

Abstract
A self-microemulsifying drug delivery system (SMEDDS) has been developed to enhance diffusion rate and oral bioavailability of valsartan. The solubility of valsartan was checked in different oils, surfactants, and cosurfactants and ternary phase diagrams were constructed to evaluate the microemulsion domain. The valsartan SMEDDS was prepared using Capmul MCM (oil), Tween 80 (surfactant), and polyethylene glycol 400 (cosurfactant). The particle size distribution, zeta potential, and polydispersity index were determined and were found to be 12.3 nm, −0.746, and 0.138, respectively. Diffusion rate of valsartan was measured by in vitro dialysis bag method using phosphate buffer pH 6.8 as diffusion media. Developed high-performance liquid chromatography method was used to determine drug content in diffusion media. Oral bioavailability of valsartan SMEDDS was checked by using rabbit model. Results of diffusion rate and oral bioavailability of valsartan SMEDDS were compared with those of pure drug solution and of marketed formulation. Diffusion of valsartan SMEDDS showed maximum drug release when compared to pure drug solution and marketed formulation. The area under curve and time showed significant improvement as the values obtained were 607 ng h/mL and 1 h for SMEDDS in comparison to 445.36 and 1.36 h for market formulation suggesting significant increase (p < 0.01) in oral bioavailability of valsartan SMEDDS.
Key words: bioavailability, particle size, SMEDDS, valsartan, zeta potential
INTRODUCTION
Valsartan is a nonpeptide, orally active, and specific angiotensin II antagonist acting on the AT1 receptor subtype. It is categorized in angiotensin receptor blocker. The Valsartan Heart Failure Trial demonstrated that the use of valsartan was associated with reduced rates of heart failure-related hospitalizations and mortality, as well as shorter duration of hospitalization (1). Valsartan is poorly soluble and the aqueous solubility is reported to be less than 1 mg/mL. The drug is rapidly absorbed following oral administration, with a bioavailability of about 23%. Peak plasma concentrations of valsartan occur 2 to 4 h after an oral dose and 94% to 97% of the drug is bound to plasma proteins (2). Rapid onset of action is desirable to provide fast relief in the treatment of heart failure. Therefore, it is necessary to enhance the aqueous solubility and dissolution rate of valsartan to obtain faster onset of action, minimize the variability in absorption, and improve its overall oral bioavailability. The various formulation strategies reported in the literature include the use of surfactants, cyclodextrin complexes, nanoparticles, solid dispersions, micronization, lipids, and permeation enhancers (3). There has been increasing focus on the utility of self-microemulsifying drug delivery system (SMEDDS) as these lipid-based formulations are reported to assist the absorption of poorly soluble drugs by reducing the inherent limitation of slow and incomplete dissolution and by facilitating the formation of a microemulsion within the intestine that is capable of maintaining otherwise a poorly soluble drug in solution (4). Some efforts have been made to enhance the solubility of valsartan to understand its effect on the bioavailability of the drug, and valsartan/hydroxypropyl-β-cyclodextrin complex has been reported to significantly increase the solubility and decrease the rate of valsartan degradation (3). A gelucire 50/13-based dispersion granule formulation has also been reported very recently (5). In addition to all these approaches, preparation of lipid-based formulation was tried to make formulation process easier. The main aim of the study was to develop valsartan SMEDDS to improve upon the solubility of the valsartan which will have some bearing on the bioavailability. The SMEDDS consists of an isotropic mixture of drug, lipid, surfactant, and typically a cosurfactant or cosolvent. When exposed to the fluids of the gastrointestinal (GI) tract, these precursor solutions spontaneously emulsify to form highly dispersed microemulsions. These dispersions commonly exhibit particle sizes below 300 nm (SEDDS) or 100 nm (SMEDDS) and have been shown to enhance the oral bioavailability of lipophilic drugs such as cyclosporine (6), halofantrine (7), ontazolast (8), and progesterone (9). The ease of dispersion and the very small particle size of the resultant colloidal microemulsion have historically been viewed as the principal reasons for their utility in the delivery of lipophilic drugs. Consequently, most of the commercially available lipidic formulations are complex mixtures of lipids, surfactants, and cosolvents/cosurfactants constructed to improve drug solubility in the formulation (and therefore increase drug payload) and also to maximize dispersion of the dose form on exposure of the capsule fill to the GI contents (10). This dispersed microemulsion is known as SMEDDS. SMEDDS produces fine droplets as observed in microemulsion when comes in contact with water under agitation. The digestive motility of the stomach and intestine provides the agitation necessary for self-emulsification in vivo (11). SMEDDS can be dispensed in form of soft or hard gelatin capsule filled. Studies of lipid based system reveals that spontaneous formation of microemulsion advantageously presents the drug in a dissolved form, and the resultant small droplet size provides a large interfacial surface area for drug release and absorption. Moreover, the oil used promotes the intestinal lymphatic transport of drugs. Many of possible mechanisms of SMEDDS include increasing membrane fluidity to facilitate transcellular absorption, opening tight junction to allow paracellular transport, inhibiting P-gp, and/or CYP450 to increase intracellular concentration and residence time by surfactants and stimulating lipoprotein/chylomicron production by lipid (12). In this study, the solubility behavior of valsartan was tested in different lipid solvents, and an optimized SMEDDS containing valsartan was formulated. The prepared SMEDDS was characterized by particle size and zeta potential values. In vitro diffusion profiles of drug in different buffer solutions were studied to observe release of drug from SMEDDS. The bioavailability of drug was also investigated by in vivo studies and compared with commercially available capsule (Valent-80®, Lupin Pvt Ltd.).
