

Abstract
Multifunctional nanomedicine is emerging as a highly integrated platform that allows for molecular diagnosis, targeted drug delivery, and simultaneous monitoring and treatment of cancer. Advances in polymer and materials science are critical for the successful development of these multi-component nanocomposites in one particulate system with such a small size confinement (<200 nm). Currently, several nanoscopic therapeutic and diagnostic systems have been translated into clinical practices. In this feature article, we will provide an up-to-date review on the development and biomedical applications of nanocomposite materials for cancer diagnosis and therapy. Overview of each functional component, i.e. polymer carriers, MR imaging agents, and therapeutic drugs will be presented. Integration of different functional components will be illustrated in several highlighted examples to demonstrate the synergy of the multifunctional nanomedicine design.
1. Introduction
Cancer is a leading cause of global health burdens. In 2007 alone, it claimed 7.9 million deaths worldwide, which was 13% of all deaths.1 In the US, more than 1.4 millions of new cancer cases were estimated for the year 2008.2 Although cancer survival rates improved over the past few decades, its mortality rates remained high in spite of the rapid decline in other diseases (e.g. cardiovascular disease). In contrast to these other diseases, cancer is a highly heterogeneous disease with diverse phenotypic expressions at different organ sites. Lack of effective diagnostic methods to detect cancer at its early onset, as well as a lack of efficacious therapy with minimal toxicity, remain the major limitations for complete eradiation of the disease.3
Medical imaging has been one of the most important tools for cancer diagnosis. Depending on which imaging modality is used, anatomical or molecular information can be obtained.4 Nuclear imaging techniques such as positron emission tomography (PET) or single photon emission computed tomography (SPECT) have excellent sensitivity and can provide biochemical information of pathological conditions. In comparison, computed tomography (CT) and magnetic resonance imaging (MRI) have high spatial and temporal resolutions and can provide superb anatomical information. Optical imaging provides novel insight into molecular and cellular processes. However, its clinical use is hampered by limited light penetration in biological tissues. Compared to other imaging modalities, MRI yields excellent soft tissue contrast, and is highly sensitive to blood flow. MR imaging of cancer can be greatly facilitated by the use of contrast agents to differentiate cancerous tissue from surrounding benign tissues. Several paramagnetic (Gd-based) MR agents5 and superparamagnetic (iron oxide) nanoparticles6-8 are currently used in the clinics and fall within the realm of “MRI-visible” nanomedicine.
In addition to cancer molecular imaging, therapeutic drug delivery is another research field that offers promises for the efficacious treatment of cancer. Current chemotherapeutics often lack specificity and efficacy because of their poor pharmacokinetics and pharmacodynamics.9-11 Severe toxic side effects in healthy tissues and patient morbidity are major drawbacks for these treatments. In the past few decades, intense research efforts have focused on the development of nanoscopic delivery systems for targeted therapy of cancer.12, 13
Various nanomedicine platforms have been established to deliver anticancer drugs and/or imaging agents to tumors. Recently, a new term “theranostic nanomedicine” was introduced to describe nanosystems that integrate diagnostic and therapeutic functions within the same platform.14, 15 Such a design may permit for the molecular diagnosis, targeted therapy, and simultaneous monitoring and treatment necessary to achieve personalized medicine for cancer.
2. Multifunctional nanomedicine
Over the past several decades, we have witnessed the explosive development of a variety of nanomedicine platforms for cancer diagnosis and treatment.12, 13 These nanoplatforms include polymer-drug conjugates, dendrimers, liposomes, polymeric micelles, polymersomes, inorganic nanoparticles (e.g. Au/Ag/Si/Fe3O4/CdSe),9, 13, 16 each with distinct chemical compositions and physical properties. Compared to traditional small molecular-based contrast agents or therapeutic drugs, these new nanomedicine platforms permit for a highly integrated design that incorporates multiple functions, such as cell targeting, imaging ultra-sensitivity, and therapy in one system.17-19 Multifunctional nanomedicine holds considerable promise as the next generation of medicine that allows for the molecular diagnosis of cancer phenotypes, customized therapy to exploit unique cancer targets, and simultaneous treatment and monitoring of therapeutic efficacy. This modular design with “theranostic” functions may prove essential in addressing the challenge of tumor heterogeneity and achieving personalized medicine for diverse cancer phenotypes.
In this Feature Article, we will review the recent progress in the development and applications of polymeric nanocomposite particles for cancer MR imaging and therapy. Fig. 1 illustrates the schematic of a nanocomposite particle with different functional components. The therapeutic component can be small molecular drugs,9 therapeutic proteins,20 small interfering RNA (siRNA),21, 22 or plasmid DNA.23, 24 Both clinically approved anticancer drugs such as doxorubicin or pre-clinical agents such as β-lapachone25 have been used in nanocomposite particles. The MR imaging component consists of contrast agents with distinctive contrast mechanisms, namely T1,5 T2,7 as well as chemical exchange saturation transfer (CEST) agents.26 The last component of the composite design is nanocarriers. Although a number of nanocarriers from inorganic frameworks such as silicon-based materials,27, 28 carbon nanotubes,29, 30 and zeolites31, 32 have been reported, we will primarily focus on biocompatible polymeric systems in the current feature article.
Fig. 1.
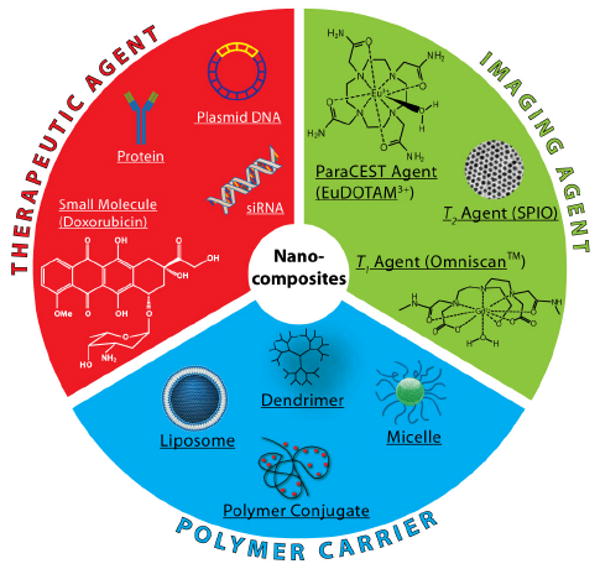
Schematic illustration of the functional components for the development of cancer nanomedicine.
2.1. Anticancer agents
A number of anticancer agents can be incorporated in nanomedicine platforms to achieve the desired therapeutic efficacy. In general, these agents can be categorized into four groups based on their structural properties: (1) small molecular anticancer drugs; (2) proteins, (3) small interfering RNA (siRNA); and (4) plasmid DNA. In this section, a brief overview of each type of cancer nanotherapeutics will be discussed.
Several excellent articles have comprehensively reviewed the development of small molecular drug-based nanomedicine.11, 33, 34 The most widely studied small molecular drugs are doxorubicin and paclitaxel. Doxorubicin has been incorporated into a number of nanocomposite platforms such as dendrimers,35, 36 polymer-drug conjugates,33, 37 liposomes,38 and polymeric micelles.39-41 Similarly, paclitaxel has also been used as a cancer therapeutic in nanocomposite particles.42, 43 Additionally, therapeutic agents for photodynamic therapy or radiation sensitization can be included. A combination of these agents can also be used to target multiple malignant processes to achieve enhanced therapeutic efficacy through synergy.44
Protein-based therapeutics are also widely used for cancer therapy.20 Currently, therapeutic proteins are mostly produced by recombinant DNA technology. Compared to small molecular anticancer drugs, protein drugs can be more specific toward cancer cells with potentially less side effects. For example, cetuximab is a monoclonal antibody that binds to the extracellular portion of the epidermal growth factor receptor (EGFR) and inhibits cell growth and proliferation.45 It is clinically used to treat lung, colorectal, and head and neck cancers. Despite their promise in oncology, protein therapeutics have been hindered in clinical applications due to their rapid clearance, enzymatic degradation, and lack of stability. Nano-delivery systems can play a key role in overcoming such limitations.46 For example, SMANCS, a conjugate of neocarzinostatin (NCS) and poly(styrene-co-maleic acid) (SMA), was developed by Maeda and coworkers in the 1980s, and has been clinically approved as one of the first polymer-protein therapeutics for cancer.47 The blood half-life of SMANCS is 10 times longer than that of NCS alone, which leads to enhanced tumor targeting via the EPR effect, resulting in an efficacious response for liver cancer therapy.48, 49
Recently, siRNA molecules have received considerable attention as novel therapeutic agents for cancer treatment. Each siRNA has a double-stranded structure that consists of 20-25 nucleotides. Sequence-specific gene silencing can be achieved with siRNA, which potentially increases its tumor specificity. The delivery of siRNA using nanoparticles is still in an early stage compared to small molecular drugs. The most common nanoplatforms for siRNA delivery are liposomes and cationic polymer-based nanoparticles. Villares et al reported the incorporation of protease-activated receptor-1 siRNA in liposomes, which were injected in mice carrying A375SM human melanoma xenografts.50 A decrease in tumor volume and metastatic lung colonies was reproted. Sonoke et al reported an increased tumor uptake and improved antitumor efficacy of a pegylated cationic liposome encapsulating an siRNA that is sequence specific for the antiapoptotic bcl-2 mRNA.51 Our laboratory recently reported the incorporation of siRNA targeting secretory clusterin (sCLU) into pegylated PEI nanoparticles.52 In breast cancer MCF-7 cells, siRNA-sCLU nanocomplexes suppressed both basal as well as IR-inducible sCLU protein expression, which led to a significant increase in IR-induced lethality over siRNA scrambled controls. The versatility of siRNA therapy opens many exciting opportunities to achieve tumor-specific response in different types of cancers.
