

Abstract
The objectives of this research were to prepare celecoxib proniosomes and evaluate the influence of proniosomal formulation on the oral bioavailability of the drug in human volunteers. A new proniosomal delivery system for a poorly water-soluble drug such as celecoxib was developed and subjected to in vitro and in vivo studies. Proniosomes were prepared by sequential spraying method, which consisted of cholesterol, span 60, and dicetyl phosphate in a molar ratio of 1:1: 0.1, respectively. The average entrapment percent of celecoxib proniosome-derived niosomes was about 95%. The prepared proniosomes showed marked enhancement in the dissolution of celecoxib as compared to pure drug powder. The bioavailability of 200 mg single dose of both celecoxib proniosomal formulation and a conventional marketed celecoxib capsule was studied in human volunteers. The obtained results show that the proniosomal formulation significantly improved the extent of celecoxib absorption than conventional capsule. The mean relative bioavailability of the proniosomal formulation to the conventional capsule was 172.06 ± 0.14%. The mean Tmax for celecoxib was prolonged when given as proniosomal capsule. There was no significant difference between the values of Kel and t1/2 for both celecoxib preparations. In conclusion, the proniosomal oral delivery system of celecoxib with improved bioavailability was established.
Key words: bioavailability, celecoxib, proniosomes
INTRODUCTION
Celecoxib is a specific cyclooxygenase-2 inhibitor (1) widely prescribed for treatment of rheumatoid arthritis and osteoarthritis and management of pain of these conditions (2). It has comparable efficacy and superior gastric tolerability and is safer when compared with conventional nonsteroidal anti-inflammatory drugs (3).
Celecoxib is a highly lipophilic, poorly soluble drug with oral bioavailability between 22% and 40% by conventional capsule dosage form (4). It is evenly distributed in vivo and has a volume of distribution of 455 ± 166 L in humans (5). This larger volume of distribution and low aqueous solubility may be related to the lipophilic nature of celecoxib and be reflective of low bioavailability. Celecoxib is extensively metabolized in humans and is excreted primarily as metabolites (4).
Many formulation approaches have been attempted to improve bioavailability of celecoxib using various solvent systems (6), complexation with β-cyclodextrins (7,8), solid dispersions (9), manipulation of the solid state of the drug (10,11), development of floating celecoxib capsule (12), and using silica–lipid hybrid microcapsules (13).
Niosomes are unilamellar or multilamellar vesicles that are made up of nonionic surfactant and can entrap amphiphilic and hydrophobic solutes (14,15). Niosomes have shown advantages as drug carriers, such as being a cheap and chemically stable alternative to liposomes, but they are associated with problems related to physical stability, such as fusion, aggregation, sedimentation, and leakage on storage (16). The proniosomes which is more stable during sterilization and storage (17) minimizes these problems by using dry, free-flowing particles that immediately form niosomal dispersion when in contact with water. Proniosomes are suitable for administration by oral or other routes (18).
Preliminary studies indicate that niosomes may increase the absorption of certain drugs from the gastrointestinal tract following oral ingestion and prolong the existence of the drug in systemic circulation (14). The encapsulation of celecoxib in lipophilic vesicular structure may be expected to enhance the oral absorption and prolong the existence of the drug in systemic circulation of the drug due to the slow release of the encapsulated drug.
Accordingly, the objective of this study is to prepare celecoxib proniosomes and evaluate the influence of proniosomal formulation on its oral bioavailability in healthy human volunteers.
MATERIALS AND METHODS
Materials
Celecoxib was a gift sample from Sedico pharmaceutical company, Egypt. Cholesterol, sorbitol and dicetyl phosphate were obtained from Fluka Biochemika Company, Sigma Germany. Span 60 (sorbitan monosterarate) was from Merk Schuchardit OHG, Germany. Chloroform for high-performance liquid chromatography (HPLC) was from RPS Chemicals Co. ltd., London, England. Ethyl alcohol absolute 99% was from United Company for Chem. and Med. Prep., Egypt. All other materials used were of pharmacopeial grade.