MATERIALS AND METHODS
Materials
Valsartan was a kind gift from Torrent Research Centre, Ahmedabad, India. Gift samples of Cremophor RH 40 (polyoxyl 40 hydrogenated castor oil) and Cremophor EL (from BASF India Ltd., Mumbai, India), Captex 200, Captex 355, Capmul C10, and Capmul MCM (from Abitec Corporation, USA), Labrafil 2125, Labrafac PG, and Plurol Olique (from Colorcon Asia, Pvt Ltd., Mumbai, India) were obtained. Tween 80 (polysorbate 80), and polyethylene glycol 400 (PEG 400) were procured form S. D. Fine Chemicals, Mumbai, India. High-performance liquid chromatography (HPLC) grade methanol and acetonitrile were purchased from Spectrochem Laboratories, India. All other chemicals were of analytical grade.
Animals
Male rabbits (weighing approximately 1.7 ± 0.3 kg) were used for the comparative bioavailability studies. The animals were maintained at a constant light (14:10 h light/day ratio), temperature (24–25°C), and humidity (60%) and were supplied with food and water ad libitum. The animal requirement was approved by the Institute Animal Ethics Committee, and all experiments were conducted as per the norms of the Committee for the Purpose of Supervision of Experiments on Animals, India.
Methods
Solubility Studies
One gram of each of solvents (oil, surfactant, and cosurfactant) was filled in 10 mL screw capped test tube. Twenty-five milligrams of valsartan was added in each tube. The tightly closed tubes were shaken for 48 h at 50 strokes per minute in water bath maintained at 30°C temperature. After visual assessment of solubility of drug, additional drug in fraction of 25 mg was subsequently added in each tube to determine maximum solubility of drug in particular solvent. After 48 h, each tube was centrifuged at 3,000 rpm for 5 min to separate excess insoluble drug. The concentration of dissolved valsartan was determined by HPLC. All the experiments were repeated thrice.
Construction of Ternary Phase Diagram
The existence of microemulsion regions was determined by using pseudoternary phase diagram. SMEDDS were diluted under agitation condition using water titration method. The mixture of oil and surfactant/cosurfactant (S/CoS) at certain weight ratios were diluted with water in a dropwise manner. The ratios of surfactant/cosurfactant were prepared in specific manner, i.e., 2:1, 1:1, and 1:2 (w/w). Each of these ratios was mixed with increasing percentage of oil, i.e., 10%, 20%, 30%, 40% up to 90% of oil to get phase diagram. Then, each mixture was titrated with water, and agitation was provided by magnetic stirrer. The formation of microemulsion regions was monitored visually for turbidity–transparency–turbidity. These values of oil, surfactant, and cosurfactant were used to determine the boundaries of microemulsion region. After the identification of microemulsion region in the phase diagrams, the microemulsion formulations were selected at desired component ratios. To determine the effect of drug addition in SMEDDS, phase diagrams were also constructed in presence of drug. In order to prepare SMEDDS, selection of microemulsion region from phase diagram was based on the fact that solution remains clear even on infinite dilution.
Preparation of SMEDDS
A series of microemulsions of SMEDDS were prepared (Table I) with varying ratios of oil, surfactant, and cosurfactant. Formulations A, B, and C were prepared using Capmul MCM as oil, Tween 80 as surfactant, and PEG 400 as cosurfactant. Similarly, formulations D, E, and F were prepared with Captex 200 P as oil, Cremophore EL as surfactant, and PEG 400 as cosurfactant. In all the formulations, the level of valsartan was kept constant to 5% of SMEDDS. Briefly, oil, surfactant, and cosurfactant were accurately weighed into glass vials according to their ratios. The amount SMEDDS should be such that it solubilizes the drug (single dose) completely. Hence, 10 mg of valsartan was dissolved in 0.2 g of SMEDDS. Then, the components were mixed by gentle stirring and vortex mixing and heated at 37°C in incubator, until valsartan perfectly has dissolved. The mixture was stored at room temperature until used.
Table I.