The last category of cancer therapeutics is plasmid DNAs for gene therapy. It is now well know that cancer arises from genetic mutations and genomic instabilities. Gene therapy has the potential to correct these abnormalities at a genetic level. Compared to siRNA, DNA is larger in size and requires delivery to the nucleus, which is a formidable challenge in gene delivery. Although viral vectors have demonstrated high transfection efficiency, safety and immunogenicity are major concerns for clinical use. Non-viral gene delivery remains an active area of research with the main focus being improvement of delivery and transfection efficiency. Recently, Abela et al reported the preparation of a virus-mimicking nanoparticle, transferrin (Tf)-cationic liposome-DNA complex (Tf-lipoplex) by encapsulating plasmid DNA cytomegalovirus-green fluorescent protein (CMV-GFP) inside the core of liposomes.53 They reported that Tf-lipoplex achieved high gene delivery efficacy in C57BL mice carrying subcutaneous LLC1 tumor xenografts. Moreover, normal tissue toxicity was low suggesting that a safe, repeated administration strategy of the particles can be used. Recently, Langer and coworkers applied a combinatorial polymer library approach to screen for optimal cationic polymers for DNA delivery.54 The large data set permits for comprehensive structure-property correlations to identify key structural motifs that allow for optimal compaction, binding affinity, and adequate dissociation of DNA from nanocarriers upon cell uptake in order to maximize delivery efficiency. These new research efforts are critical in achieving the desired therapeutic potential of DNA for cancer therapy.
2.2. MRI contrast agents
Since the introduction of MRI in the 1970's,55 its applications in clinical oncology have been and are rapidly expanding. MRI has become a vital tool in clinical cancer diagnosis because it offers superb anatomical and functional images with high spatial and temporal resolutions. Moreover, MRI does not require administration of radioactive agents or high energy electromagnetic waves as in the case of PET, SPECT and CT.56 Image contrast in MRI relies on the relaxation properties of water protons. For cancer-specific diagnosis, a targeted contrast agent is necessary to help distinguish malignant tissues from normal ones. It is important to note at this time that MRI agents produce image contrast by affecting relaxation properties of water protons57 whereas most other imaging modalities (e.g. PET, SPECT, CT, fluorescence) detect the contrast probes directly.
There are three main types of MRI contrast agents based on their different contrast mechanisms. First are the T1 agents that generate a positive image contrast by increasing longitudinal relaxation rates of surrounding water protons;57 second are the T2 agents that generate a negative image contrast by increasing the transverse relaxation rates of water;7 and finally, chemical exchange saturation transfer (CEST) agents allow for turning “on” and “off” the image contrast by an external radiofrequency (RF) pulse.58 For cancer molecular imaging applications, one major challenge is to improve the detection sensitivity of the above agents so that tumor markers can be visualized by MRI at low pathophysiological concentrations. In the sections below, we will provide a concise review of the physics and mechanisms of each type of contrast agents.
2.2.1. T1 contrast agents
Under an external magnetic field (B0), magnetic moments of precessing protons align along the direction of B0 producing a net magnetization in the same direction, or longitudinal z axis (Fig. 2). When the sample is irradiated with an RF pulse, the net magnetization can be flipped away from its original axis, a process called excitation. A net longitudinal magnetization (Mz) is zero if the RF irradiation is a 90° pulse. The net magnetization then returns to its original equilibrium state (Mz(0)), a process known as longitudinal (T1) relaxation or spin-lattice relaxation (left panel, Fig. 2).
Fig. 2.
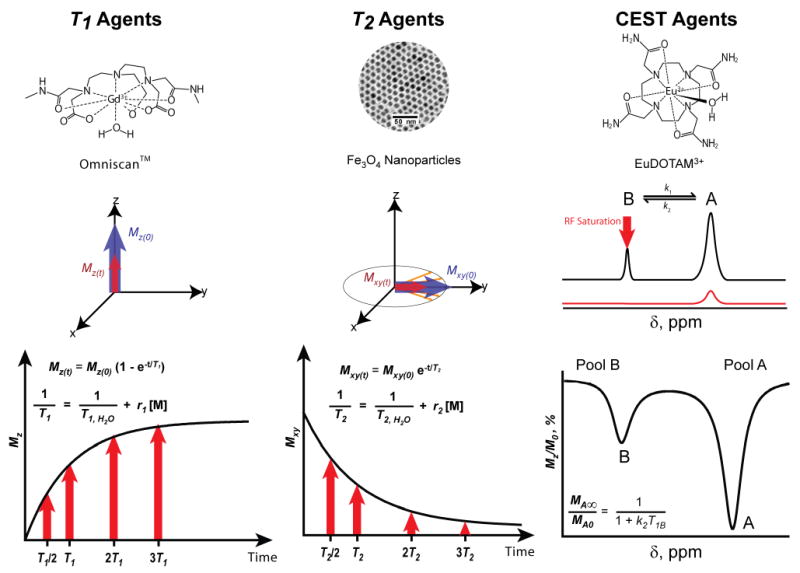
Representative chemical structures and compositions for three different types of MRI contrast agents. Left panel, the chemical structure of a T1 agent, Omniscan™, and T1 relaxation mechanism; middle panel, TEM image of a T2 agent, 9 nm Fe3O4 nanoparticles, and T2 relaxation; right panel, the chemical structure of EuDOTAM3+, a PARACEST agent. A 2-pool exchange model for CEST mechanism and a representative Z-spectrum are illustrated.
The recovery of net longitudinal magnetization is governed by Equation 1. T1 relaxation time is defined as the time required for protons to return to 63% of their original longitudinal magnetization. Its reciprocal, 1/T1, is known as the T1 relaxation rate. In biological samples, different forms of protons (mobile water, tissue-bound water) have different T1 relaxation rates. When a T1-weighted MR image is acquired, tissues containing different types of protons produce images of different signal intensity to yield image contrast.
| (1) |
| (2) |
Good T1 contrast agent must be able to shorten T1 relaxation times of surrounding water protons even at a low concentration of the agent. This ability is depicted by r1, or T1 relaxivity, of the contrast agent. Equation 2 shows the T1 relaxation rate as a function of r1 and concentration ([M]) of the contrast agent, where T1, H2O is the relaxation time of pure water. A contrast agent with high r1 values can cause surrounding protons to recover to the longitudinal magnetization rapidly producing a bright image in a T1-weighted MR image.
The most common T1 contrast agents are Gd(III)-based chelating complexes. Gd(III) ions have 7 unpaired electrons making them strongly paramagnetic. The Gd(III) ion is not naturally found in the human body and its free ionic form is highly toxic.57 Therefore, chelating ligands that can form stable complexes are necessary for the development of clinically safe MRI contrast agents. Gd(III) is known to coordinate strongly to diethylenetriamine backbones modified with carboxylic acids. A number of derivatives, both in linear and cyclic forms, have been synthesized and tested for complex associations. The first Gd(III)-complex approved for clinical use is Gd(III)-diethylene triamine pentaacetic acid (Gd-DTPA, Magnevist®) developed by Schering AG. Gd(III) has 8 coordination sites to oxygen atoms in the carboxylate groups or nitrogen atoms in the backbone and one site to water molecules. Subsequently, many derivatives of DTPA and other chelates were introduced: Gadodiamide (Gd-DTPA-BMA, Ominscan™), a cyclic chelating agent tetraaza-12-crown-4-tetraacetic acid (Gd-DOTA, or Dotarem®), GdHPDO3A (Prohance®), Gd-EOB-DTPA (Eovist®), and Gd-BOPTA (Multihance®).
In addition to Gd(III), Mn(II) was also studied as a T1 contrast agent. Mn(II) has 5 unpaired electrons and is also paramagnetic. Mn(II) complexes are not as widely used as Gd(III) due to the poor stability of Mn(II) ions and metal complexes. Thus far, MnDPDP (Teslascan™), in which Mn(II) is coordinated by dipyridoxyl diphosphate, is the only clinically approved agent. In Fig. 3, the left panel shows the molecular structures of several representative T1 agents. The integration of the derivatives of these agents into nanocarriers is discussed in Section 2.3.
Fig. 3.
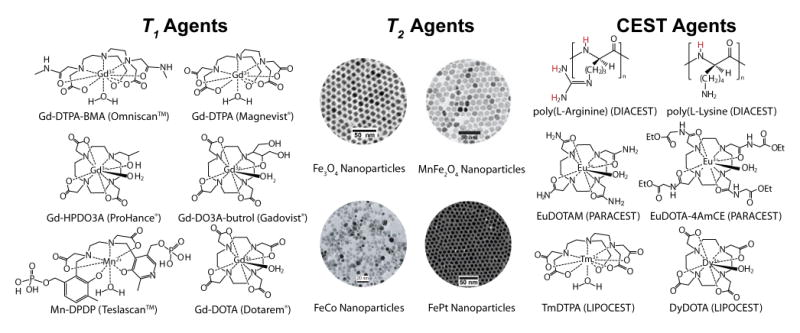
Schematic illustration of chemical structures of representative T1 agents (left), TEM images of exemplary T2 agents (middle), and chemical structures of several types of CEST agents (right). Modified from refs. 61 (MnFe2O4), 63 (FeCo), and 64 (FePt). (Permission pending)
2.1.2. T2 contrast agents
Transverse relaxation or T2 relaxation is another key mechanism for contrast agent development (middle panel, Fig. 2). Upon excitation of protons by an RF pulse (e.g. 90°), the magnetization in the xy plane is generated as transverse magnetization (Mxy). As the recovery in longitudinal magnetization takes place, the transverse magnetization begins to decrease due to spin-spin relaxation or T2 relaxation. The decrease in transverse magnetization is called decay. The time it takes for the transverse magnetization to decay to 37% of its original value is T2 time (Eq. 3). Generally, T2 relaxation is faster than that of T1. Samples in which protons have higher degree of translational and rotational freedoms (mobile waters vs. tissue bound waters) generally have longer T2 values.
| (3) |
| (4) |
T2 contrast agents are mainly superparamagnetic nanoparticles (e.g. Fe3O4).59 These agents can be strongly magnetized under an external magnetic field, which can lead to considerable distortion of the local magnetic field. T2 relaxivity (r2) is an intrinsic property of T2 contrast agents that describes its ability to affect T2 relaxation rates (Eq. 4). Unlike T1 agents, where a chemical exchange between bound and free water molecules is required for the relaxation process, T2 agents produce much stronger magnetic susceptibility, affecting a larger number of water molecules and thus, yield higher sensitivity of detection. Transverse relaxation results in signal loss (negative contrast) in T2-weighted images.