Preparation of Celecoxib Proniosomes
Proniosomes were prepared by sequential spraying method (18), where cholesterol, span 60, and dicetyl phosphate in a molar ratio of 1:1:0.1 were dissolved in 10 ml chloroform–methanol (1:1 v/v) containing 100 mg of celecoxib. The lipid mixture was introduced into a 100-ml rounded bottom flask containing 0.5 gm sorbitol powder by sequential spraying of aliquots onto the surface of sorbitol powder. The flask was attached to a rotary evaporator (Büchi, R-210/215, Labortehnik AG, CH-2930, Flawil, Switzerland) to evaporate the solvent at 100 rpm, a temperature of 60–70°C, and a reduced pressure of 600 mmHg until sorbitol appeared to be dry. This process was repeated until all surfactant solution had been applied. Evaporation was continued until the content in the flask had become a completely dry and freely flowing product. This dry product, referred to as proniosomes, was stored in a tightly closed container and was used for preparation of proniosome-derived niosomes and for further evaluation.
Preparation of Celecoxib Niosomes from Proniosomes
Proniosomes were transformed to niosomes by hydrating with 10 ml of 37°C distilled water and agitation for 2 min. The resulting niosome dispersion was used for the determination of the entrapment efficiency, particle size analysis, and morphological studies.
In Vitro Studies
Transmission Electron microscope
The morphology of hydrated niosomes prepared from proniosomes was determined using transmission electron microscopy (Jeol Jem Dos electron microscopy, Japan). The prepared sample was stained with 2% potassium phosphotungstate.
Particle Size and Particle Size Distribution
The vesicle size distribution was determined using a laser diffraction technique on a Mastersizer X Ver. 2.15; Malvern instruments Ltd. Malvern, UK. The measurements were performed at 25°C, using a 45-mm focus lens and a beam length 2.4 mm.
Determination of Celecoxib Entrapment Efficiency in Niosomes
Free celecoxib was separated from niosome-entrapped celecoxib by centrifugation (18). A 1-ml aliquot of niosome dispersion was centrifuged (Centurion Scientific Ltd) at 15,000 rpm at 4°C for 1 h. The supernatant was separated, and the niosomal residue was resuspended in distilled water and centrifuged again. This washing procedure was repeated two times to ensure that the free drug was no longer present in the voids between the niosomes. The collected supernatant fractions were diluted to 10 ml with methanol and were used for determination of the free celecoxib spectrophotometrically at λ = 254 nm as this wavelength represents the maximum absorption of celecoxib in methanol. The amount of entrapped celecoxib was obtained by subtracting the amount of free drug from the total drug incorporated in 1 ml niosomal dispersion.
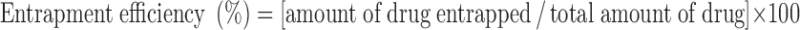 |
Dissolution Study
The dissolution behavior of celecoxib proniosomes was compared with pure celecoxib powder. The dissolution studies were performed according to the US Pharmacopeia (USP) type II apparatus (paddle method) (Varian, Inc., 1300 Weston Parkway, Cary, NC, USA). The samples (celecoxib proniosomes and pure celecoxib) corresponding to 50 mg celecoxib were placed into hard gelatin capsule. The dissolution medium was 900 phosphate buffer (pH 6.8) containing 1% sodium lauryl sulfate to maintain sink conditions (13,19). Before dissolution studies, the solubility of celecoxib in the dissolution medium was measured, where an excess amount of celecoxib was added to 5 ml phosphate buffer (pH 6.8) containing 1% (w/v) sodium lauryl sulfate. The mixture was then kept at ambient temperature for 72 h in a shaker water bath to get equilibrium. The equilibrated samples were centrifuged at 3,000 rpm for 5 min. Aliquot portions of the supernatants were taken and properly diluted with phosphate buffer (pH 6.8) for quantification of celecoxib spectrophotometrically at 254 nm. The stirring speed was 100 rpm, and the temperature was maintained at 37°C ± 1°C. The samples (3 ml) were withdrawn at various time intervals (15, 30, 45, 60, 90, 120, 180, and 210 min) using a syringe, filtered through 0.2 µm membrane filter, and analyzed by UV spectrophotometer at 254 nm. Withdrawn samples were compensated by fresh medium. The dissolution experiments were conducted in triplicate.