Compositions of Formulations
| Vehicle (%, w/w) | A | B | C | D | E | F |
|---|---|---|---|---|---|---|
| Valsartan | 5 | 5 | 5 | 5 | 5 | 5 |
| Captex 200 P | – | – | – | 10 | 10 | 10 |
| Capmul MCM | 10 | 10 | 10 | – | – | – |
| Tween 80 | 60 | 45 | 30 | – | – | – |
| Cremophore EL | – | – | – | 60 | 45 | 30 |
| PEG 400 | 30 | 45 | 60 | 30 | 45 | 60 |
A, B, C, D, E, and F indicate formulations
PEG 400 polyethylene glycol 400
In Vitro Characterization of SMEDDS
Robustness to Dilution
Robustness of SMEDDS to dilution was studied as per Date and Nagarsenker’s method with slight modification (13). SMEDDS was diluted to 10, 100, and 1,000 times with various dissolution media, viz., water, pH 1.2 buffer, pH 4.5 buffer, and pH 6.8 buffer. The diluted microemulsions were stored for 12 h and observed for any signs of phase separation or drug precipitation.
On the basis of the above test, diluted SMEDDS was used for assessment of various in vitro parameters. Diluted SMEDDS was prepared by diluting 25 μL of SMEDDS with 25 mL of water.
Determination of Microemulsion Particle Size, Polydispersity Index, and Zeta Potential
The particle size and zeta potential of the microemulsion were measured utilizing a Malvern Zetasizer 3000 (Malvern Instruments, Malvern, UK).
In Vitro Release Test
In vitro release of valsartan SMEDDS was tested by using dialysis bag method (14). Valsartan microemulsion was instilled into the dialysis bag (MWCO 12000, Hi-Media Industries Inc., USA), firmly sealed with clamp, and was placed in 1,000 mL, phosphate buffer pH 6.8 as the dissolution medium at 37°C. The revolution speed of the paddle was maintained at a rate of 50 rpm (15). The release of valsartan was also checked in hydrochloride buffer pH 1.2 and acetate buffer pH 4.5. For this, 5 mL samples were withdrawn at predetermined intervals, and the same volume of fresh dissolution medium was replenished. The release of valsartan from SMEDDS was compared with that of from marketed capsule formulation and standard drug solution. The suspension of marketed capsule formulation was prepared by milling powder from capsule with 2.5% (v/v) hydroxymethylcellulose (15 cP, S.D. Fine Chemicals, Mumbai, India) solution, and standard drug solution was prepared by dissolving drug in methanol followed by dilution in water. Samples (20 μL) were injected into HPLC to determine the released valsartan. The study was repeated three times.
Bioavailability Study
Bioavailability of valsartan SMEDDS was compared with suspension of marketed valsartan capsule (Valent-80®, Lupin Ltd., Deharadun, India). Valsartan suspension was prepared as mentioned above and diluted to a definite volume using the same vehicle afterwards. Six rabbits were allocated at random to two treatment groups and administered SMEDDS and valsartan suspension solution in a crossover design. The washout period between the two treatments was 7 days. Male rabbits (weighing approximately 1.7 ± 0.3 kg) were fasted for 12 h prior to the experiment and water was available ad lib. After oral administration of valsartan dose (5.6 mg/1.5 kg body weight), about 2 mL of blood sample was collected through retro-orbital plexus into heparinized tubes at 0, 0.25, 0.5, 1, 2, 3, 4, 5, 6, 8, 12, and 24 h. Blood samples were centrifuged at 5,000 rpm for 10 min using a high-speed centrifuging machine, and plasma samples were withdrawn and stored at −18°C.
Estimation of Valsartan by RP-HPLC
The sample preparation was done as per method of Macek et al. with some modification (16). The samples were stored in the freezer at −18°C and allowed to thaw at room temperature before processing. A 0.2 mL of plasma was transferred in 2 mL centrifuge tube. To that, 25 μL of losartan potassium (1 mg/mL) was added as an internal standard and vortex-mixed for 30 s at 2,000 rpm. Then, 1 mL of methanol was added to the tube and again vortex-mixed for 10 min at 2,000 rpm to extract out the drug. The tube was centrifuged for 10 min at 2,500 rpm, and 20 μL of the supernatant was injected into the chromatographic system. The HPLC analysis was carried out on Shimadzu HPLC system consisting of ultraviolet–visible detector (SPD-20A Prominence UV–VIS detector) and solvent delivery pump (LC-20AT Liquid Chromatography). Chromatographic column was reverse-phase C-18 column (250 × 4.6 mm, 5 μm, Hypersil BDS). The mobile phase for in vitro study consists of triethylamine buffer (pH adjusted 3.0 with orthophosphoric acid) and acetonitrile with a ratio of 45:55, and mobile phase for in vivo study consists of same buffer and acetonitrile with ratio of 60:40 pumped at a flow rate of 0.7 mL/min. The column temperature was kept at 25°C. The detector was set at 215 nm. In case of in vivo analysis, C18 column was attached with guard column cartridge (Phenomenex, Torrance, CA, USA) to prevent column blockage due to plasma component.