Superparamagnetic iron oxide (SPIO or Fe3O4) nanoparticles are the most commonly used T2 contrast agents in the clinics. Several different formulations (trade names Feridex IV, Resovist, and Lumirem) are clinically used for liver or GI tract imaging.59 Their applications in cancer molecular imaging are limited due to the high polydispersity of SPIO nanoparticles, low relaxation rates and lack of specificity in vivo. Recently, much attention has been devoted to the development of structurally well-defined superparamagnetic nanoparticles with high magnetization, and hence high r2 values. Sun et al first reported the organic synthesis of monodispersed MFe2O4 (M = Fe2+, Co2+, Mn2+) nanoparticles.60 The diameter of these nanoparticles can be fine tuned from 4 to 20 nm. Cheon and coworkers showed that by substituting a high spin d5 Mn2+ ion for d6 Fe2+ in the spinel structure, MnFe2O4 achieved higher magnetization and more sensitive MRI detection over Fe3O4 nanoparticles.61 Our laboratory recently reported that zinc-doped ferrite nanocrystals (ZnxFe3-xO4, x<0.5) enhanced magnetization by 2.5 times over Fe3O4 nanoparticles of similar size, which resulted in a significant increase in T2 relaxivity and MRI detection sensitivity.62 Other metal alloy nanoparticles (e.g. FeCo,63 FePt,64, 65 middle panel in Fig. 3) have also been synthesized with strong magnetization properties. For example, the 7 nm FeCo nanocrystals exhibit Ms at 215 emu/g with superb r1 and r2 relaxivities.63 One of the major considerations for the use of novel magnetic nanoparticles is to ensure that the metal compositions and their degradation products do not result in both acute and chronic toxicity.
2.1.3. CEST agents
In recent years, a new contrast mechanism based on chemical exchange saturation transfer (CEST) has been reported.66 CEST MRI makes use of the chemical exchange phenomena between bulk water protons (δ = 4.78 ppm) and exchangeable protons with a different chemical shift.67 To illustrate the CEST mechanism (right panel, Fig. 2), bulk water protons are considered as pool A and exchangeable protons on MR contrast agents as pool B. A proton from each pool exchanges reversibly and is governed by forward and reverse exchange rates k1 and k2, respectively. When water protons in pool B are irradiated with a saturation pulse, their net magnetization will become zero and hence, generate no NMR signal (red curve). Due to chemical exchange, a saturation transfer from pool B to pool A takes place causing a decrease in NMR signal of bulk water protons in pool A. Longitudinal relaxation of the protons return both systems to their equilibrium or steady state. The steady-state intensity (MA∞/MA0) of protons in pool A is expressed in Equation 5, where T1B is the longitudinal relaxation time of water protons in pool B.67 In MRI, CEST images are the result of signal losses and appear darkened as compared to images acquired without a pre-saturation RF pulse. Given that CEST contrast can be turned “on” and “off” under otherwise identical imaging conditions, the contrast images can provide a much improved detection accuracy of CEST agents. Several studies reported CEST imaging of biological molecules such as glycogen,68 an endogenous lysine-rich protein (LRP) as a reporter gene,69 and proteins and peptides.70 CEST imaging was also applied to MRI of a hyperpolarized Xe noble gas.71, 72 These CEST probes are classified as DIACEST because of their diamagnetic nature.73
| (5) |
CEST imaging can also be applied to paramagnetic metal chelates that result in PARACEST agents (right panel, Fig. 3).74 Gd3+ complexes are not good PARACEST agents because Gd3+ ions are isotropic and are not chemical shift agents. Other metal ions in the lanthanide family are better candidates because of their anisotropic distribution of electrons in the f orbitals creating a large chemical shift difference between bound water (pool B) and bulk water (pool A).26 Europium (III) DOTA(glycine ethyl ester)4 was first tested for PARACEST imaging by Zhang et al.74 The protons of bound water molecules appeared at about +50 ppm away from bulk water protons. Complexes of Tm3+ and Dy3+ were able to shift the bound water peaks to +500 ppm and -720 ppm, respectively.75 The full list of lanthanide metal complexes for PARACEST imaging can be found in a comprehensive review by Zhang et al.58 Exchange rates of water molecules can be fine-tuned by chemical modifications of the chelating agents. Compared to CEST agents based on organic functional groups (e.g. amide, urea), PARACEST agents provide larger chemical shifts that allow for faster exchange rates to increase imaging sensitivity. Various PARACEST agents have been elegantly developed as environmentally sensitive probes to detect pH and cancer-specific enzymes.76, 77
Recently, Aime and coworkers reported the development of LIPOCEST agents based on liposomes with encapsulation of lanthanide agents (e.g. Tm-DOTMA).78 In this design, the encapsulated Ln complex exhibits fast water exchange and therefore is not a PARACEST agent by itself. However, it shifts the intra-liposomal water peak away from the bulk water peak, which can be used as an antenna to initiate the CEST effect. In this case, water exchange is not determined by the characteristics of the Ln agent, but rather the water permeability of the liposomal bilayer. Using the intra-liposomal water resonance as the antenna for RF excitation, a significant CEST effect was detected at LIPOCEST concentrations of ∼90 pM. This ultra-sensitivity and the potential of establishing multi-chromatic detection of LIPOCEST particles make them exciting candidates as “multi-colored” MR probes for molecular diagnosis of cancer (see Section 3).
2.3. Polymeric nanocarriers
Nanocarriers serve as vehicles that can carry drugs and/or MRI contrast agents and transport them to tumor sites. The size of nanocarriers usually ranges from 10-200 nm to avoid rapid kidney secretion (<10 nm) or reticuloendothelial system (RES) filtration elimination (>200 nm).79 Nanocarriers consist of either naturally occurring macromolecules such as chitosan80, 81 or synthetic macromolecules such as pegylated phospholipids.82 Biocompatibility and biodegradability are essential criteria for the clinical use of these platforms. Several inorganic nanocarriers such as silica-based nanoparticles27, 28 and zeolites32 have been investigated. However, questions regarding their biocompatibility and biodegradability have often surfaced and have hindered the clinical translation of these materials.
In this article, we will discuss polymeric nanocarriers that are categorized based on three drug-incorporation mechanisms (Fig. 4). The first includes polymeric carriers that use covalent chemistry for direct drug conjugation. This group can be further categorized into linear polymers, hyperbranched polymers, and dendrimers. The second group of nanocarriers involves hydrophobic interactions between the drugs and nanocarriers. This group includes polymeric micelles from amphiphilic block copolymers. Nanocarriers in the final group offer a water-filled depot for hydrophilic drug encapsulation. Liposomes and polymersomes are the main examples of this group. This section will discuss a general overview of each group with specific examples that are being explored for cancer imaging and therapy.
Fig. 4.
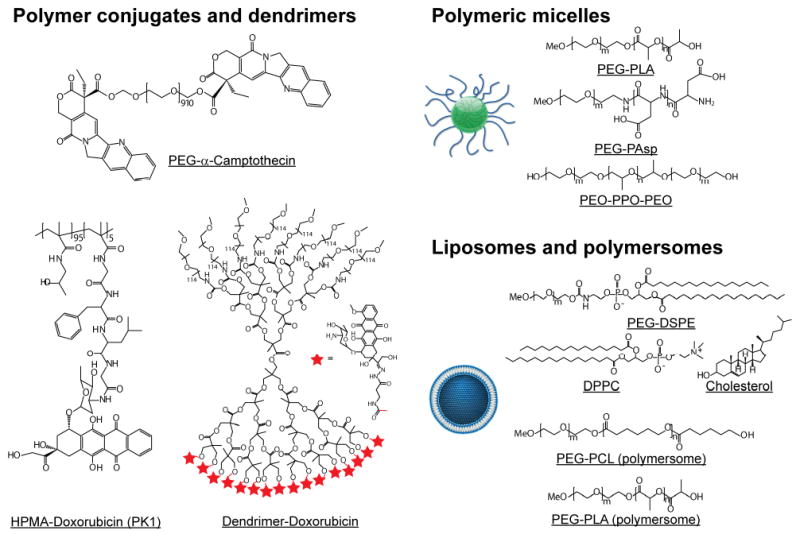
Schematic illustration of chemical structures of representative polymer conjugates and dendrimers (left), polymeric micelles (top right), and liposomes and polymersomes (bottom right).
