were too small. If a small cluster of genotypes indicative of non-IBD occurred near the end of the analyzed region, it would probably simply curtail the discovered IBD segment rather than result in an additional short segment of discovered IBD.
Table 4.
Investigation of Non-HBD Gaps < 1 cM between HBD Segments Detected with BEAGLE
| Gap No. | Platform on which Gap Was Found | Size of Gap (cM) | Disappeared when Doubling the Number of Runs | No. of Heterozygous Genotypes on Platform with Gap | No. of Heterozygous Genotypes on Alternate Platform |
|---|---|---|---|---|---|
| 1 | Affymetrix | 0.17 | No | 3 | 0 |
| 2 | Affymetrix | 0.63 | No | 3 | 0 |
| 3 | Illumina | 0.38 | No | 3 | 0 |
| 4 | Illumina | 0.27 | No | 5 | 2 |
| 5 | Illumina | 0.58 | Yes | 0 | 0 |
The HBD-discovery analysis also allows examination of the success or otherwise of the genotype error modeling, as heterozygous genotypes are inconsistent with HBD. In the Illumina data, there were 12 HBD segments containing one heterozygous genotype, whereas there were no HBD segments containing more than one heterozygous genotype. In the Affymetrix data, there were 18 HBD segments containing one heterozygous genotype, there were four Affymetrix HBD segments containing two heterozygous genotypes, and there was one Affymetrix HBD segment containing three heterozygous genotypes. Thus, our method is successful in extending HBD across genotype errors, provided that the overall weight of evidence for HBD is sufficiently high. We expect that the error modeling is also working successfully in the IBD detection, although it is more difficult to verify this.
The genotype error modeling allows the HBD to extend across some double heterozygotes in an individual. These double heterozygotes may represent correlated genotype errors in some cases. The genotypes were called with BEAGLECALL, which makes use of LD.16 Using LD when calling genotypes improves overall genotype accuracy, but it could yield correlated genotype errors at tightly linked markers in a sample. In order to check whether correlated genotype errors are a plausible explanation, we looked at the corresponding genotypes as called by CHIAMO17 (Affymetrix data) or GenCall (Illumina data). Because these methods do not utilize LD, they are less likely to produce correlated genotype errors (although their overall error rate is higher). We found that correlated genotype error could explain some, but not all, of the five clusters of heterozygous genotypes in HBD segments that we found in the Affymetrix HBD data, but it does not explain either of the heterozygous clusters found in the Illumina HBD segments. The model of IBD and HBD is not designed to allow for correlated genotype errors. However, these are fairly rare events, and thus ignoring them should be acceptable in most applications.
We looked for individuals with high genomic levels of HBD with the use of the Illumina 550K data. The highest level of genomic HBD (percentage of autosomal SNPs with P(HBD) > 0.5) was 7.1%. Two individuals had > 5%, six had > 3%, and seven had > 1% genomic HBD. Of 1373 individuals, 435 had some HBD detected (i.e., posterior probability of HBD > 0.5 somewhere in their genome). The mean amount of HBD was 4.7 per 10,000 individuals (per position). When the individuals with > 1% HBD were removed, the mean amount of HBD dropped to 2.6 per 10,000. One individual was HBD for almost all of chromosome 13 (there was some heterozygosity in the first few hundred SNPs but not for the remainder of the chromosome) but was not HBD elsewhere in the genome. This is presumably a cell-line artifact.28 Figure 9 shows the genomic patterns of HBD for the five individuals with the highest levels of HBD (> 3%), excluding the individual with the chromosome 13 artifact. From the figure, one can see that the HBD and non-HBD boundaries are quite clear (on this scale), with the probabilities quickly moving from 0 to 1. This pattern is also seen in IBD data (not shown).
Figure 9.
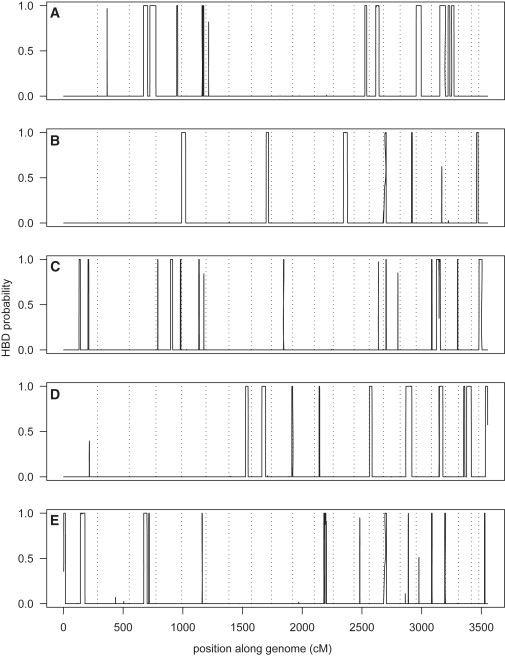
Genomic HBD Probabilities from BEAGLE for Five Individuals with the Highest Genomic Levels of HBD
Individual (A) has 7.1% of autosomal SNPs with P(HBD) > 0.5, (B) has 3.2%, (C) has 3.6%, (D) has 6.4%, (E) has 3.5%. Results are from data on the Illumina 550K platform. The dotted vertical lines are the chromosome boundaries.
