

Abstract
Objective
Pulmonary hypertension is a characteristic feature of acute respiratory distress syndrome (ARDS) and contributes to mortality. Administration of sildenafil in ambulatory patients with pulmonary hypertension improves oxygenation and ameliorates pulmonary hypertension. Our aim was to determine whether sildenafil is beneficial for patients with ARDS.
Design
Prospective, open-label, multicenter, interventional cohort study.
Setting
Medical-surgical ICU of two university hospitals.
Patients
Ten consecutive patients meeting the NAECC criteria for ARDS.
Interventions
A single dose of 50 mg sildenafil citrate administered via a nasogastric tube.
Main results
Administration of sildenafil in patients with ARDS decreased mean pulmonary arterial pressure from 25 to 22 mmHg (P = 0.022) and pulmonary artery occlusion pressure from 16 to 13 mmHg (P = 0.049). Systemic mean arterial pressures were markedly decreased from 81 to 75 mmHg (P = 0.005). Sildenafil did not improve pulmonary arterial oxygen tension, but resulted in a further increase in the shunt fraction.
Conclusion
Although sildenafil reduced pulmonary arterial pressures during ARDS, the increased shunt fraction and decreased arterial oxygenation render it unsuitable for the treatment of patients with ARDS.
Electronic supplementary material
The online version of this article (doi:10.1007/s00134-010-1754-3) contains supplementary material, which is available to authorized users.
Keywords: Sildenafil, ARDS, Hypoxemia, Pulmonary hypertension, ICU
Introduction
With an incidence of 24 per 100,000 in The Netherlands, acute respiratory distress syndrome (ARDS) is a common cause of respiratory insufficiency requiring mechanical ventilation and admission to an intensive care unit (ICU) [1]. Both direct (e.g., pneumonia, inhalation injury) and indirect lung injury (e.g., sepsis, acute pancreatitis) can induce ARDS [2], which is characterized by pulmonary edema (resulting from increased vascular permeability), atelectasis and a ventilation-perfusion mismatch. As a result, impaired gas exchange and hypoxemia develop. Due to hypoxia, the pulmonary arteries constrict, a phenomenon known as hypoxic pulmonary vasoconstriction [3]. Thrombo-embolic occlusions in the pulmonary microvasculature [4], increased production of vasoconstrictive agents such as endothelin-1 [5] and thromboxane A2 [6], and pulmonary edema further augment pulmonary hypertension, which is a hallmark of ARDS [7]. The presence of pulmonary hypertension contributes to mortality [8]. Current treatment of ARDS is supportive. Specific therapeutic strategies are lacking.
Inhalation of nitric oxide (NO) dilates the pulmonary vasculature and enhances perfusion to ventilated lung regions, thereby improving oxygenation in patients with ARDS [9, 10]. However, NO inhalation as an adjunctive therapy in ARDS is controversial, since a considerable number of patients are unresponsive, and NO also has adverse effects, including renal dysfunction [11, 12].
Impaired vasoreactivity is likely to explain unresponsiveness to inhaled NO. Under physiological conditions, NO initiates the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP) in pulmonary endothelial cells. Activation of cGMP-dependent protein kinases induces relaxation of smooth muscle cells and thus vasodilation. A family of enzymes called phosphodiesterases (PDE) inactivates cGMP by converting it to GMP [13]. During sepsis, pulmonary cGMP release may be impaired, as suggested in an animal model of endotoxemia, thereby accounting for the pulmonary vasounresponsiveness to NO [14]. Additionally, increased PDE activity may contribute to enhanced degradation of cGMP [15, 16]. Inhibition of PDE may therefore improve or even replace inhalational NO therapy in ARDS. In accordance, previous animal studies have demonstrated beneficial effects of PDE inhibitors in experimental ARDS by attenuating endotoxin-induced vasomotor dysfunction [17–19]. Besides influencing vasoreactivity, PDE inhibitors also have antiinflammatory properties, decreasing pulmonary edema, neutrophil infiltration, reactive oxygen species and levels of proinflammatory mediators in experimental acute lung injury [17–19]. These preclinical results suggest that inhibiting PDE may be a potential therapeutic target in ARDS.