RESULT AND DISCUSSION
Solubility Studies
Solubility of valsartan was checked in a number of solvents and presented in graphical manner in Fig. 1. Based on the solubility data, Capmul MCM was selected as oil phase, Tween 80 as surfactant, and PEG 400 as cosurfactant since these solvents showed better solubility.
Fig. 1.

Graph showing solubility of valsartan in different solvents
Pseudoternary Phase Diagram Study
In order to form self-emulsifying o/w and w/o microemulsions, an oil, a blend of two surfactants, and an aqueous phase were used. These four component systems can be best described by pseudoternary phase diagram where a constant ratio of two of the components was used and other two were varied (17). To determine optimum concentration of oil, surfactant, and cosurfactant, phase diagrams were constructed. SMEDDS forms microemulsion when titrated with water under agitation condition. The particle size of microemulsion is less than 100 nm and as the energy required to form microemulsion is very low, it is a thermodynamically spontaneous process (17). This process is facilitated by presence of surfactant. The surfactant forms a layer around oil globule in such a way that polar head lies toward aqueous and nonpolar tail pull out oil and thereby reduces surface tension between oil phase and aqueous phase (18). Another factor affecting formation of microemulsion is the ratio of surfactant and cosurfactant. The lipid mixtures with different surfactant, cosurfactant, and oil ratios lead to the formation of SMEDDS with different properties (19). Since surfactant and cosurfactant adsorb at interface and providing mechanical barrier to coalescence, selection of oil, surfactant, and cosurfactant and mixing ratio to S/CoS play important role in microemulsion formation (20). Six different formulations were prepared using different oils, surfactants, and cosurfactants in varying ratio. SMEDDS A, B, and C were formulated using same excipients with three different S/CoS ratios of 2:1, 1:1, and 1:2 and diluted to get microemulsion region (Fig. 2). Microemulsion regions were observed visually. Initially, the concentration of oil taken was maximum, i.e., 90%, and amount of S/CoS was kept to minimum, i.e., 10%. Gradually, oil concentration was decreased and that of S/CoS was increased. It was observed during these experiments that high concentration of oil forms poor emulsion with entrapment of very less amount of water upon dilution. Another observation was that as concentration of S/CoS increases, the time estimated to form microemulsion decreases. A series of microemulsions were prepared at different concentrations of oil and S/CoS, but concentration of oil was found to be a rate-limiting factor (Fig. 2), and in all cases, high oil concentration resulted in poor emulsion region. Hence, it was decided to keep the oil concentration less than 10%.
Fig. 2.

Phase diagram prepared with the following components: oil—Capmul MCM, surfactant—Tween 80, and cosurfactant—PEG 400. S/CoS ratio of a is 2:1, b is 1:1, and c is 1:2
Other important factors affecting microemulsions were concentration and ratio of S/CoS. In the present study, three S/CoS ratios, 2:1, 1:1, and 1:2, were tried (Fig. 2). The black region in Fig. 2 shows self-microemulsifying (SMEDDS) region having particle size less than 100 nm, whereas gray region indicates formation of self-emulsifying (SEDDS) domain with particle size 100 to 300 nm (7). Since phase diagrams were constructed after infinite dilution of SMEDDS, Table II gives clear picture for composition of formed microemulsion region.
Table II.