Assessment of False Discovery Rates
Using the results from the false-positive and power analyses, as well as from the detection study, we can estimate false discovery rates for the UK European population. We expect that false discovery rates would be similar in other outbred European populations, such as individuals of European descent in the USA. Rates for the Illumina data were estimated as described in Material and Methods, and results are shown in Table 5. These estimates are approximate, because they are calculated from limited data. It is also important to note that the composite individuals constructed for the false-positive analysis may be more difficult to phase than a typical individual because of the reduction in IBD sharing with other individuals in the sample. This could affect false-positive rates for BEAGLE and for GERMLINE (but not for PLINK, because it does not utilize LD information). We see that BEAGLE has a low false discovery rate (< 1%), even for segments 1–2 cM in length. In contrast, GERMLINE and PLINK have much higher false discovery rates for the small segment sizes, and PLINK has a high false discovery rate even for large segments. For a 50% false discovery rate (half of reported segments are false), one would consider segments of a length of ≥ 3 cM for GERMLINE and of any length (≥ 1 cM) for BEAGLE or PLINK. If a lower false discovery rate, such as 10%, were desired, one might consider segments of a length of ≥ 4 cM for GERMLINE and of any length (≥ 1 cM) for BEAGLE. The default 5 cM threshold for GERMLINE seems reasonable, but one could reduce it to 3 or 4 cM for this type of data. It is important to consider power as well as false discovery rate. If two methods have the same false discovery rate, one would prefer the method with the higher power. With BEAGLE, one can have high confidence even in very small (1 cM) reported IBD segments. Additionally, BEAGLE's power to detect segments over 2 cM in length is quite high.
Table 5.
Estimated False Discovery Rates, per Locus, for Northern European Samples Genotyped on the Illumina 550K Array
| IBD Size |
|
False Discovery Rate |
||||
|---|---|---|---|---|---|---|
| BEAGLE | GERMLINE | PLINK (Default) | PLINK (Relaxed) | |||
| 1-2 cM | 2.7e-4 | 0.004 | 0.86 | N/Ab | 0.37 | |
| 2-3 cM | 9.7e-5 | 0 | 0.60 | 0.23 | 0.46 | |
| 3-4 cM | 5.3e-5 | 0 | 0.12 | 0.33 | 0.37 | |
| 4-5 cM | 3.2e-5 | 0 | 0 | 0.33 | 0.30 | |
Estimated rate of true IBD of this size, per locus.
When false-positive rate and power are both 0, the false discovery rate is undefined.
Discussion
We have presented a new method for detection of recent IBD or HBD in unrelated individuals. Our method is implemented in the freely available BEAGLE software package (version 3.2). The method is, to our knowledge, the first method that fully accounts for LD, which allows increased resolution of detection and enables much more IBD to be detected. For example, when using the Illumina 550K data, only 21% of the total length of IBD that we found was contained in segments of length > 4 cM. Greater power to detect IBD will lead to increased power for IBD mapping, as well as to increased accuracy from IBD-based genotype imputation and phasing.6
Current IBD resolution may be somewhat restricted by the relative scarcity of low-frequency variants on existing SNP arrays. We found that increasing the number of SNPs in a region, by combining the Illumina 550K and Affymetrix 500K panels, did not greatly increase the amount of IBD detected. However, rare variants can provide greater evidence for IBD than common variants. Thus, analysis of sequence data, or of data from panels with more rare SNPs as well as the common SNPs, will probably further improve resolution and power to detect small IBD segments.
We found that power to detect HBD is much higher than power to detect IBD of the same segment length. This is because phase uncertainty can reduce power to detect IBD, but there is no phase uncertainty in HBD regions, because HBD regions consist of homozygous genotypes. In a large sample, an iterative approach will be possible, in which detected IBD will be used to obtain highly accurate haplotypes for the individuals with IBD, which will allow improved detection of IBD, further improving the resolution of our method.