2.3.1 Polymer-drug conjugates
The use of polymers as carriers dated back to the 1950's when Jatzkewits reported N-vinylpyrrolidine conjugates of glycyl-L-leucine-mescaline as a drug depot.83 Since then, there has been an enormous increase in research involving polymeric therapeutics for cancer therapy. Anticancer drugs are oftentimes covalently conjugated to polymeric carriers. The choice of polymers for drug conjugation is very critical. Generally, they are water-soluble polymers with available functional groups for covalent attachment of the drugs. The simplest form of polymer-drug conjugates is the attachement of poly(ethylenne glycol) or PEG to drugs, a process known as pegylation. A number of protein therapeutics were modified by PEG to improve proteins' solubility and reduce immunogenicity.84, 85 Several challenges in pegylation of proteins include possibilities of crosslinking the proteins, alteration of protein charges as a result of chemical transformations, and a loss in bioactivity of the proteins.33 Currently, numerous pegylated proteins are used in the clinical treatment of cancer. For example, SMANCS was approved for treatment of hepatocellular carcinoma in Japan in 1993 and PEG-L-asparaginase (ONCASPAR) is used to treat acute lymphoblastic leukemia.86 In addition to pegylation of proteins, small molecular drugs were also pegylated as a way to improve their pharmacokinetics for cancer therapy. For example, PEG–camptothecin (PROTHECAN) recently entered clinical trials for cancer therapy.87
Conjugation of small molecular drugs to polymers can improve their pharmacokinetics and tumor accumulation.88 The most common polymeric system for delivering small molecular drugs is N-(2-hydroxypropyl) methacrylamide (HPMA). HPMA was first developed by Kopacek and coworkers as a plasma expander.89 Since then, a variety of HPMA-drug conjugates were synthesized and tested. Early collaborative works from two pioneers in HPMA-drug conjugates, Kopacek and Duncan, led to two doxorubicin-based formulations that entered clinical testing for cancer therapy.89, 90 They reported an increase of four to five folds of the maximum tolerated dose (MTD = 320 mg/m2) of the conjugates compared to that of free doxorubicin. Additionally, an increased blood circulation half-life was also observed over the free drug. Phase I/II clinical trials of HPMA copolymer-Gly-Phe-Leu-Gly-doxorubicin containing galactosamine (PK2; FCE28069) showed an increased MTD to 160 mg/m2.91 More recently, other small molecular drugs such as paclitaxel92 and camptothecin93 were conjugated to HPMA.
Non-linear polymers, especially dendrimers, are also of great interests for drug delivery. Dendrimers were first synthesized in the late 1970's.94 They are well defined hyperbranched macromolecules with very narrow polydispersity and a high density of functional groups.95 Anticancer drugs were generally covalently attached to the branches of dendrimers. Lai et al reported a conjugation of doxorubicin to polyamidoamine (PAMAM) dendrimers via pH-sensitive linkers.96 The dendrimers showed improved toxicity in human oral squamous cell carcinoma (Ca9-22). Minko and coworkers covalently conjugated paclitaxel with PAMAM G4 hydroxyl-terminated dendrimer.97 They compared the toxicity of the drug-dendrimer conjugates in A2780 human ovarian carcinoma cells. The dendrimer-drug conjugates were reported to increase cytotoxicity by 10-fold compared to non-conjugated drug. Several other research groups also reported drug-conjugated dendrimers and their cell-killing effects.98, 99
In addition to therapeutic drug delivery, a similar polymer conjugate strategy was applied to the development of polymer-based MR contrast agents for cancer diagnosis. Lu and coworkers have extensively investigated the development of HPMA and poly(glutamic acid) (PGA) as polymer conjugates for T1 MRI contrast agents.100 Both MR imaging agents and anticancer drugs were integrated into the same PGA-based polymer platform as multifunctional nanocomposite particles.101, 102 In another study, they reported that HPMA conjugates with higher molecular weight had prolonged blood circulation half-lives and led to higher tumor accumulation.103 A polymer conjugate containing a PARACEST agent, Eu3+-complex, has recently been reported by Sherry and coworkers with improved imaging sensitivity.104
A large number of dendrimers conjugated to T1 agents have been reported.105-107 Kobayashi et al have extensively investigated the use of dendrimeric MRI agents for blood pool imaging and lymphangiography.108 The attachment of PARACEST agents to dendrimers has not been widely investigated. In 2007, Pikkemaat et al described the synthesis of Yb(III)-DOTAM-functionalized poly(propylene imine) dendrimers and used them for pH mapping.109
2.3.2. Polymeric micelles
Polymeric micelles offer another potential platform for the delivery of anticancer drugs and MRI contrast agents. Micelles are nanoscopic core-shell particles composed of amphiphilic block copolymers (Fig. 4). Micelle cores are hydrophobic and can serve as a natural carrier for hydrophobic drugs, imaging agents, or both in the same particle. Hydrophobic polymers such as poly(D,L-lactic acid) (PLA) and poly(ε-caprolactone) (PCL) are typically used to form micelle cores. The shells of the micelles are mostly PEG-based polymers that solubilize the particles and improve their pharmacokinetics. A number of excellent comprehensive review articles concerning polymeric micelles for cancer drug delivery are available.11, 110, 111 Generally, small molecular drugs are loaded into micelles through non-covalent hydrophobic interactions. Kataoka and coworkers reported covalent conjugation of doxorubicin with the carboxylic acid groups of poly(ethylene glycol)-poly(L-aspartic acid) to increase drug loading in the micelles.112 Since then, they have expanded their interests to other anticancer drugs such as cisplatin,113 paclitaxel,43 and plasmid DNA.114
In addition to diblock copolymers (e.g. PEG-PLA, PEG-PCL), triblock copolymers such as Pluronic® (PEO-PPO-PEO) are also used as micelle carriers. A number of pluronic polymers with different PEO and PPO lengths are available. Kabanov and coworkers have developed Pluronic® micelles for doxorubicin delivery and reported its ability to overcome multidrug resistance in various cancer models.115
For MRI applications, we recently reported SPIO-loaded polymeric micelles as ultra-sensitive MR probes.19, 62, 116 Hydrophobic SPIO nanoparticles were encapsulated inside micelle cores forming highly stable MRI contrast agents. Additionally, an increased transverse relaxivity was observed by increasing the number of SPIO nanoparticles in a micelle. Recently, Nakamura et al designed poly(ethylene glycol)-poly(L-aspartic acid) micelles as smart T1 contrast agents.117 DTPA chelating ligands were conjugated to aspartic acid in the copolymer. Once the Gd-bound polymer formed micelles in the presence of a cationic polymer, the T1 relaxivity was significantly decreased due to the lack of water access. The authors proposed that the intact micelles can passively target tumors and a positive contrast can be observed when micelles dissociate inside tumors.
2.3.3. Liposomes and polymersomes
Liposomes and polymersomes are vesicular nanostructures that are self-assembled from amphiphilic phospholipids and block copolymers, respectively.118, 119 As a result of their inner hydrophilic compartment, these nanostructures are more suitable for delivery of water-soluble agents such as therapeutic proteins or DNAs. Poorly soluble drugs can be entrapped within the hydrophobic bilayer membrane, but the loading capacity is limited due to membrane destabilization effects and may result in unstable structures.120
Among all the nanomedicine platforms, liposomes have demonstrated the most clinical success, with several FDA-approved formulations for cancer treatment.13, 119 Stealth liposomes, where hydrophilic polymers such as PEG have been conjugated on the liposomal surface, considerably prolonged blood circulation times. Effective passive targeting to solid tumors through the EPR effect has been noted in numerous studies.121 Successful liposomes include the clinically approved doxorubicin-containing PEG-liposomes (Doxil®/Caelyx®). These clinical successes make liposomes a very attractive platform for multifunctional nanomedicine development.
Compared to therapeutic delivery, the use of liposomes for the delivery of MRI contrast agents is less advanced clinically but nonetheless an area of active research. Initially, Gd-metal complexes were encapsulated inside the aqueous compartment of the liposome. Unger et al reported the use of Gd-DTPA-loaded liposomes for MR imaging of hepatic metastases.122 They have shown that the liposomes significantly enhanced MRI contrast of the tumors compared to background signal from liver. Nonencapsulated Gd-DTPA failed to produce a visible contrast between tumors and liver. In a separate study, Allen and coworkers developed liposomes that contain both gadolinium complex and iohexol.123 The resulting nanoparticles showed both MRI and CT sensitivities as a multimodal imaging agent. Recently, Aime and coworkers reported the development of liposomes with encapsulation of lanthanide agents (e.g. Tm-DOTMA) as CEST probes for molecular imaging applications.124, 125
3. Integrated nanocomposite particles
All the components of nanocomposite particles have been discussed based on their roles in cancer imaging and drug delivery. Integration of these components into one nanomedicine platform has the potential to achieve both diagnostic information and therapeutic treatment. Here we provide a few highlighted examples of different types of multifunctional nanomedicine.
Paramagnetic macromolecular conjugates
As described previously, polymer conjugates represent one of the most extensively studied nanocarriers.33, 37, 126 More recently, polymer conjugates have been investigated in diagnostic imaging applications, including PET, CT, and MRI.103, 108, 127, 128 Recently, Lu and coworkers reported an integration of therapeutic and diagnostic functionalities in a polymer conjugate system.101, 102 In these studies, poly(L-glutamic acid) (PGA) was used as the polymer carrier, and a T1 agent (Gd-DO3A) and mesochlorin e6 (Mce6), a photosensitizer, were incorporated for MR imaging and photodynamic therapy (PDT), respectively (Fig. 5a). PGA-(Gd-DO3A) was also tested as a control for MRI sensitivity without the PDT activity. The pharmacokinetics of the polymer conjugates were investigated in MDA-MB-231 breast tumor xenografts using T1-weighted MRI. Data show that pegylated polymer conjugates (PEG-PGA-(Gd-DO3A)-Mce6) had prolonged blood circulation half-lives and lower liver uptake than non-pegylated conjugate platforms (PGA-(Gd-DO3A)-Mce6). MRI images of a tumor treated with PEG-PGA-(Gd-DO3A)-Mce6 before and after the injection showed contrast enhancement (Fig. 5d). Quantitative image analysis was able to show that PEG-PGA-(Gd-DO3A)-Mce6 had higher tumor accumulation than its non-pegylated counterparts (Fig. 5b). Based on this information, MRI-guided photodynamic therapy was performed on tumor-bearing mice. Efficacious response in tumors treated with PEG-PGA-(Gd-DO3A)-Mce6 was observed where the tumor size decreased in the first 30 days following treatment (Fig. 5c). In comparison, tumors treated with PGA-(Gd-DO3A)-Mce6 and PGA-(Gd-DO3A) showed much less response with tumor size increasing over time.
Fig. 5.