Compositions of Microemulsion After Dilution
| Formulation | S/CoS ratio | Weight/weight percentage component in microemulsion formulation | |||
|---|---|---|---|---|---|
| Oil | S | CoS | Aqueous | ||
| SMEDDS A | 2:1 | 5 | 13.66 | 6.33 | 75 |
| SMEDDS B | 1:1 | 10 | 17.5 | 17.5 | 55 |
| SMEDDS C | 1:2 | 4 | 3 | 6 | 87 |
SMEDDS self-microemulsifying drug delivery system, S/CoS surfactant/cosurfactant, S surfactant, CoS cosurfactant
It is clear from Table II and Fig. 2a that after dilution, SMEDDS A contains 5% of oil, 13.33% of surfactant, 6.66% cosurfactant, and 75% of water. Alternatively, it can also be concluded that 20% of S/CoS mixture with ratio of 2:1 could solubilize 5% of oil in 75% of water. When equal amount of cosurfactant added (S/CoS mix 1:1), microemulsion region increased and oil solubilized up to 10% with surfactant, cosurfactant, and water concentration of 17.5%, 17.5%, and 55%, respectively (Fig. 2b). When cosurfactant concentration was further increased up to S/CoS mix 1:2, Fig. 2c revealed that oil concentration was decreased to 4% with 3% of surfactant, 6% of cosurfactant, and remaining 87% of water. On comparing all three cases, it can be concluded that equal amount of surfactant and cosurfactant is required to get optimum concentration of oil. Moreover, since the drug is lipid soluble, oil concentration is important factor to entrap required amount of drug dose. It is also clear from diagram (Fig. 2b) that formulation B covers maximum microemulsion region (black region) as compared to formulations A and C. If concentration of surfactant is kept high as in formulation A (Fig. 2a), it gives limited microemulsion region but the microemulsion formed is of small particles size (17.8) with zeta potential value of −12.1. In many self-microemulsion studies, it is reported (21) that zeta potential plays an important role. Increase of repulsive forces between microemulsion droplets prevents coalescence of microemulsion droplets. If S/CoS ratio decreased, it results in milky, white emulsion. This problem can be evaluated by increasing ratio up to 1:1. The phase diagram clearly shows that SMEDDS prepared with S/CoS ratio of 1:1 (Fig. 2b), cover maximum self-microemulsifying region as compared to other two ratios, and the time required to form microemulsion is less than 2 min. In the case with S/CoS ratio (1:2) when concentration of cosurfactant (PEG 400) was increased, the microemulsion region was found to be decreased, and time required to form microemulsion was increased by more than 2 min (Fig. 2c). After comparing the three conditions, S/CoS ratio 1:1 was considered as optimal ratio to form rapid and clear microemulsion. SMEDDS D, E, and F were prepared with S/CoS ratio of 2:1, 1:1, and 1:2, respectively (Fig. 3). Phase diagram study shows that SMEDDS D, E, and F forms only self-emulsification region (gray region).
Fig. 3.

Phase diagram prepared with the following components: oil—Captex 200 P, surfactant—Cremophore EL, and cosurfactant—PEG 400. S/CoS ratio of a is 2:1, b is 1:1, and c is 1:2
In Vitro Characterization of SMEDDS
Robustness to Dilution
Diluted SMEDDS did not show any precipitation or phase separation on storage in various dilution media. This revels that all media were robust to dilution.
Particle Size Analysis and PDI Determination
There is a relationship between the droplet size and the concentration of the surfactant being used. In some cases, increasing the surfactant concentration could lead to droplets with smaller mean droplet size such as in the case of a mixture of saturated C8–C10 polyglycolized glycerides. This could be explained by the stabilization of the oil droplets as a result of the localization of the surfactant molecules at the oil–water interface. On the other hand, in some cases, the mean droplet size may increase with increasing surfactant concentrations. This phenomenon could be attributed to the interfacial disruption elicited by enhanced water penetration into the oil droplets mediated by the increased surfactant concentration and leading to ejection of oil droplets into the aqueous phase (19). The particle size determination following self-microemulsification is a critical factor to evaluate a self-microemulsion system as droplet size is reported to have an effect on drug absorption. The smaller is the droplet size, the larger is the interfacial surface area provided for drug absorption (22).
The optimization of SMEDDS was based on microemulsion domain obtained and particle size of SMEDDS. The mean particle size and PDI for all the SMEDDS have been summarized in Table III. The results show that particle sizes of SMEDDS D, E, and F were more than 100 nm and with higher PDI. Polydispersity is the ratio of standard deviation to the mean droplet size. This signifies the uniformity of droplet size within the formulation. The higher the value of polydispersity, the lower is the uniformity of the droplet size in the formulation (23). The polydispersity values of SMEDDS A, B, and C are 0.127, 0.138, and 0.202, respectively, which indicates uniformity of droplet size within the formulation. In contrast to that, these values are increased for SMEDDS D, E, and F to 0.342, 0.442, and 0.386, respectively, which indicates nonuniformity of particles in microemulsion. A less solubility of drug in solvents may be the reason behind this. This leads to precipitation of drug and thereby increases particle size of SMEDDS. SMEDDS with increased particle size causes agglomeration of globules and suffers with instability of system. Moreover, pseudoternary phase diagram also revealed that these three formulations do not fall under self-microemulsion region; hence, they were dropped for further study. SMEDDS A, B, and C were found having particle size less than 100 nm which fulfill the criteria of microemulsion and low PDI shows uniformity of particles. Therefore, SMEDDS A, B, and C were considered for further in vitro and in vivo studies.
Table III.