Sildenafil selectively inhibits the isoenzyme PDE type 5 (PDE5), which is specific for cGMP [20]. It has been demonstrated that sildenafil increases exercise capacity during severe hypoxia in healthy volunteers [21] and improves gas exchange in patients with pulmonary hypertension [22] by selectively dilating arteries in well-ventilated areas of the lung [23]. We hypothesized that sildenafil administered to patients with ARDS would improve hypoxemia by regulation of vascular tone, thereby decreasing the shunt fraction.
Materials and methods
Patients
This prospective, interventional cohort study was approved by the Ethics Committee of the VU University Medical Center. Written informed consent was obtained from the patients' next of kin. We enrolled ten patients within 1 week after establishing the diagnosis of ARDS; they were older than 18 years and admitted to the mixed medical/surgical ICUs of two university hospitals. ARDS was diagnosed according to the North American European Consensus Conference (NAECC) definition of ARDS [24]: partial arterial oxygen tension/fraction of inspired oxygen (PaO2/FiO2) (P/F) ratio of less than 200 mmHg, bilateral consolidations on a chest X-ray and the absence of hydrostatic pulmonary edema, which was defined as a pulmonary artery occlusion pressure (PAOP) ≤18 mmHg. In addition, a risk factor associated with ARDS had to be present. We excluded patients with coronary artery disease, bilateral pneumonia (patients with an initial presentation of bilateral infiltrates), preexistent lung fibrosis and hepatic insufficiency, as well as patients who were treated with nitrates, other PDE inhibitors, prostacyclins or more than 300 mg of hydrocortisone (or its equivalent of other steroids) per day. Bilateral pneumonia and lung fibrosis were exclusion criteria because of a presumed absence of vasoreactivity. We also excluded patients in whom nasogastric feeding (i.e., administration of medication) was not possible because of an underlying condition.
Study protocol
Demographics, reason for admission to the ICU and medical history were taken from the chart. To assess disease severity, the Acute Physiology and Chronic Health Evaluation (APACHE II) score was calculated using the worst variables in a 24-h timeframe preceding enrollment [25]. When not already in place, a pulmonary artery catheter was inserted. In all patients, central venous pressure (CVP), pulmonary arterial pressure (PAP) and cardiac output (CO) were measured by a continuous cardiac output monitoring balloon-tip thermodilution pulmonary artery catheter (Baxter Edwards Critical Care, Irvine, CA) inserted via the internal jugular or subclavian vein. Pulmonary artery occlusion pressure (PAOP) was obtained from graphic recordings at end-expiration at the level of the heart without discontinuation of PEEP. All pressures were obtained after calibration, zeroing to atmosphere and using the midchest level as reference. Arterial blood pressure was continuously monitored with an intraarterial catheter. Hemodynamic variables were recorded by a patient data monitoring system.
One tablet of 50 mg of sildenafil was ground and dissolved in 20 ml of 0.9% NaCl. After baseline measurements (t = 0), the ground sildenafil was administered as a single dose through the nasogastric tube. Hemodynamic variables were recorded at t = 30, 60, 90, 120, 150, 180, 210, 240 and 360 min after the sildenafil was administrated. At the same time points, arterial blood was drawn to determine the levels of sildenafil and desmethylsildenafil. Blood gas analysis of both arterial and mixed venous blood was performed (Rapid Lab 865, Bayer Diagnostics, Dublin, Ireland). The shunt fraction (Q
s/Q
t) was calculated using a standard formula, 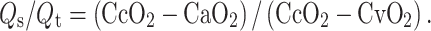 Ideally, ventilator settings were not changed, and dosages of vasopressors were not increased unless deemed necessary by the discretion of the treating physician during the 6 h of monitoring.
Ideally, ventilator settings were not changed, and dosages of vasopressors were not increased unless deemed necessary by the discretion of the treating physician during the 6 h of monitoring.
Measurements of sildenafil levels
Serum concentrations of sildenafil and its metabolite desmethylsildenafil were measured using a validated liquid-gas chromatography mass-spectrometry method [26]. The calibration curve exhibited a linear range of 0.05–625 ng/ml with a 2-ng/ml limit for detection for sildenafil and a range of 0.05–280 ng/ml for desmethylsildenafil with a 3-ng/ml detection limit. The intra- and inter-day precision was within 6% for both compounds. Intra- and inter-day accuracy ranged from 92–103%.