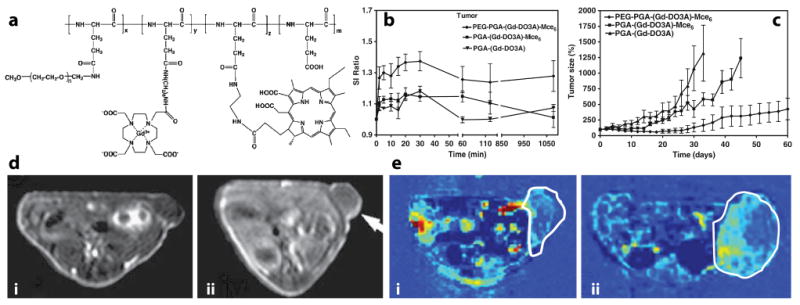
(a) Chemical structure of PEG-PGA-(Gd-DO3A)-Mce6. (b) Signal intensity ratio (SI ratio = SNRpost-inj/SNRpre-inj) in tumors as a function of time for MRI-sensitive polymer conjugates. (c) Antitumor efficacy of photodynamic therapy in MDA-MB-231 xenograft-bearing mice. (d) Spin-echo images of mice treated with PEG-PGA-(Gd-DO3A)-Mce6 before (i) and at 18 hrs after (ii) injection. (e) Representative maps of vascular flow leakage rate in tumor-bearing mice after injection of PEG-PGA-(Gd-DO3A)-Mce6 (i), and PGA-(Gd-DO3A) (ii). Modified from ref. 101. (Permission pending).
In addition to using MRI information to guide PDT therapy, the authors also applied dynamic contrast enhancement (DCE) MRI to monitor the treatment response. DCE-MRI has been widely used to study tumor perfusion properties in diagnostic radiology, in particular monitoring of anti-angiogenic therapy of tumors.129, 130 Using the same MRI-sensitive polymer conjugates, the authors showed that tumors treated with PEG-PGA-(Gd-DO3A)-Mce6 had reduced vascular permeability compared to those treated with the other two agents (Fig. 5e). These results were further validated by decreased microvessel density from histological analysis. This study represents an elegant design of a multifunctional polymer conjugate system that allows for MRI-guided PDT therapy and post-therapy assessment of PDT efficacy in one platform.
Superparamagnetic nanocomposite particles
Compared to paramagnetic chelates (e.g. Gd-DTPA), superparamagnetic nanoparticles (e.g. Fe3O4) can create substantial disturbances in the local magnetic field leading to a rapid dephasing of protons and higher sensitivity for MR detection (Fig. 2). The increased sensitivity allowed for molecular imaging of cancer-specific markers such as transferrin,131 folate132 and Her-2/neu.133, 134 For multifunctional nanomedicine development, simultaneous incorporation of SPIO agents and drug molecules in nanocomposite nanoparticles is a challenging task due to the requirement of materials compatibility among the different components.
Different approaches have been explored for the development of superparamagnetic nanocomposite particles. Recently, Jon and coworkers reported the development of drug-loaded superparamagnetic iron oxide nanoparticles.135 The nanocomposites were prepared by first thermally crosslinking the polymer shell of the SPIO nanoparticles. This was achieved by using poly(3-(trimethoxysilyl)propyl methacrylate-r-PEG methyl ether methacrylate-r-N-acryloxysuccinimide. Upon heating, the trimethoxysilyl segment crosslinked to form a stable layer protecting the Fe3O4 nanoparticle core. The carboxylic acid groups from the acryloxyl segment were present at the surface of the crosslinked SPIO nanoparticles providing a negatively charged electrostatic site for incorporation of doxorubicin (DOX), a positively charged anticancer drug (Fig. 6a). A 2 wt% drug loading was achieved. Drug release was shown to be pH-dependent where 60% of drug was released within 1 hr at pH 5.1 while it took 4 hrs for the comparable amount to be released at pH 7.4. MRI of the particles was evaluated using Lewis lung carcinoma (LLC) tumor-bearing mice. T2-w MR images were taken before and 4.5 hr after the injection (Fig. 6b). The images showed a darkened image contrast indicating tumor accumulation of DOX-SPIO nanoparticles. Antitumor efficacy studies in LLC tumor-bearing mice showed 38% tumor growth inhibition compared to control groups (Fig. 6d), as supported by pictures of excised tumors 19 days after treatment (Fig. 6c).
Fig. 6.
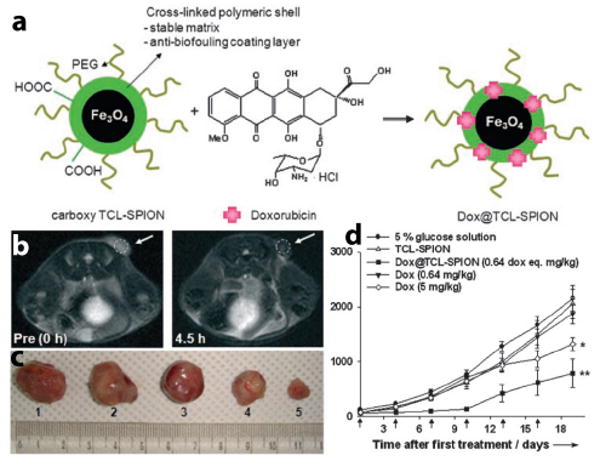
(a) Schematic preparation of DOX-SPIO nanoparticles. (b) T2-w MR images of LLC-tumor-bearing mice before and 4.5 hr after the DOX-SPIO injection. (c) Images of excised tumors from the mice 19 days after treatment. (d) Tumor-volume relationship of LLC tumors for different treatment groups (*P<0.005, **P<0.01, n=5–7). Modified from ref. 135. (Permission pending)
In a different approach, our laboratory established a polymeric micelle platform for the delivery of anticancer drugs and SPIO agents (Fig. 7a).19 Amphiphilic block copolymers (e.g., poly(ethylene glycol)-poly(D,L-lactic acid), PEG-PLA) were used to form core-shell nanoparticles. Hydrophobic SPIO nanoparticles and DOX were loaded inside the hydrophobic core of micelles (Fig. 7b-c). Previous studies have shown that a high loading of SPIO, up to 50wt% of micelles can be achieved, and furthermore, SPIO clustering led to considerable enhancement of T2 relaxivity on a per Fe basis over single SPIO micelles.116 In addition, DOX loading was increased from 3 to 12 wt% when SPIO was introduced, demonstrating synergy in loading both agents in the same micelle core. To achieve cancer targeting functionality, the surface of micelles was functionalized with a cyclic RGDfK pentapeptide (cRGD) to target αvβ3 integrin receptors that are over-expressed in the tumor vasculature.136, 137
Fig. 7.
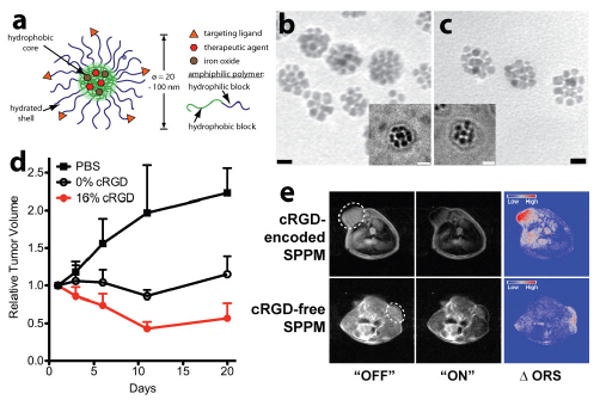
(a) Schematic of a multifunctional polymeric micelle with cRGD surface functionalization and DOX and SPIO loading. (b, c) TEM images of cRGD-encoded and cRGD-free micelles showing the clusters of SPIO nanoparticles in the micelle core. The insets are corresponding cryo-TEM images. (d) Tumor growth inhibition for different micelle groups in A549 human lung tumor-bearing mice. (e) Off-resonance saturation MR images of A549 tumors treated with cRGD-encoded and cRGD-free micelles respectively. Modified from refs. 19 and 110. (Permission pending)
Antitumor efficacy and MR imaging studies were carried out in subcutaneous A549 human lung tumor-bearing mice (Fig. 7d).110 cRGD-encoded micelles via intravenous administration (4 mg DOX/kg dose) showed a significant tumor growth inhibition compared to non-cRGD micelles at the same DOX dose. This was primarily due to the αvβ3-targeted accumulation of cRGD-micelles as demonstrated by MRI (Fig. 7e). Conventionally, a T2*-weighted method serves as the gold standard for SPIO imaging. However, this method is prone to image artifacts due to magnetic susceptibility effects from SPIO and tissue interfaces. Recently, we reported the use of an off-resonance saturation (ORS) method to improve the detection accuracy of SPIO particles.138 The ORS method allows for the turning “on” and “off” of SPIO contrast by the application of an RF pulse, which can significantly improve image accuracy. For example, one hour after micelle injection, ORS images showed a clear accumulation of cRGD-encoded micelles in A549 tumors (top panel, Fig. 7e). In comparison, less ORS contrast was observed with cRGD-free micelles. Quantitative image analysis indicated that contrast over noise ratio (CNR) of the tumor was almost twice as much for cRGD-encoded micelles over cRGD-free micelles. Prussian blue staining of Fe in tumor tissues showed micelles closely associated with αvβ3 expressing tumor vasculature for cRGD-encoded micelles. In contrast, cRGD-free SPPM showed accumulation in the tumor parenchyma in a diffusive pattern, consistent with passive targeting of SPPM to solid tumors through the EPR effect.
The above studies demonstrate the “theranostic” potential of superparamagnetic nanocomposite particles. Tumor accumulation of SPIO-loaded nanoparticles can be non-invasively measured by MR imaging and these data correlate well with the therapeutic response of these nanoparticles in preclinical animal tumor models. Potentially, the proposed platform can be used to provide an initial pharmacokinetic assessment of the tumor targeting efficiency of nanoparticles, as well as a quick surrogate marker for subsequent therapeutic responses. In addition, resistance to treatment (e.g. down regulation of targeted receptors) can also be monitored with repeated applications, which can be used to adjust the treatment regimen (e.g. using a different targeting ligand) to overcome resistance while minimizing toxic side effects.