In Vivo Evaluation of Proniosomes
Study Design
The studies were carried out to compare the pharmacokinetics of celecoxib proniosomal capsules to marketed celecoxib capsules (Celebrex®, Pfizer, Egypt) following administration of a single oral dose of 200 mg of each, using a nonblind, two-treatment, two-period, randomized, crossover design with a washout period of 2 weeks between the two phases. The study was approved by the University Protection of Human Subjects Committee, and the protocol complies with the declarations of Helsinki and Tokyo for humans. Six healthy male volunteers between 24 and 43 years old and weighing from 60 to 84 kg participated in the study after giving informed written consent. All were judged to be healthy and were not receiving any medication during the study period. The volunteers were randomly selected to receive two capsules (100 mg/capsule) of either Celebrex® or celecoxib proniosomal capsule. Both products were administered with 150 ml of water in the morning after a 12-h overnight fast. Food and drinks were withheld for at least 2 h after dosing. Blood samples (4 ml) were collected in heparinized tubes for the measurement of celecoxib at 30 min predose and at 1, 2, 3, 4, 5, 6, 12, 24, and 48 h postdose. The blood samples were centrifuged at 3,000 rpm for 10 min, and the plasma was transferred to separate glass tubes to be kept frozen until analysis.
Analysis of Plasma Levels of Celecoxib
A simple and rapid HPLC assay was developed for determination of celecoxib in human plasma. The HPLC system consisted of a Shimadzu LC-10 AD VP Liquid Chromatograph using ODS-H Optimal Column (150 × 3.9 mm) and a model SPD-10A VP UV–VIS detector (Shimadzu, Japan) equipped with a Rheodyne injector (Model 7161, Catati, California, USA equipped with 20 µl injector loop). Peak areas were determined with a C-R6A Chromatopac Shimadzu integrator. The wavelength was set at 254 nm. The mobile phase used methanol/distilled water at a proportion of 70:30. The mobile phase was prepared daily and degassed by a DGU-12A Shimadzu degasser. Analysis was run at a flow rate of 1 ml/min.
Standard Solutions
Standard solutions of celecoxib and internal standard (propyl paraben) were prepared by dissolving 10 mg of each in 100 ml of methanol. The working standard solution for each was prepared by taking 10 ml from the above solutions in 100 ml methanol (10 µg/ ml).
Calibration Curve
Standard samples were prepared by transferring aliquots of standard solution into centrifuge tubes provided with tight sealing polyethylene caps, containing 1 ml of blank plasma to yield final celecoxib concentrations of 50, 100, 200, 300, 500, 1,000, and 1,500 ng/ml. Internal standard solution was added to each tube to yield a concentration of 10 µg/ml. The final volume in each tube was completed to 6 ml with ether. After mixing by vortex for 20 s, the mixture was centrifuged for 10 min at 3,000 rpm. The upper layer was transferred to another tube filtered through 0.45 µm millipore filter and evaporated to dryness under nitrogen stream at ambient temperature, and the residue was reconstituted in 400 µl of the mobile phase; 20 µl was injected on the column for analysis. Under the conditions described, the retention times of celecoxib and propyl paraben were 6.13 and 3.10 min, respectively. A standard curve was constructed by plotting the peak area ratio of celecoxib to propyl paraben versus celecoxib concentration in plasma. All assays were performed in triplicate.
Plasma Analysis
One milliliter of the different plasma samples obtained from the volunteers was assayed as described above without the addition of celecoxib.