Statistical analysis
All data are summarized as median (range) or absolute numbers where appropriate. Measurements were compared to t = 0 using the Wilcoxon signed rank test. The Spearman correlation coefficient was used to express relations using baseline and t = 30 min. A P value <0.05 was considered statistically significant. Statistical analysis was performed using SPSS 14.0 (SPSS, Chicago, IL) and Prism 5.0 (GraphPad Software, San Diego, CA).
Results
Between 1 December 2005 and 1 December 2007, 21 ARDS patients were assessed for eligibility. Eleven patients were excluded: three were treated with another PDE inhibitor (enoximone), three had contraindications to enteral medications, and one patient suffered from pulmonary hemorrhage and was excluded for safety reasons. In three patients, consent was not provided. One patient was excluded after enrollment because findings on a CT scan performed on the day after the study protocol had been carried out rendered him ineligible. The radiological images indicated atelectasis, not ARDS.
Patient characteristics and baseline hemodynamic and respiratory parameters are displayed in Table 1. Sepsis and pneumonia were the most frequent causes of ARDS. All patients had severe pulmonary injury with pulmonary hypertension and an increased pulmonary shunt fraction. The ICU mortality rate was 10%.
Table 1.
Baseline measurements
| General | n = 10 |
|---|---|
| Age (years) | 62.5 (40–78) |
| Sex (m/f) | 7/3 |
| APACHE II | 18.5 (9–29) |
| Cause of ARDS | |
| Sepsis | 4 |
| Pneumonia | 2 |
| Pleural empyema | 1 |
| Post CABG | 2 |
| Post open-repair AAA | 1 |
| Hemodynamic parameters | |
| Heart rate (/min) | 84 (64–108) |
| MAP (mmHg) | 81 (68–115) |
| CVP (mmHg) | 12 (5–15) |
| MPAP (mmHg) | 25 (23–32) |
| PAOP (mmHg) | 16 (8–18) |
| CO (l/min) | 6.85 (4.80–11.70) |
| CI (l/min*m2) | 3.66 (2.68–5.44) |
| SVRI (dyne*s/cm5) | 1,632 (1,058–2,425) |
| PVRI (dyne*s/cm5) | 234 (139–478) |
| Vasopressors (yes/no) | 7/3 |
| Respiratory parameters | |
| PaO2/FiO2 ratio | 144 (122–200) |
| PaO2 (mmHg) | 84 (72–103) |
| PEEP (cmH2O) | 10 (8–18) |
| SvO2 | 0.65 (0.57–0.72) |
| A-a gradient (mmHg) | 319 (223–362) |
| Q s/Q t | 24 (18–31) |
Data are median (range) or absolute numbers: APACHE Acute Physiology and Chronic Health Evaluation; CABG coronary artery bypass grafting; AAA abdominal aortic aneurysm; MAP mean arterial pressure; CVP central venous pressure; MPAP mean pulmonary artery pressure; PAOP pulmonary artery occlusion pressure; CO cardiac output; CI cardiac index; SVRI systemic vascular resistance index; PVRI pulmonary vascular resistance index; PEEP positive end expiratory pressure; SvO 2 mixed venous saturation; Q s/Q t shunt fraction
Effects on hemodynamic parameters
All hemodynamic effects were most apparent after 30 min. The mean pulmonary arterial pressure (MPAP) declined after sildenafil from 25 (23–32) mmHg at baseline to 22 (18–26) mmHg at t = 30 and remained at this level until t = 180 (P ≤ 0.022) (Fig. 1). The PAOP decreased from 16 (8–18) mmHg at baseline to 13 (8–18) mmHg at t = 30 (P = 0.049) and declined further to 12.5 (7–21) mmHg at t = 60 (P = 0.027). Sildenafil induced vasodilation that was not selective for the pulmonary vasculature, but decreased the systemic mean arterial pressure (MAP) from 81 (68–115) to 75 (66–98) mmHg at t = 30 (P = 0.005) and remained below baseline levels for 3.5 h (Fig. 1). In four of the seven patients requiring vasopressors, the dose was increased to maintain a mean arterial pressure of at least 65 mmHg. This typically occurred 30–60 min after sildenafil administration. Sildenafil had a rapid effect on the systemic vascular resistance index (SVRI), decreasing from 1,688 (1,058–2,425) dyne*s/cm5 at baseline to 1,400 (917–1,986) dyne*s/cm5 at t = 30 (P = 0.012). The pulmonary vascular resistance index (PVRI) declined from 215 (139–478) dyne*s/cm5 at baseline to 170 (71–418) dyne*s/cm5 only after 90 min to remain at this level until t = 210 (P ≤ 0.047). The cardiac index (CI) was 3.4 (2.7–5.4) at baseline, which did not change over time (at t = 30, CI was 3.7 (2.6–5.6) and at t = 90: 3.9 (2.1–5.5), NS). The left ventricular stroke work index was not affected; however, the right ventricular stroke work index decreased (P ≤ 0.038). The ratios of PVRI to SVRI and of MPAP to MAP did not change over time.