By creating composite individuals, we constructed a data set on which to compare false-positive rates of IBD segment detection. We found that PLINK and GERMLINE had much higher rates of false-positive results than BEAGLE, as well as having lower power, and, consequently, their false discovery rates were very high for small to moderately sized segments (< 5 cM for PLINK and < 3 cM for GERMLINE). For large IBD segments (> 5 cM), we expect that all methods would have good power and low false discovery rates, but for small segments, BEAGLE is clearly superior in both respects. In an outbred population, a high proportion of the detectable (with BEAGLE, or a similarly high-powered method) IBD will be found in small segments. Thus, being able to detect small segments of IBD will greatly increase the usefulness of IBD-based approaches.
In this work, we controlled false-positive IBD detection by use of a very stringent prior distribution. Indeed, our prior probabilities of IBD are lower than the rates of IBD that we found in a UK European population. Our false discovery rates are extremely small, and thus, depending on the application, one might wish to increase the prior probability of IBD somewhat, in order to achieve an increase in power. For example, by increasing the prior probability of HBD from 1 in 10,000 to 1 in 1000, we were able to increase the power to detect HBD segments of a length of 1 cM from 22% to 50%.
It is computationally expensive to apply our IBD-detection method to all pairs of individuals in large samples. Running BEAGLE for IBD detection with ten runs on approximately 13,000 pairs on chromosome 1 Illumina 550K data took 600 hr of computing time. PLINK took 5 min to do the same analysis. GERMLINE took < 7 min to analyze all 887,778 pairs, not including the phasing time, which took around 3 hr with BEAGLE. Thus, running PLINK or GERMLINE on a genome-wide data set is feasible, whereas at present BEAGLE IBD can be applied only to a restricted number of pairs (such as several thousand pairs over the whole genome) or to candidate regions. It is possible to apply the BEAGLE HBD detection on a genome-wide scale (e.g., we applied it to data with approximately 500K SNPs and 1500 individuals), because the number of individuals is much fewer than the number of all pairs of individuals. For those problems for which BEAGLE is computationally feasible, the greatly improved accuracy and power justifies the increased computing requirements. We expect that it will be possible to apply our IBD detection method comprehensively over the whole genome for a large data set if we develop a computationally efficient prefilter, so that IBD probabilities are calculated only on pairs of individuals for which the data are suggestive of IBD at a given location. We are currently working on such a filter, with encouraging preliminary results.
Web Resources
The URLs for data presented herein are as follows:
BEAGLE, http://www.stat.auckland.ac.nz/∼bbrowning/beagle/beagle.html
BEAGLECALL, http://www.stat.auckland.ac.nz/∼bbrowning/beaglecall/beaglecall.html
European Genotype Archive (repository of Wellcome Trust Case Control Consortium genotype data), http://www.ebi.ac.uk/ega/
Welcome Trust Case Control Consortium, http://www.wtccc.org.uk
Acknowledgments
This study makes use of data generated by the Wellcome Trust Case Control Consortium and the Wellcome Trust Sanger Institute. The Illumina 550K genotype data for individuals in the 1958 British Birth Cohort was generated by the Wellcome Trust Sanger Institute. A full list of the investigators who contributed to the generation of the Wellcome Trust Case Control Consortium data is available from www.wtccc.org.uk. Funding for the Wellcome Trust Case Control Consortium project was provided by the Wellcome Trust under award 076113.
This work was supported by New Zealand Marsden Fund award 07-UOA-175 and by National Institutes of Health (NIH) awards R01GM075091 and R01HG004960. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, the New Zealand Marsden Fund, or the Wellcome Trust.
References
- 1.Browning S.R. Estimation of pairwise identity by descent from dense genetic marker data in a population sample of haplotypes. Genetics. 2008;178:2123–2132. doi: 10.1534/genetics.107.084624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson, S., Merriman, B., Chen, Z., Ogdie, M., Stone, J., and Strom, S. (2006). Applications of Pedigree-Free Identity-By-Descent Mapping to Localizing Disease Genes [abstract 1530]. Presented at the annual meeting of The American Society of Human Genetics, October 11, 2006, New Orleans, LA, USA. Available from http://www.ashg.org/genetics/ashg06s/.