Pharmacokinetic Analysis
Celecoxib pharmacokinetics parameters were determined by noncompartmental kinetics (20). The elimination rate constant (Kel) was estimated by least square regression of plasma concentration–time data points in the terminal log-linear region of the curves. Half life (t1/2) was calculated as 0.693 divided by Kel. The area under the plasma concentration–time curve from zero to the last measurable plasma concentration at time t (AUC0–t) was calculated using linear trapezoidal rule. The area under the curve from zero to infinity, 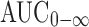 was calculated as
was calculated as 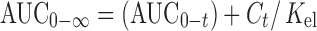 , where Ct is the last measured concentration at the time. Peak plasma concentration (Cmax) and the time to peak concentration (Tmax) were obtained directly from the individual plasma concentration versus time curve. The absorption rate constant (Ka) was determined from the plasma concentration–time data by Wagner Nelson method (21).
, where Ct is the last measured concentration at the time. Peak plasma concentration (Cmax) and the time to peak concentration (Tmax) were obtained directly from the individual plasma concentration versus time curve. The absorption rate constant (Ka) was determined from the plasma concentration–time data by Wagner Nelson method (21).
Statistical Analysis
An analysis of variance (ANOVA) was performed for untransformed data for the pharmacokinetic parameters Cmax, AUC0–t, 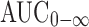 , Ka, and t1/2 using the software SPSS 11.0 (SPSS Inc., Chicago, USA). The values of Tmax were analyzed using Wilcoxon signed rank test for the paired samples. A statistically significant difference was considered at P value <0.05.
, Ka, and t1/2 using the software SPSS 11.0 (SPSS Inc., Chicago, USA). The values of Tmax were analyzed using Wilcoxon signed rank test for the paired samples. A statistically significant difference was considered at P value <0.05.
RESULTS AND DISCUSSION
In Vitro Evaluation of Proniosomes
Transmission Electron Microscopy
Figure 1 shows the produced niosomes prepared from proniosomes. As observed, they are well-identified perfect spheres, and they exist in dispersed and aggregate collections.
Fig. 1.

Transmission electron microphotograph of celecoxib proniosome-derived niosomes stained with 2% potassium phosphotungstate
Size Distribution
Particle size analysis of the proniosome-derived niosomes shows that the average ± SD (µm) of particle size of 90% of the particle is 1 ± 0.12 µm with polydispersity index of 0.1167. The polydispersity index was calculated according to Bhavana et al., (22). A polydispersity index of 1 indicates large variations in particle size; a reported value of 0 means that size variation is absent (23). The obtained low value of polydispersity index of proniosome-derived niosomes indicates a limited variation in particle size.
Entrapment Efficiency
The entrapment was expressed as a percentage of the total amount of celecoxib used in preparation of the proniosomes. The mean entrapment efficiency of proniosome-derived niosomes was 95.14 ± 1.31%. The high entrapment efficiency is probably due to the lipophilic character of celecoxib. The concentration of celecoxib was changed from 50 to 100 mg celecoxib to verify that the drug concentration was not significantly altering the behavior of the niosome preparation. The entrapment efficiency of niosomes prepared by both celecoxib concentrations showed no significant difference, remaining at about 95%.
Dissolution Study
Celecoxib has been reported to have aqueous solubility of 3–7 µg/ml (5). Therefore, to maintain sink conditions, phosphate buffer (pH 6.8) with 1% sodium lauryl sulfate was used as the dissolution medium for the in vitro dissolution studies of celecoxib (13,19). The solubility of celecoxib in the dissolution medium at room temperature was 106 ± 2.64 µg/ml (mean ± SD, n = 3) as measured spectrophotometrically at 254 nm.
The dissolution profiles of celecoxib from proniosomes and pure drug are illustrated in Fig. 2. The pure celecoxib appeared to exhibit higher and faster release in the first 30 min than from the proniosomes. The pure drug showed 31% release after 30 min, and the proniosomes showed only 22%. After 60 min, the celecoxib release from proniosomes had reached about 48% and increased to reach about 75% after 210 min, while pure celecoxib reached 38% and 49% after 60 and 210 min, respectively.