Fig. 1.

Boxplot of hemodynamic variables during the study period displayed as median, interquartile range (box) and range (whiskers). An asterisk indicates statistically significant change as compared to t = 0 (Wilcoxon signed ranks test)
Effects on respiratory parameters
Sildenafil administration tended to decrease PaO2 from 87 (72–100) mmHg at baseline to 70 (54–131) mmHg at t = 30 (P = 0.074), slowly returning to baseline level after 210 min. The P/F-ratio decreased concordantly from 144 (122–200) at baseline to 115 (77–218) at t = 30 (P = 0.074), returning to baseline at t = 150. The levels of PEEP and FiO2 were not changed during the observation period. Simultaneously with the decrement of PaO2, the shunt fraction increased from 24 (18–31)% to 31 (21–42)% (P ≤ 0.037), returning to baseline at t = 120 (Fig. 2). The alveolo-arterial (A-a) gradient was not significantly affected (data not shown). Sildenafil did not alter levels of PaCO2 (data not shown). There was no statistically significant correlation between decrease in shunt fraction and decrease in MAP, nor between decrease in shunt fraction and decrease in MPAP.
Fig. 2.

Boxplot of shunt fraction during the study period displayed as median, interquartile range (box) and range (whiskers). An asterisk indicates statistically significant change as compared to t = 0 (Wilcoxon signed ranks test)
Levels of sildenafil
The maximum plasma concentration (C max) of sildenafil was reached between t = 30 and t = 120, and ranged from 107 to 975 ng/ml. The C max of desmethylsildenafil, the active metabolite of sildenafil, was found to be between t = 30 and t = 60, and varied between 23 and 191 ng/ml. Correlation coefficients were calculated for the relation between sildenafil/desmethylsildenafil plasma concentrations at t = 30 and MPAP, PAOP, MAP and P/F ratio (Fig. 3, see electronic supplement). However, neither an increase in C max of sildenafil nor an increase in C max of desmethylsildenafil correlated significantly with a decrease in the MPAP, PAOP, MAP or P/F-ratio.
Discussion
In this study, we evaluated the effect of 50 mg of sildenafil administered as a single dose on pulmonary vascular tone and oxygenation in patients with ARDS. The major findings of the study were that sildenafil resulted in attenuation of pulmonary arterial pressures and pulmonary vascular resistance, and to a lesser degree of systemic arterial pressures and resistance. However, the observed profound increase in shunt fraction, as well as a marked decrease of PaO2, may render sildenafil unsuitable for the treatment of ARDS.