- 4.Albrechtsen A., Sand Korneliussen T., Moltke I., van Overseem Hansen T., Nielsen F.C., Nielsen R. Relatedness mapping and tracts of relatedness for genome-wide data in the presence of linkage disequilibrium. Genet. Epidemiol. 2009;33:266–274. doi: 10.1002/gepi.20378. [DOI] [PubMed] [Google Scholar]
- 5.Wijsman E.M., Amos C.I. Genetic analysis of simulated oligogenic traits in nuclear and extended pedigrees: summary of GAW10 contributions. Genet. Epidemiol. 1997;14:719–735. doi: 10.1002/(SICI)1098-2272(1997)14:6<719::AID-GEPI28>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 6.Kong A., Masson G., Frigge M.L., Gylfason A., Zusmanovich P., Thorleifsson G., Olason P.I., Ingason A., Steinberg S., Rafnar T. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchini J., Cutler D., Patterson N., Stephens M., Eskin E., Halperin E., Lin S., Qin Z.S., Munro H.M., Abecasis G.R., Donnelly P., International HapMap Consortium A comparison of phasing algorithms for trios and unrelated individuals. Am. J. Hum. Genet. 2006;78:437–450. doi: 10.1086/500808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gusev A., Lowe J.K., Stoffel M., Daly M.J., Altshuler D., Breslow J.L., Friedman J.M., Pe'er I. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson, S., Merriman, B., Chen, Z., and Jen, J. (2005). Detecting identical-by-descent DNA intervals between affected distant relatives using high-density SNP genotyping [abstract 44]. Presented at the annual meeting of The American Society of Human Genetics, October 27, 2005, Salt Lake City, UT, USA. Available from http://www.ashg.org/genetics/ashg05s/.
- 10.Miyazawa H., Kato M., Awata T., Kohda M., Iwasa H., Koyama N., Tanaka T., Huqun, Kyo S., Okazaki Y., Hagiwara K. Homozygosity haplotype allows a genomewide search for the autosomal segments shared among patients. Am. J. Hum. Genet. 2007;80:1090–1102. doi: 10.1086/518176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houwen R.H.J., Baharloo S., Blankenship K., Raeymaekers P., Juyn J., Sandkuijl L.A., Freimer N.B. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat. Genet. 1994;8:380–386. doi: 10.1038/ng1294-380. [DOI] [PubMed] [Google Scholar]
- 12.Te Meerman G.J., Van der Meulen M.A., Sandkuijl L.A. Perspectives of identity by descent (IBD) mapping in founder populations. Clin. Exp. Allergy. 1995;25(Suppl 2):97–102. doi: 10.1111/j.1365-2222.1995.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 13.Leibon G., Rockmore D.N., Pollak M.R. A SNP streak model for the identification of genetic regions identical-by-descent. Stat. Appl. Genet. Mol. 2008;7 doi: 10.2202/1544-6115.1340. article16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas A., Camp N.J., Farnham J.M., Allen-Brady K., Cannon-Albright L.A. Shared genomic segment analysis. Mapping disease predisposition genes in extended pedigrees using SNP genotype assays. Ann. Hum. Genet. 2008;72:279–287. doi: 10.1111/j.1469-1809.2007.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Power C., Elliott J. Cohort profile: 1958 British birth cohort (National Child Development Study) Int. J. Epidemiol. 2006;35:34–41. doi: 10.1093/ije/dyi183. [DOI] [PubMed] [Google Scholar]
- 16.Browning B.L., Yu Z. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am. J. Hum. Genet. 2009;85:847–861. doi: 10.1016/j.ajhg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPeek M.S., Sun L. Statistical tests for detection of misspecified relationships by use of genome-screen data. Am. J. Hum. Genet. 2000;66:1076–1094. doi: 10.1086/302800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abney M., Ober C., McPeek M.S. Quantitative-trait homozygosity and association mapping and empirical genomewide significance in large, complex pedigrees: fasting serum-insulin level in the Hutterites. Am. J. Hum. Genet. 2002;70:920–934. doi: 10.1086/339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leutenegger A.L., Prum B., Génin E., Verny C., Lemainque A., Clerget-Darpoux F., Thompson E.A. Estimation of the inbreeding coefficient through use of genomic data. Am. J. Hum. Genet. 2003;73:516–523. doi: 10.1086/378207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Browning S.R. Multilocus association mapping using variable-length Markov chains. Am. J. Hum. Genet. 2006;78:903–913. doi: 10.1086/503876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheet P., Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson, E.A. (2008). Analysis of data on related individuals through inference of identity by descent. Technical Report 539. Department of Statistics, University of Washington.
- 26.Rabiner L.R. A Tutorial on Hidden Markov-Models and Selected Applications in Speech Recognition. Proc. IEEE. 1989;77:257–286. [Google Scholar]
- 27.Baum, L.E. (1972). An inequality and associated maximization technique in statistical estimation for probabilistic functions on Markov processes. Inequalities-III; Proceedings of the third symposium on inequalities University of California Los Angeles, 1969. New York: Academic Press; Los Angeles: University of California Los Angeles. pp. 1–8.
- 28.Simon-Sanchez J., Scholz S., Fung H.C., Matarin M., Hernandez D., Gibbs J.R., Britton A., de Vrieze F.W., Peckham E., Gwinn-Hardy K. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 2007;16:1–14. doi: 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]