Fig. 2.

Dissolution profiles of celecoxib proniosomes and pure celecoxib in 900 ml phosphate buffer (pH 6.8) containing 1% sodium lauryl sulfate (mean ± SD; n = 3)
These results pointed to initial slow release of celecoxib from proniosomes. This slow release is due to the small percent of un-entrapped drug and the slow release of the entrapped drug from proniosome-derived niosomes. This can be due to the fact that the presence of cholesterol at a high percentage reduces the leakage or permeability of encapsulating material by decreasing the niosomal membrane fluidity (24). Also at 25°C, the molecules of span 60 are in ordered gel state. The subsequent increase in celecoxib release from proniosomes after 1 h may be due to the presence of charged lipid dicetyl phosphate within the niosomal bilayer which increases the permeability of the bilayer and also is responsible for increase in the curvature and decrease in the size of the vesicles which provides the maximum surface area exposed to the dissolution medium resulting into the higher release (22).
In Vivo Evaluation of Proniosomes
The average plasma celecoxib concentration–time curves after a single oral dose of 200 mg celecoxib as proniosomal capsules and Celebrex® capsules as a reference drug product for celecoxib are shown in Fig. 3. The pharmacokinetic parameters of celecoxib were calculated from individual curves, and the mean values are presented in Table I.
Fig. 3.

Mean plasma concentration–time curves of celecoxib (±SD) in human volunteers (n = 6) after oral administration of 200 mg as single oral dose of proniosomal formulation and Celebrex® capsules
Table I.
Pharmacokinetic Parameters of Celecoxib in Human Volunteers (n = 6) After Administration of 200 mg as Single Oral Dose of Proniosomal Formulation and Celebrex® Capsules
| PK parameter | Conventional capsule | Proniosomal capsule |
|---|---|---|
| C max (ng/ml) | 705.17 ± 31.89 | 842.5 ± 34.72* |
| AUC0–48 (ng.h/ml) | 6,761.58 ± 612.73 | 11,681.21 ± 523.90* |
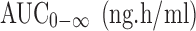
|
7,210.58 ± 689.02 | 12,406.21 ± 469.11*a |
| T max (h) | 2.5 ± 0.55 | 4 ± 0.00** |
| K a (h−1) | 1.52 ± 0.17 | 0.73 ± 0.05* |
| K el (h−1) | 0.051 ± 0.01 | 0.059 ± 0.002 |
| t 1/2 (h) | 13.33 ± 1.98 | 11.75 ± 0.55 |
Each value represents the mean ± standard division of six subjects
*P < 0.05 based on ANOVA, statistically significant difference between conventional and proniosomal capsules
**P < 0.05 based on Wilcoxon signed rank test, statistical significant difference between conventional and proniosomal capsules
Statistical analysis of the pharmacokinetic parameters Cmax, AUC0–48, 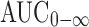 , and Tmax data obtained for proniosomal preparation showed significantly (P < 0.05) higher values and significantly lower values for absorption rate constant (Ka) compared with Celebrex® capsule. There was no significant difference between the values of Kel and t1/2 for both celecoxib preparations.
, and Tmax data obtained for proniosomal preparation showed significantly (P < 0.05) higher values and significantly lower values for absorption rate constant (Ka) compared with Celebrex® capsule. There was no significant difference between the values of Kel and t1/2 for both celecoxib preparations.
The average value of Cmax was 842.50 and 705.17 ng/ml after oral administration of proniosomal and conventional capsules, respectively. This result indicated that proniosomes significantly increase the Cmax of celecoxib. The mean Tmax values for proniosomal formulation and the reference celecoxib capsule were 4 and 2.5 h, respectively; the difference between the mean Tmax values was found to be statistically significant (P < 0.05) when analyzed by Wilcoxon’s signed-rank test. The delayed occurrence of Tmax with the proniosomal formulation is due to the slow release of the drug from proniosome-derived niosomes which is reflected on the significantly lower values of absorption rate constant Ka of proniosomal formulation (0.73 h−1) in comparison with Ka of the reference celecoxib capsule (1.52 h−1). Therefore, this slow release can prolong the localization of the drug at the site of absorption and can affect the extent of drug absorption.