Multi-chromatic CEST nanoparticles
Liposomes are one of the most established nanoplatforms with several formulations (e.g. Doxil®) already in clinical use for cancer treatment. Their proven safety and biocompatibility make it attractive for the development of multifunctional nanomedicine.
Aime and coworkers have recently reported the development of LIPOCEST agents by encapsulating lanthanide complexes inside the aqueous compartment of liposomes (Fig. 8a, see also Section 2.2).78, 124, 125 Encapsulation of a fast exchanging lanthanide complex was able to induce a change in the chemical shift of intra-liposomal water, which became exchangeable protons for CEST imaging (pool B as in Fig. 2 right panel). Since 106-109 mobile water protons can be trapped inside each liposome (50-200 nm in diameter), this large pool of water protons results in a significantly increased sensitivity for MRI detection. In a representative LIPOCEST system where ∼100 mM of TmDOTMA- was entrapped in liposomes from a mixture of POPC:DPPG:cholesterol (55:5:40 w/w/w) lipids, Aime and coworkers demonstrate detection of LIPOCEST particles at 90 pM of concentration (Fig. 8b).78 In addition to ultrasensitivity, the other exciting feature of LIPOCEST agents is their multi-chromatic properties. In this regard, the chemical shift of intra-liposomal water protons can be controlled by varying the type of lanthanide ions (Fig. 8c), type of chelating ligands (Fig. 8d) and/or concentration of Ln complexes. Thus, a variety of LIPOCEST agents can be prepared where each LIPOCEST particle can be activated by an appropriate water frequency, which will lead to a collection of “multi-colored” MRI contrast agents. Potentially, each LIPOCEST probe can be encoded with a cancer-specific ligand (monoclonal antibodies or phage peptides) to produce a library of molecular probes that allow for simultaneous “finger printing” of different phenotypes of cancer. The LIPOCEST composition that provides the best match for tumors can be functionalized with therapeutic agents to deliver targeted therapy of cancer. Although still at an early stage, this multi-chromatic feature and integrated therapeutic design of LIPOCEST particles have the potential to achieve personalized medicine to improve the safety and efficacy of cancer chemotherapy.
Fig. 8.
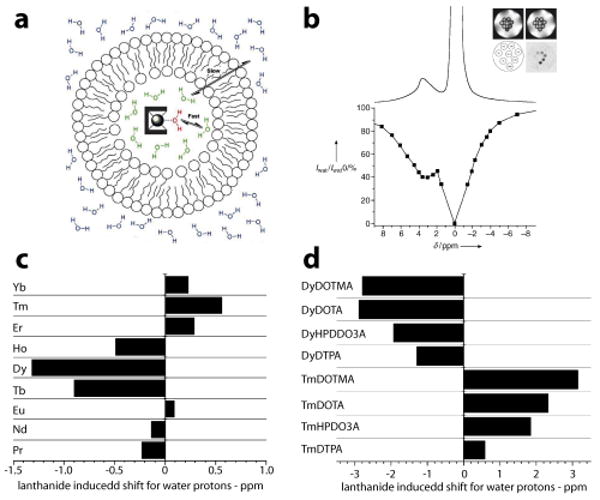
(a) Schematic of a liposome filled with a water shift reagent, [Tm(DOTMA)]. Water exchange rate across the liposomal membrane is the key determinant factor for LIPOCEST effect. (b) 1H NMR and Z-spectrum of [Tm(DOTMA)] LIPOCEST agent. (c) Lanthanide induced shift for water protons in 100 mM aqueous solution of [Ln-DTPA]2− complexes, and (d) influence of chelating ligands on lanthanide induced shift. Modified from Refs. 57, 67, and 78. (Permission Pending)
4. Conclusions
Multifunctional nanomedicine is a rapidly evolving field for cancer imaging and intervention. Significant advances in cancer biology, materials science, and imaging technology now make it possible to apply the integrated design principles to address the formidable challenges presented by cancer therapy. Multi-component nanomedicine with modular designs that can be personalized to individual patients and deliver therapy based on diagnostic information represent a new tool to combat cancer. Despite the therapeutic promise, many scientific and technological challenges still remain. For example, fundamental understanding of the molecular interactions among therapeutic drugs, MR contrast agents, and polymer carriers at the nanoscale will be critical for the assembly of these components into efficacious nanocomposite particles. The influence of one structural component on the performance of the others must be carefully investigated to ensure synergy in the integrated design. This will be particularly true for MRI functionalities to achieve adequate imaging sensitivity for cancer-specific diagnosis. A mechanistic understanding of the subtle relaxation behaviors and their dependence on nanostructural parameters will be essential to guide the incorporation strategies of different types of MRI contrast agents. In addition to the highlighted examples in the current feature article, more and more nanocomposite particles with integration of multiple functionalities will emerge to address the challenges of tumor heterogeneity and adaptive resistance in cancer therapy. Ultimately, successful development of these “nanotheranostics” may one day provide us with magic bullets for cancer therapy.
Acknowledgments
We thank the National Institutes of Health (NIH EB005394 and CA129011) for financial support to JG. C. Khemtong thanks Department of Defense Breast Cancer Research Program for a Multidisciplinary Postdoctoral Award (W81XWH-06-1-0751). This is CSCN042 from the Program in Cell Stress and Cancer Nanomedicine, Simmons Comprehensive Cancer Center, UT Southwestern Medical Center.
References
- 1.WHO. Cancer. 2008 www.who.int.
- 2.Jemal A, Siegel R, Ward E, Hao YP, Xu JQ, Murray T, Thun MJ. Ca-Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Smith RA, Cokkinides V, Eyre HJ. CA Cancer J Clin. 2005;55:31–44. doi: 10.3322/canjclin.55.1.31. quiz 55-36. [DOI] [PubMed] [Google Scholar]
- 4.Weissleder R, Pittet MJ. Nature. 2008;452:580–589. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 6.Jun YW, Choi JS, Cheon J. Chem Commun. 2007:1203–1214. doi: 10.1039/b614735f. [DOI] [PubMed] [Google Scholar]
- 7.Laurent S, Forge D, Port M, Roch A, Robic C, Elst LV, Muller RN. Chem Rev. 2008;108:2064–2110. doi: 10.1021/cr068445e. [DOI] [PubMed] [Google Scholar]
- 8.Thorek DL, Chen AK, Czupryna J, Tsourkas A. Ann Biomed Eng. 2006;34:23–38. doi: 10.1007/s10439-005-9002-7. [DOI] [PubMed] [Google Scholar]
- 9.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 10.Otsuka H, Nagasaki Y, Kataoka K. Adv Drug Deliv Rev. 2003;55:403–419. doi: 10.1016/s0169-409x(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 11.Sutton D, Nasongkla N, Blanco E, Gao J. Pharm Res. 2007;24:1029–1046. doi: 10.1007/s11095-006-9223-y. [DOI] [PubMed] [Google Scholar]
- 12.Ferrari M. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 13.Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nature Nanotechnology. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 14.Sumer B, Gao JM. Nanomedicine-Uk. 2008;3:137–140. doi: 10.2217/17435889.3.2.137. [DOI] [PubMed] [Google Scholar]
- 15.Warner S. Scientist. 2004;18:38–39. [Google Scholar]
- 16.Trewyn BG, Giri S, Slowing II, Lin VSY. Chem Commun. 2007:3236–3245. doi: 10.1039/b701744h. [DOI] [PubMed] [Google Scholar]
- 17.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. Acs Nano. 2008;2:889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarthy JR, Weissleder R. Adv Drug Deliver Rev. 2008;60:1241–1251. doi: 10.1016/j.addr.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nasongkla N, Bey E, Ren JM, Ai H, Khemtong C, Guthi JS, Chin SF, Sherry AD, Boothman DA, Gao JM. Nano Lett. 2006;6:2427–2430. doi: 10.1021/nl061412u. [DOI] [PubMed] [Google Scholar]
- 20.Leader B, Baca QJ, Golan DE. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 21.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Proc Natl Acad Sci U S A. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo S, Huang F, Guo P. Gene Ther. 2006;13:814–820. doi: 10.1038/sj.gt.3302716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang HL, Tang GP, Wang QQ, Li D, Shen FP, Zhou J, Yu H. Chem Commun. 2006:2382–2384. doi: 10.1039/b601130f. [DOI] [PubMed] [Google Scholar]
- 24.Zuber G, Zammut-Italiano L, Dauty E, Behr JP. Angew Chem Int Ed Engl. 2003;42:2666–2669. doi: 10.1002/anie.200250446. [DOI] [PubMed] [Google Scholar]
- 25.Blanco E, Bey EA, Dong Y, Weinberg BD, Sutton DM, Boothman DA, Gao J. J Control Release. 2007;122:365–374. doi: 10.1016/j.jconrel.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherry AD, Woods M. Annu Rev Biomed Eng. 2008;10:391–411. doi: 10.1146/annurev.bioeng.9.060906.151929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu J, Liong M, Zink JI, Tamanoi F. Small. 2007;3:1341–1346. doi: 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- 28.Slowing II, Trewyn BG, Lin VSY. J Am Chem Soc. 2007;129:8845–8849. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]