The reductions of pulmonary arterial pressures and vascular resistance are in line with previous studies evaluating the effect of sildenafil on ambulatory patients with idiopathic pulmonary hypertension or pulmonary hypertension due to lung fibrosis. However, in these studies, reducing pulmonary hypertension was accompanied by an increase in exercise capacity or elevated PaO2 [22, 23, 27]. This increase in PaO2 was explained by an effect called preferential vasodilation in which sildenafil is thought to selectively cause vasodilation in the lung, emphasized by a decrease in the ratio of the pulmonary to systemic vascular resistance index [22]. In contrast, in our study, sildenafil decreased both pulmonary and arterial pressure and resistance, without affecting the ratio of the pulmonary to systemic vascular resistance index or the ratio of the pulmonary to systemic arterial pressure. In other words, we did not find preferential vasodilation, which may explain the deterioration in shunt fraction and PaO2. An explanation for these contrasting results may be the duration of disease. Sildenafil-induced preferential vasodilation was observed in patients with pulmonary hypertension due to chronic pulmonary fibrosis and hence chronic hypoxia [23]. Chronic hypoxia not only results in vasoconstriction, but also leads to vascular remodeling with thickening of the medial layer, eventually even obliterating the endovascular lumen [28]. Once medial thickening has occurred, the vasoreactivity in diseased lung areas is restricted [29], which may allow for preferential vasodilation in well-ventilated areas, thereby decreasing the shunt fraction. In contrast, in ARDS, duration of disease was less than 7 days. This time span does not allow for intimal thickening to occur. In this study, sildenafil may have resulted in general pulmonary vasodilation, increasing blood flow through both well-ventilated as well as hypoventilated (consolidated) areas, thereby reducing hypoxic vasoconstriction and increasing the shunt fraction, which in turn resulted in a lower oxygenation. Impaired vasoreactivity in chronic pulmonary hypertension as opposed to normal vasoreactivity in acute pulmonary hypertension (i.e., ARDS) may be the discriminating factor in the effect of sildenafil on oxygenation.
A compromised pulmonary blood flow may have contributed to the deterioration of oxygenation. Although both pulmonary blood pressure and pulmonary resistance decreased after sildenafil administration, we did not assess the net effect of these parameters on pulmonary blood flow. However, the clearance of CO2 was not affected, rendering this hypothesis unlikely. Notably, most of the patients were on vasopressor therapy, which may have rendered these patients at increased risk of an increasing shunt fraction after sildenafil administration. Although the effect of sildenafil on worsening of oxygenation or shunt fraction was the same in patients treated with vasopressor therapy compared to those without vasopressors, the number of patients was too small to draw conclusions about the applicability of sildenafil in specific patient groups.
Right ventricular dysfunction is a common finding in ARDS [30], contributing to prolonged mechanical ventilation [31] and to mortality [32]. It has been postulated that right ventricular failure during ARDS should prompt adjustments to ventilatory settings [33, 34], including prone positioning [35]. As sildenafil decreased right ventricular afterload in this study, thereby decreasing the right ventricular stroke work index, this compound may be an alternative strategy for ARDS patients for whom right ventricular failure is thought to contribute to a detrimental course. It should be noted that we included only patients who strictly met the NAECC criteria for ARDS, including a pulmonary artery occlusion wedge pressure of <18 mmHg. It can be hypothesized that sildenafil may benefit ARDS patients with more severe pulmonary hypertension and concomitant higher pulmonary resistance by reducing right ventricular afterload and hence increasing right ventricular function.
In several studies with ambulatory patients, sildenafil did not have an effect on systemic arterial blood pressure [22, 23]. In our study, however, MAP decreased markedly after 50 mg of sildenafil together with a reduced SVRI. This effect may be attributed to the fact that seven out of ten patients required vasopressor support, four because of sepsis. During sepsis, increased NO release leads to vasodilation [36]. Increased levels of NO may lead to elevated levels of cGMP in the systemic circulation, which are not being degraded because of PDE inhibition by sildenafil. As a consequence, this may lead to more overt vasodilation in critically ill or septic patients when compared to ambulatory patients.
We did not find a correlation between hemodynamic or respiratory changes and the plasma concentrations of sildenafil or desmethylsildenafil. Although impaired gastric voiding was an exclusion criterion, we cannot rule out intestinal malabsorbtion. However, as sildenafil resulted in distinct hemodynamic and respiratory effects, malabsorbtion probably did not account for the absence of a correlation between effects and drug levels in this study. Rather, the sample size may have been too small to account for the great variability in pharmacodynamic properties of drugs generally found in critically ill patients [37, 38].