Mean Particle Size and Polydispersity Index and Zeta Potential
| Formulation | Mean particle size (nm) | Polydispersity index | Zeta potential |
|---|---|---|---|
| SMEDDS A | 17.8 ± 1.9 | 0.127 | −12.1 |
| SMEDDS B | 12.3 ± 2.1 | 0.138 | −0.746 |
| SMEDDS C | 97.1 ± 2.2 | 0.202 | −4.65 |
| SMEDDS D | 111.9 ± 1.9 | 0.341 | −0.249 |
| SMEDDS E | 135.4 ± 2.7 | 0.442 | −0.381 |
| SMEDDS F | 157.5 ± 1.8 | 0.386 | −0.563 |
SMEDDS self-microemulsifying drug delivery system
Zeta Potential Measurement
The surfactant (Tween 80) and cosurfactant (PEG 400) used in this study are nonionic which do not contribute any charge to the microemulsion particle. Lu et al. and Cui et al. reported stable SMEDDS using same excipients (24,25). This indicates that negative charge particle do not affect the stability of microemulsion. SMEDDS B reports negative zeta potential value −0.746.
So considering the above in vitro characteristic, it can be concluded that SMEDDS B with S/CoS ratio 1:1 generates maximum microemulsion region and forms rapid microemulsion with lesser particle size 12.3, low PDI 0.138, and zeta potential −0.746. Therefore, SMEDDS B was considered as optimal self-microemulsifying system for in vitro and bioavailability studies.
In Vitro Release Study
In vitro release of valsartan was checked for SMEDDS B, conventional capsule (Valent-80®), and standard drug solution by using dialysis bag method. The release of valsartan from these dosage forms was evaluated in buffers of pH 1.2, 4.5, and 6.8. The data showed that release of valsartan was faster in phosphate buffer of pH 6.8 than other two media (Fig. 4). The pH-dependent solubility of drug can be responsible for higher release. The release pattern shows that drug release form SMEDDS B is faster than other two, conventional capsule and standard drug solution. Moreover, SMEDDS B release more than 90% drug within 1 h while release rate is very slow in case of conventional capsule suspension, i.e., only 58% and same for standard drug suspension is only 19% within first hour. The factors affecting drug release may be (a) SMEDDS with reduced particle size provides more surface area to release drug from solvents and thereby increases drug release rate and (b) oil phase of SMEDDS may act as carrier molecules which itself does not diffuse through the barrier but allow drug molecules to get diffused form membrane of dialysis bag. Although exact mechanism is not known, it is confirmed that any of these factors affect the bioavailability of drug.
Fig. 4.

In vitro release of valsartan in different media
Bioavailability Study
In vivo pharmacokinetic behaviors of valsartan with SMEDDS and marketed formulation (Valent 80®) were studied in rabbit. Mean plasma concentration was plotted as a function of time as shown in Fig. 5. The noncompartment model is used to evaluate pharmacokinetic parameters of valsartan absorption which are summarized in Table IV. The linear trapezoidal rule is used to calculate the area under curve (AUC0→t).
Fig. 5.

Plasma concentration–time profile of valsartan SMEDDS and capsule suspension
Table IV.
Pharmacokinetic Parameters for SMEDDS B and Capsule Suspension
| Parameters | SMEDDS B | Capsule suspension |
|---|---|---|
| t max (h) | 1 ± 0.44 | 1.36 ± 0.39 |
| C max (ng/mL) | 112.61 ± 9.13 | 69.24 ± 3.99 |
| AUC0→t (ng h/mL) | 607.93 ± 45.06 | 445.36 ± 70.50 |
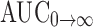 (ng h/mL) (ng h/mL) |
1,124.57 ± 79.66 | 893.72 ± 116.56 |
| AUMC0→t (ng h/mL) | 4,752.96 ± 102.70 | 3,848.13 ± 265.20 |
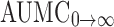 (ng h/mL) (ng h/mL) |
37,933.75 ± 1,609.08 | 33,804.48 ± 1,761.19 |
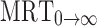 (h) (h) |
33.73 ± 1.11 | 37.82 ± 1.40 |
| Relative bioavailability (%) | 178.70 | – |
SMEDDS self-microemulsifying drug delivery system, t
max time of peak concentration, C
max peak of maximum concentration, AUC
0→t area under the concentration time profile curve until last observation, 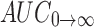 area under the concentration time profile curve extrapolated to infinity, AUMC
0→t area under moment curve computed to the last observation,
area under the concentration time profile curve extrapolated to infinity, AUMC
0→t area under moment curve computed to the last observation, 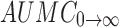 area under moment curve extrapolated to infinity,
area under moment curve extrapolated to infinity, 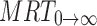 mean residence time
mean residence time
Relative bioavailability was calculated using following formulae:
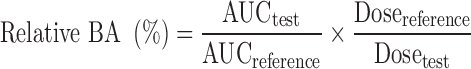 |
Plasma concentration Cmax and AUC0→t are significantly increased for SMEDDS B than those for the capsule suspension. Tmax is decreased for SMEDDS B and it was 1 h for SMEDDS and 1.36 h for capsule formulation. Relative bioavailability is increased 1.78-fold. The results of  were compared using t test, and it was found that it is highly significant (p < 0.01) when SMEDDS B and capsule formulation were compared. The consistency in the intrinsic properties of drug may be contributing factor. Increased bioavailability of SMEDDS may due to its lymphatic transport through transcellular pathway (23). It is also reported that the long-chain oils promote lipoprotein synthesis and subsequent lymphatic absorption (26). The main rate-limiting barrier for drug absorption/diffusion is the single layer of intestinal epithelial cell. High content of surfactants in SMEDDS could increase the permeability by disturbing the cell membrane (27). It should be noted that the surfactant with best enhancement ability requires both hydrophilic and lipophilic domains reaching a balance with intermediate values of HLB such as Tween 80 used in our study, having a polyoxyethylene and intermediate hydrocarbon chain. Its structural characteristics impart both lipophilic and hydrophilic properties to the surfactant, allowing it to partition between lipid and protein domains. Surfactant also demonstrated a reversible effect on the opening of tight junction; it may interact with the polar head groups of the lipid bilayers, modifying hydrogen bonding and ionic forces between these groups. It may also insert itself between the lipophilic tails of the bilayers, resulting in a disruption of the lipid-packing arrangement (12). On the basis of in vitro and in vivo correlation, it can be assumed that increase in release profile of valsartan from SMEDDS can lead to increase of bioavailability of valsartan.