- 29.Hartman KB, Laus S, Bolskar RD, Muthupillai R, Helm L, Toth E, Merbach AE, Wilson LJ. Nano Lett. 2008;8:415–419. doi: 10.1021/nl0720408. [DOI] [PubMed] [Google Scholar]
- 30.Sitharaman B, Kissell KR, Hartman KB, Tran LA, Baikalov A, Rusakova I, Sun Y, Khant HA, Ludtke SJ, Chiu W, Laus S, Toth E, Helm L, Merbach AE, Wilson LJ. Chem Commun. 2005:3915–3917. doi: 10.1039/b504435a. [DOI] [PubMed] [Google Scholar]
- 31.Csajbok E, Banyai I, Vander Elst L, Muller RN, Zhou WZ, Peters JA. Chem-Eur J. 2005;11:4799–4807. doi: 10.1002/chem.200500039. [DOI] [PubMed] [Google Scholar]
- 32.Zarkovic N, Zarkovic K, Kralj M, Borovic S, Sabolovic S, Blazi MP, Cipak A, Pavelic K. Anticancer Res. 2003;23:1589–1595. [PubMed] [Google Scholar]
- 33.Duncan R. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 34.Kabanov AV, Batrakova EV, Miller DW. Adv Drug Deliver Rev. 2003;55:151–164. doi: 10.1016/s0169-409x(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 35.Ke WL, Zhao YS, Huang RQ, Jiang C, Pei YY. J Pharm Sci-Us. 2008;97:2208–2216. doi: 10.1002/jps.21155. [DOI] [PubMed] [Google Scholar]
- 36.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Frechet JMJ, Dy EE, Szoka FC. P Natl Acad Sci USA. 2006;103:16649–16654. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nori A, Kopecek J. Adv Drug Deliv Rev. 2005;57:609–636. doi: 10.1016/j.addr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Gabizon A, Shmeeda H, Barenholz Y. Clin Pharmacokinet. 2003;42:419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 39.Batrakova EV, Li S, Li YL, Alakhov VY, Elmquist WF, Kabanov AV. J Control Release. 2004;100:389–397. doi: 10.1016/j.jconrel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Kataoka K, Matsumoto T, Yokoyama M, Okano T, Sakurai Y, Fukushima S, Okamoto K, Kwon GS. J Control Release. 2000;64:143–153. doi: 10.1016/s0168-3659(99)00133-9. [DOI] [PubMed] [Google Scholar]
- 41.Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA, Gao J. Angew Chem Int Ed Engl. 2004;43:6323–6327. doi: 10.1002/anie.200460800. [DOI] [PubMed] [Google Scholar]
- 42.D'Souza GGM, Wang T, Rockwell K, Torchilin VP. Pharm Res. 2008;25:2567–2572. doi: 10.1007/s11095-008-9674-4. [DOI] [PubMed] [Google Scholar]
- 43.Hamaguchi T, Matsumura Y, Suzuki M, Shimizu K, Goda R, Nakamura I, Nakatomi I, Yokoyama M, Kataoka K, Kakizoe T. Brit J Cancer. 2005;92:1240–1246. doi: 10.1038/sj.bjc.6602479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hongrapipat J, Kopeckova P, Liu J, Prakongpan S, Kopecek J. Mol Pharmaceut. 2008;5:696–709. doi: 10.1021/mp800006e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 46.Putney SD, Burke PA. Nat Biotechnol. 1998;16:153–157. doi: 10.1038/nbt0298-153. [DOI] [PubMed] [Google Scholar]
- 47.Maeda H, Ueda M, Morinaga T, Matsumoto T. J Med Chem. 1985;28:455–461. doi: 10.1021/jm00382a012. [DOI] [PubMed] [Google Scholar]
- 48.Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H. Clin Pharmacokinet. 2003;42:1089–1105. doi: 10.2165/00003088-200342130-00002. [DOI] [PubMed] [Google Scholar]
- 49.Maeda H, Sawa T, Konno T. J Control Release. 2001;74:47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 50.Villares GJ, Zigler M, Wang H, Melnikova VO, Wu H, Friedman R, Leslie MC, Vivas-Mejia PE, Lopez-Berestein G, Sood AK, Bar-Eli M. Cancer Res. 2008;68:9078–9086. doi: 10.1158/0008-5472.CAN-08-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonoke S, Ueda T, Fujiwara K, Sato Y, Takagaki K, Hirabayashi K, Ohgi T, Yano J. Cancer Res. 2008;68:8843–8851. doi: 10.1158/0008-5472.CAN-08-0127. [DOI] [PubMed] [Google Scholar]
- 52.Sutton D, Kim S, Shuai X, Leskov K, Marques JT, Williams BR, Boothman DA, Gao J. Int J Nanomedicine. 2006;1:155–162. doi: 10.2147/nano.2006.1.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abela RA, Qian J, Xu L, Lawrence TS, Zhang M. Cancer Gene Ther. 2008;15:496–507. doi: 10.1038/cgt.2008.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Green JJ, Langer R, Anderson DG. Accounts Chem Res. 2008;41:749–759. doi: 10.1021/ar7002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lauterbur PC. Nature. 1973;242:191–192. [Google Scholar]
- 56.Jaffer FA, Weissleder R. JAMA. 2005;293:855–862. doi: 10.1001/jama.293.7.855. [DOI] [PubMed] [Google Scholar]
- 57.Aime S, Crich SG, Gianolio E, Giovenzana GB, Tei L, Terreno E. Coordin Chem Rev. 2006;250:1562–1579. [Google Scholar]
- 58.Zhang SR, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. Accounts Chem Res. 2003;36:783–790. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- 59.Wang YXJ, Hussain SM, Krestin GP. Eur Radiol. 2001;11:2319–2331. doi: 10.1007/s003300100908. [DOI] [PubMed] [Google Scholar]
- 60.Sun SH, Zeng H, Robinson DB, Raoux S, Rice PM, Wang SX, Li GX. J Am Chem Soc. 2004;126:273–279. doi: 10.1021/ja0380852. [DOI] [PubMed] [Google Scholar]
- 61.Lee JH, Huh YM, Jun Y, Seo J, Jang J, Song HT, Kim S, Cho EJ, Yoon HG, Suh JS, Cheon J. Nature Medicine. 2007;13:95–99. doi: 10.1038/nm1467. [DOI] [PubMed] [Google Scholar]
- 62.Barcena C, Sra AK, Chaubey GS, Khemtong C, Liu JP, Gao J. Chem Commun. 2008:2224–2226. doi: 10.1039/b801041b. [DOI] [PubMed] [Google Scholar]
- 63.Seo WS, Lee JH, Sun XM, Suzuki Y, Mann D, Liu Z, Terashima M, Yang PC, McConnell MV, Nishimura DG, Dai HJ. Nature Materials. 2006;5:971–976. doi: 10.1038/nmat1775. [DOI] [PubMed] [Google Scholar]
- 64.Chen M, Liu JP, Sun S. J Am Chem Soc. 2004;126:8394–8395. doi: 10.1021/ja047648m. [DOI] [PubMed] [Google Scholar]
- 65.Sun SH, Murray CB, Weller D, Folks L, Moser A. Science. 2000;287:1989–1992. doi: 10.1126/science.287.5460.1989. [DOI] [PubMed] [Google Scholar]
- 66.Ward KM, Aletras AH, Balaban RS. J Magn Reson. 2000;143:79–87. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- 67.Woods M, Donald EWC, Sherry AD. Chem Soc Rev. 2006;35:500–511. doi: 10.1039/b509907m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Zijl PC, Jones CK, Ren J, Malloy CR, Sherry AD. Proc Natl Acad Sci U S A. 2007;104:4359–4364. doi: 10.1073/pnas.0700281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gilad AA, McMahon MT, Walczak P, Winnard PT, Raman V, van Laarhoven HWM, Skoglund CM, Bulte JWM, van Zijl PCM. Nat Biotechnol. 2007;25:217–219. doi: 10.1038/nbt1277. [DOI] [PubMed] [Google Scholar]
- 70.Zhou JY, Payen JF, Wilson DA, Traystman RJ, van Zijl PCM. Nat Med. 2003;9:1085–1090. doi: 10.1038/nm907. [DOI] [PubMed] [Google Scholar]
- 71.Schroder L, Lowery TJ, Hilty C, Wemmer DE, Pines A. Science. 2006;314:446–449. doi: 10.1126/science.1131847. [DOI] [PubMed] [Google Scholar]
- 72.Garcia S, Chavez L, Lowery TJ, Han SI, Wemmer DE, Pines A. J Magn Reson. 2007;184:72–77. doi: 10.1016/j.jmr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 73.Zhou JY, van Zijl PCM. Prog Nucl Mag Res Sp. 2006;48:109–136. [Google Scholar]
- 74.Zhang SR, Winter P, Wu KC, Sherry AD. J Am Chem Soc. 2001;123:1517–1518. doi: 10.1021/ja005820q. [DOI] [PubMed] [Google Scholar]
- 75.Zhang S, Wu K, Sherry AD. J Am Chem Soc. 2002;124:4226–4227. doi: 10.1021/ja017133k. [DOI] [PubMed] [Google Scholar]
- 76.Ali MM, Woods M, Suh EH, Kovacs Z, Tircso G, Zhao P, Kodibagkar VD, Sherry AD. J Biol Inorg Chem. 2007;12:855–865. doi: 10.1007/s00775-007-0240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoo B, Pagel MD. J Am Chem Soc. 2006;128:14032–14033. doi: 10.1021/ja063874f. [DOI] [PubMed] [Google Scholar]
- 78.Aime S, Castelli DD, Terreno E. Angew Chem Int Edit. 2005;44:5513–5515. doi: 10.1002/anie.200501473. [DOI] [PubMed] [Google Scholar]