Implications of the study
Although levels of PO2 have not been associated with adverse outcome in ARDS, it can be hypothesized that prolonged administration of sildenafil may prolong the need of ventilatory support when oxygenation levels are targeted. Results of the study do not justify the use of sildenafil to improve oxygenation. In patients in whom right ventricular dysfunction is a therapeutic target, sildenafil may be of use. The effect of sildenafil on mortality in ARDS patients with right ventricular failure, however, remains to be determined.
Conclusion
Administration of sildenafil to patients with ARDS ameliorates pulmonary hypertension, thereby decreasing right ventricular afterload, but has deleterious effects on shunt fraction, oxygenation and systemic blood pressure. In spite of the promising features of sildenafil found in preclinical studies, results of this study do not justify a routine place for sildenafil in the treatment of ARDS patients. Whether sildenafil may be beneficial in ARDS patients with more severe pulmonary hypertension remains to be determined.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figures 3a and 3b: Dose-effect curves of sildenafil and desmethylsildenafil, respectively, on the MAP, MPAP, PAOP and P/F-ratio expressed as mean ± SEM. Spearman correlation coefficients for t = 30 min are displayed in each figure. (DOC 187 kb)
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
This article is discussed in the editorial available at: doi:10.1007/s00134-010-1771-2.
References
- 1.Wind J, Versteegt J, Twisk J, van der Werf TS, Bindels AJGH, Spijkstra JJ, Girbes ARJ, Groeneveld ABJ. Epidemiology of acute lung injury and acute respiratory distress syndrome in The Netherlands: a survey. Respir Med. 2007;101:2091–2098. doi: 10.1016/j.rmed.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 3.Morrell ED, Tsai BM, Crisostomo PR, Hammoud ZT, Meldrum DR. Experimental therapies for hypoxia-induced pulmonary hypertension during acute lung injury. Shock. 2006;25:214–226. doi: 10.1097/01.shk.0000191380.44972.46. [DOI] [PubMed] [Google Scholar]
- 4.Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS. Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia—a review. Crit Care Med. 2006;34:871–877. [PubMed] [Google Scholar]
- 5.Kuklin V, Kirov M, Sovershaev M, Andreasen T, Ingebretsen OC, Ytrehus K, Bjertnaes L. Tezosentan-induced attenuation of lung injury in endotoxemic sheep is associated with reduced activation of protein kinase C. Crit Care. 2005;9:R211–R217. doi: 10.1186/cc3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishitsuka Y, Moriuchi H, Hatamoto K, Yang C, Takase J, Golbidi S, Irikura M, Irie T. Involvement of thromboxane A2 (TXA2) in the early stages of oleic acid-induced lung injury and the preventive effect of ozagrel, a TXA2 synthase inhibitor, in guinea-pigs. J Pharm Pharmacol. 2004;56:513–520. doi: 10.1211/0022357023150. [DOI] [PubMed] [Google Scholar]
- 7.Moloney ED, Evans TW. Pathophysiology and pharmacological treatment of pulmonary hypertension in acute respiratory distress syndrome. Eur Respir J. 2003;21:720–727. doi: 10.1183/09031936.03.00120102. [DOI] [PubMed] [Google Scholar]
- 8.Roberts JDJ, Fineman JR, Morin FC, Shaul PW, Rimar S, Schreiber MD, Polin RA, Zwass MS, Zayek MM, Gross I, Heymann MA, Zapol WM. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. The Inhaled Nitric Oxide Study Group. N Engl J Med. 1997;336:605–610. doi: 10.1056/NEJM199702273360902. [DOI] [PubMed] [Google Scholar]