were compared using t test, and it was found that it is highly significant (p < 0.01) when SMEDDS B and capsule formulation were compared. The consistency in the intrinsic properties of drug may be contributing factor. Increased bioavailability of SMEDDS may due to its lymphatic transport through transcellular pathway (23). It is also reported that the long-chain oils promote lipoprotein synthesis and subsequent lymphatic absorption (26). The main rate-limiting barrier for drug absorption/diffusion is the single layer of intestinal epithelial cell. High content of surfactants in SMEDDS could increase the permeability by disturbing the cell membrane (27). It should be noted that the surfactant with best enhancement ability requires both hydrophilic and lipophilic domains reaching a balance with intermediate values of HLB such as Tween 80 used in our study, having a polyoxyethylene and intermediate hydrocarbon chain. Its structural characteristics impart both lipophilic and hydrophilic properties to the surfactant, allowing it to partition between lipid and protein domains. Surfactant also demonstrated a reversible effect on the opening of tight junction; it may interact with the polar head groups of the lipid bilayers, modifying hydrogen bonding and ionic forces between these groups. It may also insert itself between the lipophilic tails of the bilayers, resulting in a disruption of the lipid-packing arrangement (12). On the basis of in vitro and in vivo correlation, it can be assumed that increase in release profile of valsartan from SMEDDS can lead to increase of bioavailability of valsartan.
CONCLUSION
SMEDDS of valsartan was prepared and optimized by using in vitro parameters like particle size, polydispersity index, zeta potential, in vitro release, and bioavailability studies. Optimal SMEDDS contains Capmul MCM as oil phase, Tween 80 as a surfactant, and PEG 400 as cosurfactant. The combination of all three components, i.e., oil/surfactant/cosurfactant in the ratio of 10:45:45, formulates SMEDDS with lower particle size 12.3, PDI 0.138, and zeta potential −0.746. This optimized SMEDDS showed good in vitro release which is increased more than 90% when compared with marketed formulation and drug suspension. In vivo study revealed significant improvement in extent of absorption of valsartan in rabbit to 1.78-fold compared to with conventional capsule formulation. Our study illustrated the potential use of self-microemulsified drug delivery system to dispense lipid-soluble drug by oral route.
Acknowledgment
We are thankful to Torrent Pharmaceuticals, Ahmedabad for providing Valsartan as a gift.