The individual 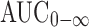 values for the proniosomal capsule were compared to those for the Celebrex® capsule to determine the relative bioavailability. The mean relative bioavailability of the proniosomal formulation to the conventional capsule was 172.06 ± 0.14%. This result indicated that 72.06% increase in the oral bioavailability of celecoxib was achieved by the proniosomal formulation. Similar results were reported by Xiao et al. (25), who studied the effect of oral proliposome on silymarin bioavailability.
values for the proniosomal capsule were compared to those for the Celebrex® capsule to determine the relative bioavailability. The mean relative bioavailability of the proniosomal formulation to the conventional capsule was 172.06 ± 0.14%. This result indicated that 72.06% increase in the oral bioavailability of celecoxib was achieved by the proniosomal formulation. Similar results were reported by Xiao et al. (25), who studied the effect of oral proliposome on silymarin bioavailability.
CONCLUSION
The results reported here indicate that proniosomal capsule formulation can be successfully used to enhance the bioavailability of celecoxib.
References
- 1.Geis GS. Update on clinical development with celecoxib, a new specific COX-2 inhibitor: what can we expect? Scand J Rheumatol. 1999;28:31–37. doi: 10.1080/030097499750042407. [DOI] [PubMed] [Google Scholar]
- 2.Simon LS, Lanza FL, Lipsky PE. Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase-2 inhibitor. Arthritis Rheum. 1998;41:1591–1602. doi: 10.1002/1529-0131(199809)41:9<1591::AID-ART9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 3.Tindall E. Celecoxib for the treatment of pain and inflammation: the preclinical and clinical results. J Am Osteopath Assoc. 1999;99:S13–S17. doi: 10.7556/jaoa.1999.99.11.S13. [DOI] [PubMed] [Google Scholar]
- 4.Paulson SK, Hribar JD, Liu NW, Hajdu EJ, Bible RH, Piergies A, Karim A. Metabolism and excretion of celecoxib in healthy male volunteers. Drug Metab Dispos. 2000;28:308–314. [PubMed] [Google Scholar]
- 5.Paulson SK, Vaughn MB, Jessen SM, Lawal Y, Gresk CJ, Yan B, Maziasz TJ, Cook CS, Karim A. Pharmacokinetics of celecoxib after oral administration in dogs and humans; effect of food and site of absorption. J Pharmacol Exp Ther. 2001;297(2):638–645. [PubMed] [Google Scholar]
- 6.Neelam S, Sonu B. Solubility enhancement of Cox-2 inhibitors using various solvent systems. AAPS PharmSciTech 2003;4(3):Article 33. [DOI] [PMC free article] [PubMed]
- 7.Narender R, Tasneem R, Ramakrishna S, Chowdary K, Prakash V. Β-cyclodextrin complexes of celecoxib: molecular-modeling, characterization, and dissolution studies. AAPS PharmSciTech. 2004;6(1):Article 7. [DOI] [PMC free article] [PubMed]
- 8.Nagarsenker MS, Joshi MS. Celecoxib-cyclodextrin systems: characterization and evaluation of in vitro and in vivo advantage. Drug Dev Ind Pharm. 2005;31(2):169–178. doi: 10.1081/DDC-200047795. [DOI] [PubMed] [Google Scholar]
- 9.Dixit R, Nagarsenker M. In vitro and in vivo advantage of celecoxib surface solid dispersion and dosage form development. Indian J Pharm Sci. 2007;69(3):370–377. doi: 10.4103/0250-474X.34545. [DOI] [Google Scholar]