- 9.Bigatello LM, Hurford WE, Kacmarek RM, Roberts JDJ, Zapol WM. Prolonged inhalation of low concentrations of nitric oxide in patients with severe adult respiratory distress syndrome. Effects on pulmonary hemodynamics and oxygenation. Anesthesiology. 1994;80:761–770. doi: 10.1097/00000542-199404000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Rossaint R, Falke KJ, Lopez F, Slama K, Pison U, Zapol WM. Inhaled nitric oxide for the adult respiratory distress syndrome. N Engl J Med. 1993;328:399–405. doi: 10.1056/NEJM199302113280605. [DOI] [PubMed] [Google Scholar]
- 11.Adhikari NK, Burns KE, Friedrich JO, Granton JT, Cook DJ, Meade MO. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systematic review and meta-analysis. BMJ. 2007;334:779. doi: 10.1136/bmj.39139.716794.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krafft P, Fridrich P, Fitzgerald RD, Koc D, Steltzer H. Effectiveness of nitric oxide inhalation in septic ARDS. Chest. 1996;109:486–493. doi: 10.1378/chest.109.2.486. [DOI] [PubMed] [Google Scholar]
- 13.Rabe KF, Tenor H, Dent G, Schudt C, Nakashima M, Magnussen H. Identification of PDE isozymes in human pulmonary artery and effect of selective PDE inhibitors. Am J Physiol. 1994;266:L536–L543. doi: 10.1152/ajplung.1994.266.5.L536. [DOI] [PubMed] [Google Scholar]
- 14.Kurrek MM, Zapol WM, Holzmann A, Filippov G, Winkler M, Bloch KD. In vivo lipopolysaccharide pretreatment inhibits cGMP release from the isolated-perfused rat lung. Am J Physiol. 1995;269:L618–L624. doi: 10.1152/ajplung.1995.269.5.L618. [DOI] [PubMed] [Google Scholar]
- 15.Holzmann A, Manktelow C, Weimann J, Bloch KD, Zapol WM. Inhibition of lung phosphodiesterase improves responsiveness to inhaled nitric oxide in isolated-perfused lungs from rats challenged with endotoxin. Intensive Care Med. 2001;27:251–257. doi: 10.1007/s001340000774. [DOI] [PubMed] [Google Scholar]
- 16.Boer C, Groeneveld ABJ, Scheffer GJ, de Lange JJ, Westerhof N, Sipkema P. Induced nitric oxide impairs relaxation but not contraction in endotoxin-exposed rat pulmonary arteries. J Surg Res. 2005;127:197–202. doi: 10.1016/j.jss.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Coimbra R, Melbostad H, Loomis W, Porcides RD, Wolf P, Tobar M, Hoyt DB. LPS-induced acute lung injury is attenuated by phosphodiesterase inhibition: effects on proinflammatory mediators, metalloproteinases, NF-kappaB, and ICAM-1 expression. J Trauma. 2006;60:115–125. doi: 10.1097/01.ta.0000200075.12489.74. [DOI] [PubMed] [Google Scholar]
- 18.Sheridan BC, McIntyre RC, Meldrum DR, Fullerton DA. Phosphodiesterase inhibition overcomes pulmonary vasomotor dysfunction in acute lung injury. J Surg Res. 1997;71:145–149. doi: 10.1006/jsre.1997.5139. [DOI] [PubMed] [Google Scholar]
- 19.Howell RE, Jenkins LP, Howell DE. Inhibition of lipopolysaccharide-induced pulmonary edema by isozyme-selective phosphodiesterase inhibitors in guinea pigs. J Pharmacol Exp Ther. 1995;275:703–709. [PubMed] [Google Scholar]
- 20.Klein A, Zils U, Bopp C, Gries A, Martin E, Gust R. Low-dose phosphodiesterase inhibition improves responsiveness to inhaled nitric oxide in isolated lungs from endotoxemic rats. J Surg Res. 2007;138:224–230. doi: 10.1016/j.jss.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Ghofrani HA, Reichenberger F, Kohstall MG, Mrosek EH, Seeger T, lschewski H, Seeger W, Grimminger F. Sildenafil increased exercise capacity during hypoxia at low altitudes and at Mount Everest base camp: a randomized, double-blind, placebo-controlled crossover trial. Ann Intern Med. 2004;141:169–177. doi: 10.7326/0003-4819-141-3-200408030-00005. [DOI] [PubMed] [Google Scholar]
- 22.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 23.Ghofrani HA, Wiedemann R, Rose F, Schermuly RT, Olschewski H, Weissmann N, Gunther A, Walmrath D, Seeger W, Grimminger F. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet. 2002;360:895–900. doi: 10.1016/S0140-6736(02)11024-5. [DOI] [PubMed] [Google Scholar]