Notations
- AUC
area under curve
- BA
bioavailability
- CD
cyclodextrin
- Cmax
maximum concentration
- GI
gastrointestinal
- HLB
hydrophilic lipophilic balance
- HPLC
high-performance liquid chromatography
- MRT
mean residence time
- o/w
oil in water
- PDI
polydispersity index
- S/CoS
surfactant/cosurfactant
- SMEDDS
self-microemulsifying drug delivery system
- Tmax
maximum time
- w/o
water in oil
References
- 1.Smith DG, Cerulli A, Frech FH. Use of valsartan for the treatment of heart-failure patients not receiving ACE inhibitors: a budget impact analysis. Clin Ther. 2005;27(6):951. doi: 10.1016/j.clinthera.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Brookman LJ, Rolan PE, Benjamin IS, Palmer KR, Wyld P, Lloyd PJ, et al. Pharmacokinetics of valsartan in patients with liver disease. Clin Pharmacol Ther. 1997;62(3):272–8. doi: 10.1016/S0009-9236(97)90029-1. [DOI] [PubMed] [Google Scholar]
- 3.Cappello B, Di Maio C, Iervolino M, Miro A. Improvement of solubility and stability of valsartan by hydroxypropyl-beta-cyclodextrin. J Incl Phenom Macrocycl Chem. 2006;54:289–94. doi: 10.1007/s10847-005-9004-y. [DOI] [Google Scholar]
- 4.Charman WN. Lipid, lipophilic drugs and oral drug delivery-some emerging concepts. J Pharm Sci. 2000;89(8):967–78. doi: 10.1002/1520-6017(200008)89:8<967::AID-JPS1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 5.Shrivastava AR, Ursekar B, Kapadia CJ. Design, optimization, preparation and evaluation of dispersion granules of valsartan and formulation into tablets. Curr Drug Deliv. 2009;6(1):28–37. doi: 10.2174/156720109787048258. [DOI] [PubMed] [Google Scholar]
- 6.Klauser RM, Irshik H, Kletzmayr J, Sturm I, Brunner W, Woloszczuk W, et al. Pharmacokinetic cyclosporine a profiles under long-term neoral treatment in renal transplant recipients: does fat intake still matter? Transplant Proc. 1997;29:3137–40. doi: 10.1016/S0041-1345(97)00814-2. [DOI] [PubMed] [Google Scholar]
- 7.Khoo SM, Humberstone AJ, Porter CJH, Edwards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167:155–64. doi: 10.1016/S0378-5173(98)00054-4. [DOI] [Google Scholar]
- 8.Jayaraj AA, Keirns JJ. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water soluble LTB4 inhibitor. J Pharm Sci. 1998;87:164–9. doi: 10.1021/js970300n. [DOI] [PubMed] [Google Scholar]
- 9.Solomon LJ, Seager H, Pouton CW. Influence of lipolysis on drug absorption from the gastrointestinal tract. Adv Drug Deliv Rev. 1997;25:33–46. doi: 10.1016/S0169-409X(96)00489-9. [DOI] [Google Scholar]
- 10.Porter CJ, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–12. doi: 10.1023/B:PHAM.0000036914.22132.cc. [DOI] [PubMed] [Google Scholar]
- 11.Kommuru TR, Gurley B, Khan MA, Reddy JK. Self-emulsifying drug delivery system (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212:233–46. doi: 10.1016/S0378-5173(00)00614-1. [DOI] [PubMed] [Google Scholar]
- 12.Wei W, Yang W, Li Q. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Euro J Pharm Biopharm. 2006;63(3):288–94. doi: 10.1016/j.ejpb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329:166–72. doi: 10.1016/j.ijpharm.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 14.Kang BK, Lee JS, Chon SK, Jeong SY, Yuk SH, Khang G, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274:65–73. doi: 10.1016/j.ijpharm.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 15.Centre for Drug Evaluation and Research. http://www.accessdata.fda.gov.
- 16.Macek J, Klıma J, Ptacek P. Rapid determination of valsartan in human plasma by protein precipitation and high-performance liquid chromatography. J Chromatogr B. 2006;832:169–72. doi: 10.1016/j.jchromb.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 17.Constantinides PP. Lipid microemulsion for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–72. doi: 10.1023/A:1016268311867. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45:89–121. doi: 10.1016/S0169-409X(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 19.Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:73–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Patel AR, Vavia RR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007;9(3):E344–52. doi: 10.1208/aapsj0903041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355:269–76. doi: 10.1016/j.ijpharm.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 22.Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm. 2000;50:179–88. doi: 10.1016/S0939-6411(00)00089-8. [DOI] [PubMed] [Google Scholar]
- 23.Baboota S, Shakeel F, Ahuja A, Ali J, Shafiq S. Design, development and evaluation of novel nanoemulsion formulations for transdermal potential of celecoxib. Acta Pharm. 2007;57:315–32. doi: 10.2478/v10007-007-0025-5. [DOI] [PubMed] [Google Scholar]
- 24.Lu JL, Wang JC, Zhao SX, Liu XY, Zhao H, Zhang X, et al. Self-microemulsifying drug delivery system (SMEDDS) improves anticancer effect of oral 9-nitrocamptothecin on human cancer xenografts in nude mice. Euro J Pharm Biopharm. 2008;69:899–907. doi: 10.1016/j.ejpb.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 25.Cui J, Yu B, Zhao Y, Zhu W, Li H, Lou H, et al. Enhancement of oral absorption of curcumin by self-microemulsifying drug delivery systems. Int J Pharm. 2009;371:148–55. doi: 10.1016/j.ijpharm.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Charman WN, Stella VJ. Transport of lipophilic molecules by the intestinal lymphatic system. Adv Drug Deliv Rev. 1991;7:1–14. doi: 10.1016/0169-409X(91)90046-F. [DOI] [Google Scholar]
- 27.Swenson ES, Curatolo WJ. Means to enhance penetration. Adv Drug Deliv Rev. 1992;8:39–42. doi: 10.1016/0169-409X(92)90015-I. [DOI] [Google Scholar]