- 10.Gupta VR, Mutalik S, Patel MM, Jani GK. Spherical crystals of celecoxib to improve solubility, dissolution rate and micromeritic properties. Acta Pharm. 2007;57(2):173–184. doi: 10.2478/v10007-007-0014-8. [DOI] [PubMed] [Google Scholar]
- 11.Guzman HR, Tawa M, Zhang Z, Ratanabanangkoon P, Shaw P, Gardner CR, Chen H, Moreau JP, Almarsson O, Remenar JF. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J Pharm Sci. 2007;96(10):2686–2702. doi: 10.1002/jps.20906. [DOI] [PubMed] [Google Scholar]
- 12.Javed A, Shweta A, Alka A, Anil K, Rakesh K, Roop K. Formulation and development of floating capsules of celecoxib: in vitro and in vivo evaluation. AAPS PharmSciTech. 2007;8(4):Article 119. [DOI] [PMC free article] [PubMed]
- 13.Angel T, Spomenka S, Andrew K, Thomas R, Clive A. Silica-lipid hybrid (SLH) microcapsules: a novel oral delivery system for poorly soluble drugs. J Control Release. 2009;134:62–70. doi: 10.1016/j.jconrel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Azmin MN, Florence AT, Handjani-Vila RM, Stuart FB, Vanlerberghe G, Whittaker JS. The effects of nonionic surfactant vesicles (niosomes) entrapment on the absorption and distribution of methotrexate in mice. J Pharm Pharmacol. 1985;37:237–242. doi: 10.1111/j.2042-7158.1985.tb05051.x. [DOI] [PubMed] [Google Scholar]
- 15.Baillie AJ, Florence AT, Hume LR, Muirhead GT, Rogerson A. The preparation and properties of niosomes-non-ionic surfactant vesicles. J Pharm Pharmacol. 1985;37:863–868. doi: 10.1111/j.2042-7158.1985.tb04990.x. [DOI] [PubMed] [Google Scholar]
- 16.Namdeo A, Jain NK. Niosomal delivery of 5-fluorouracil. J Microencapsul. 1999;16:731–740. doi: 10.1080/026520499288672. [DOI] [PubMed] [Google Scholar]
- 17.Blazek-Welsh AI, Rhodes DG. SEM imaging predicts quality of niosomes from maltodextrin-based proniosomes. Pharm Res. 2001;18:656–661. doi: 10.1023/A:1011037527889. [DOI] [PubMed] [Google Scholar]
- 18.Hu C, Rhodes DG. Proniosomes: a novel drug carrier preparation. Int J Pharm. 1990;185:23–35. doi: 10.1016/S0378-5173(99)00122-2. [DOI] [PubMed] [Google Scholar]
- 19.Kora PR, Sekuboyina VS. Influence of hydrophilic polymer on celecoxib complexation with hydroxypropyl β-cyclodextrin. AAPS PharmSciTech 2006;7(3):Article 79. [DOI] [PMC free article] [PubMed]
- 20.Gilbaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker; 1982. pp. 409–417. [Google Scholar]
- 21.Shargel L, Wu-pong S, Yu A. Applied biopharmaceutics &pharmacokinetics. 5th ed. the McGraw-Hill Companies; 2005;171-3.
- 22.Bhavana V, Ajay JK, Jain NK. Proniosome based transdermal delivery of levonorgestrel for effective contraception. J Control Release. 1998;54:149–165. doi: 10.1016/S0168-3659(97)00100-4. [DOI] [PubMed] [Google Scholar]
- 23.Raymond M, Josbert M, Marcel H, Adriënne P, Grietje M, Gert S. Liposome-encapsulated prednisolone phosphate inhibits growth of established tumors in mice. Neoplasia. 2005;7(2):118–127. doi: 10.1593/neo.04340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Betageri GV. Liposomal encapsulation and stability of dideoxyinosine triphosphate. Drug Dev Ind Pharm. 1993;19:532–539. [Google Scholar]
- 25.Xiao Y, Song Y, Chen Z, Ping Q. Preparation of silymarin proliposome: a new way to increase oral bioavailability of silymarin in beagle dogs. Int J Pharm. 2006;319:162–168. doi: 10.1016/j.ijpharm.2006.03.037. [DOI] [PubMed] [Google Scholar]