- 24.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 25.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–829. doi: 10.1097/00003246-198510000-00009. [DOI] [PubMed] [Google Scholar]
- 26.Vos RM, Chahbouni A, Sinjewel A, Swart EL. Quantitative analysis of sildenafil and desmethylsildenafil in human serum by liquid chromatography−mass spectrometry with minimal sample pretreatment. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876:283–287. doi: 10.1016/j.jchromb.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 27.Lepore JJ, Maroo A, Bigatello LM, Dec GW, Zapol WM, Bloch KD, Semigran MJ. Hemodynamic effects of sildenafil in patients with congestive heart failure and pulmonary hypertension: combined administration with inhaled nitric oxide. Chest. 2005;127:1647–1653. doi: 10.1378/chest.127.5.1647. [DOI] [PubMed] [Google Scholar]
- 28.Pak O, Aldashev A, Welsh D, Peacock A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur Respir J. 2007;30:364–372. doi: 10.1183/09031936.00128706. [DOI] [PubMed] [Google Scholar]
- 29.Shimoda LA, Sham JS, Sylvester JT. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol Res. 2000;49:549–560. [PubMed] [Google Scholar]
- 30.Zapol WM, Snider MT. Pulmonary hypertension in severe acute respiratory failure. N Engl J Med. 1977;296:476–480. doi: 10.1056/NEJM197703032960903. [DOI] [PubMed] [Google Scholar]
- 31.Vieillard-Baron A, Schmitt JM, Augarde R, Fellahi JL, Prin S, Page B, Beauchet A, Jardin F. Acute cor pulmonale in acute respiratory distress syndrome submitted to protective ventilation: incidence, clinical implications and prognosis. Crit Care Med. 2001;29:1551–1555. doi: 10.1097/00003246-200108000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Monchi M, Bellenfant F, Cariou A, Joly LM, Thebert D, Laurent I, Dhainaut JF, Brunet F. Early predictive factors of survival in the acute respiratory distress syndrome. A multivariate analysis. Am J Respir Crit Care Med. 1998;158:1076–1081. doi: 10.1164/ajrccm.158.4.9802009. [DOI] [PubMed] [Google Scholar]
- 33.Vieillard-Baron A. Is right ventricular function the one that matters in ARDS patients? Definitely yes. Intensive Care Med. 2009;35:4–6. doi: 10.1007/s00134-008-1308-0. [DOI] [PubMed] [Google Scholar]
- 34.Mekontso Dessap A, Charron C, Devaquet J, Aboab J, Jardin F, Brochard L, Vieillard-Baron A. Impact of acute hypercapnia and augmented positive end-expiratory pressure on right ventricle function in severe acute respiratory distress syndrome. Intensive Care Med. 2009;35:1850–1858. doi: 10.1007/s00134-009-1569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vieillard-Baron A, Charron C, Caille V, Belliard G, Page B, Jardin F. Prone positioning unloads the right ventricle in severe ARDS. Chest. 2007;132:1440–1446. doi: 10.1378/chest.07-1013. [DOI] [PubMed] [Google Scholar]
- 36.Vincent JL, Zhang H, Szabo C, Preiser JC. Effects of nitric oxide in septic shock. Am J Respir Crit Care Med. 2000;161:1781–1785. doi: 10.1164/ajrccm.161.6.9812004. [DOI] [PubMed] [Google Scholar]
- 37.Strack van Schijndel RJM, Wierdsma NJ, van Heijningen EMB, Weijs PJM, de Groot SDW, Girbes ARJ. Fecal energy losses in enterally fed intensive care patients: an explorative study using bomb calorimetry. Clin Nutr. 2006;25:758–764. doi: 10.1016/j.clnu.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Scaglione F, Paraboni L. Pharmacokinetics/pharmacodynamics of antibacterials in the intensive care unit: setting appropriate dosing regimens. Int J Antimicrob Agents. 2008;32:294–301. doi: 10.1016/j.ijantimicag.2008.03.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures 3a and 3b: Dose-effect curves of sildenafil and desmethylsildenafil, respectively, on the MAP, MPAP, PAOP and P/F-ratio expressed as mean ± SEM. Spearman correlation coefficients for t = 30 min are displayed in each figure. (DOC 187 kb)