- 79.Li SD, Huang L. Mol Pharm. 2008;5:496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 80.Kim JH, Kim YS, Park K, Kang E, Lee S, Nam HY, Kim K, Park JH, Chi DY, Park RW, Kim IS, Choi K, Chan Kwon I. Biomaterials. 2008;29:1920–1930. doi: 10.1016/j.biomaterials.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 81.Min KH, Park K, Kim YS, Bae SM, Lee S, Jo HG, Park RW, Kim IS, Jeong SY, Kim K, Kwon IC. J Control Release. 2008;127:208–218. doi: 10.1016/j.jconrel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 82.Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. Science. 2002;298:1759–1762. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 83.Jatzkewitz H. Z Naturforsch. 1955;10:27–31. [Google Scholar]
- 84.Delgado C, Francis GE, Fisher D. Crit Rev Ther Drug. 1992;9:249–304. [PubMed] [Google Scholar]
- 85.Davis FF. Adv Drug Deliver Rev. 2002;54:457–458. doi: 10.1016/s0169-409x(02)00021-2. [DOI] [PubMed] [Google Scholar]
- 86.Nandy P, Periclou AP, Avramis VI. Anticancer Res. 1998;18:727–737. [PubMed] [Google Scholar]
- 87.Greenwald RB, Choe YH, McGuire J, Conover CD. Adv Drug Deliv Rev. 2003;55:217–250. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 88.Duncan R, Spreafico F. Clin Pharmacokinet. 1994;27:290–306. doi: 10.2165/00003088-199427040-00004. [DOI] [PubMed] [Google Scholar]
- 89.Sprincl L, Exner J, Sterba O, Kopecek J. Journal of Biomedical Materials Research. 1976;10:953–963. doi: 10.1002/jbm.820100612. [DOI] [PubMed] [Google Scholar]
- 90.Duncan R, Kopecek J. Adv Polym Sci. 1984;57:51–101. [Google Scholar]
- 91.Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, Doran J, Young AM, Burtles S, Kerr DJ, Clin CCRPII. J Clin Oncol. 2002;20:1668–1676. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- 92.Meerum Terwogt JM, ten Bokkel Huinink WW, Schellens JH, Schot M, Mandjes IA, Zurlo MG, Rocchetti M, Rosing H, Koopman FJ, Beijnen JH. Anticancer Drugs. 2001;12:315–323. doi: 10.1097/00001813-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 93.Schoemaker NE, van Kesteren C, Rosing H, Jansen S, Swart M, Lieverst J, Fraier D, Breda M, Pellizzoni C, Spinelli R, Grazia Porro M, Beijnen JH, Schellens JH, ten Bokkel Huinink WW. Br J Cancer. 2002;87:608–614. doi: 10.1038/sj.bjc.6600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crespo L, Sanclimens G, Pons M, Giralt E, Royo M, Albericio F. Chem Rev. 2005;105:1663–1681. doi: 10.1021/cr030449l. [DOI] [PubMed] [Google Scholar]
- 95.Gao C, Yan D. Prog Polym Sci. 2004;29:183–275. [Google Scholar]
- 96.Lai PS, Lou PJ, Peng CL, Pai CL, Yen WN, Huang MY, Young TH, Shieh MJ. J Control Release. 2007;122:39–46. doi: 10.1016/j.jconrel.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 97.Khandare JJ, Jayant S, Singh A, Chandna P, Wang Y, Vorsa N, Minko T. Bioconjug Chem. 2006;17:1464–1472. doi: 10.1021/bc060240p. [DOI] [PubMed] [Google Scholar]
- 98.Bertin PA, Smith D, Nguyen ST. Chem Commun. 2005:3793–3795. doi: 10.1039/b504643b. [DOI] [PubMed] [Google Scholar]
- 99.Kolhe P, Khandare J, Pillai O, Kannan S, Lieh-Lai M, Kannan RM. Biomaterials. 2006;27:660–669. doi: 10.1016/j.biomaterials.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 100.Lu ZR, Ye FR, Vaidya A. J Control Release. 2007;122:269–277. doi: 10.1016/j.jconrel.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vaidya A, Sun Y, Feng Y, Emerson L, Jeong EK, Lu ZR. Pharm Res. 2008;25:2002–2011. doi: 10.1007/s11095-008-9608-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vaidya A, Sun Y, Ke T, Jeong EK, Lu ZR. Magn Reson Med. 2006;56:761–767. doi: 10.1002/mrm.21009. [DOI] [PubMed] [Google Scholar]
- 103.Lu ZR. Pharm Res. 2007;24:1170–1171. doi: 10.1007/s11095-007-9281-9. [DOI] [PubMed] [Google Scholar]
- 104.Wu Y, Zhou Y, Ouari O, Woods M, Zhao P, Soesbe TC, Kiefer GE, Sherry AD. J Am Chem Soc. 2008;130:13854–13855. doi: 10.1021/ja805775u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ali MM, Woods M, Caravan P, Opina ACL, Spiller M, Fettinger JC, Sherry AD. Chem-Eur J. 2008;14:7250–7258. doi: 10.1002/chem.200800402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shukla R, Thomas TP, Peters J, Kotlyar A, Myc A, Baker JR., Jr Chem Commun. 2005:5739–5741. doi: 10.1039/b507350b. [DOI] [PubMed] [Google Scholar]
- 107.Wang SJ, Brechbiel M, Wiener EC. Invest Radiol. 2003;38:662–668. doi: 10.1097/01.rli.0000084887.47427.75. [DOI] [PubMed] [Google Scholar]
- 108.Kobayashi H, Kawamoto S, Star RA, Waldmann TA, Tagaya Y, Brechbiel MW. Cancer Research. 2003;63:271–276. [PubMed] [Google Scholar]
- 109.Pikkemaat JA, Wegh RT, Lamerichs R, van de Molengraaf RA, Langereis S, Burdinski D, Raymond AYF, Janssen HM, de Waal BFM, Willard NP, Meijer EW, Grull H. Contrast Media Mol I. 2007;2:229–239. doi: 10.1002/cmmi.149. [DOI] [PubMed] [Google Scholar]
- 110.Blanco E, Kessinger CW, Sumer BD, Gao J. Exp Biol Med. doi: 10.3181/0808-MR-250. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Torchilin VP. Adv Drug Deliv Rev. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 112.Yokoyama M, Okano T, Sakurai Y, Ekimoto H, Shibazaki C, Kataoka K. Cancer Res. 1991;51:3229–3236. [PubMed] [Google Scholar]
- 113.Nishiyama N, Kataoka K. J Control Release. 2001;74:83–94. doi: 10.1016/s0168-3659(01)00314-5. [DOI] [PubMed] [Google Scholar]
- 114.Akagi D, Oba M, Koyama H, Nishiyama N, Fukushima S, Miyata T, Nagawa H, Kataoka K. Gene Therapy. 2007;14:1029–1038. doi: 10.1038/sj.gt.3302945. [DOI] [PubMed] [Google Scholar]
- 115.Kabanov AV, Batrakova EV, Alakhov VY. Adv Drug Deliver Rev. 2002;54:759–779. doi: 10.1016/s0169-409x(02)00047-9. [DOI] [PubMed] [Google Scholar]
- 116.Ai H, Flask C, Weinberg B, Shuai X, Pagel MD, Farrell D, Duerk J, Gao JM. Advanced Materials. 2005;17:1949–+. [Google Scholar]
- 117.Nakamura E, Makino K, Okano T, Yamamoto T, Yokoyama M. J Control Release. 2006;114:325–333. doi: 10.1016/j.jconrel.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 118.Discher DE, Ahmed F. Annu Rev Biomed Eng. 2006;8:323–341. doi: 10.1146/annurev.bioeng.8.061505.095838. [DOI] [PubMed] [Google Scholar]
- 119.Torchilin VP. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 120.Liu J, Lee H, Huesca M, Young A, Allen C. Cancer Chemother Pharmacol. 2006;58:306–318. doi: 10.1007/s00280-005-0161-x. [DOI] [PubMed] [Google Scholar]
- 121.Gabizon AA. Cancer Invest. 2001;19:424–436. doi: 10.1081/cnv-100103136. [DOI] [PubMed] [Google Scholar]
- 122.Unger EC, Winokur T, Macdougall P, Rosenblum J, Clair M, Gatenby R, Tilcock C. Radiology. 1989;171:81–85. doi: 10.1148/radiology.171.1.2928550. [DOI] [PubMed] [Google Scholar]
- 123.Zheng JZ, Liu JB, Dunne M, Jaffray DA, Allen C. Pharm Res. 2007;24:1193–1201. doi: 10.1007/s11095-006-9220-1. [DOI] [PubMed] [Google Scholar]
- 124.Aime S, Castelli DD, Lawson D, Terreno E. J Am Chem Soc. 2007;129:2430–+. doi: 10.1021/ja0677867. [DOI] [PubMed] [Google Scholar]
- 125.Terreno E, Cabella C, Carrera C, Castelli DD, Mazzon R, Rollet S, Stancanello J, Visigalli M, Aime S. Angew Chem Int Edit. 2007;46:966–968. doi: 10.1002/anie.200604027. [DOI] [PubMed] [Google Scholar]
- 126.Megeed Z, Cappello J, Ghandehari H. Adv Drug Deliv Rev. 2002;54:1075–1091. doi: 10.1016/s0169-409x(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 127.Hamoudeh M, Kamleh MA, Diab R, Fessi H. Adv Drug Deliv Rev. 2008;60:1329–1346. doi: 10.1016/j.addr.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 128.Rabin O, Manuel Perez J, Grimm J, Wojtkiewicz G, Weissleder R. Nat Mater. 2006;5:118–122. doi: 10.1038/nmat1571. [DOI] [PubMed] [Google Scholar]
- 129.Ocak I, Baluk P, Barrett T, McDonald DM, Choyke P. Front Biosci. 2007;12:3601–3616. doi: 10.2741/2337. [DOI] [PubMed] [Google Scholar]
- 130.Barrett T, Kobayashi H, Brechbiel M, Choyke PL. Eur J Radiol. 2006;60:353–366. doi: 10.1016/j.ejrad.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 131.Hogemann-Savellano D, Bos E, Blondet C, Sato F, Abe T, Josephson L, Weissleder R, Gaudet J, Sgroi D, Peters PJ, Basilion JP. Neoplasia. 2003;5:495–506. doi: 10.1016/s1476-5586(03)80034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sonvico F, Mornet S, Vasseur S, Dubernet C, Jaillard D, Degrouard J, Hoebeke J, Duguet E, Colombo P, Couvreur P. Bioconjug Chem. 2005;16:1181–1188. doi: 10.1021/bc050050z. [DOI] [PubMed] [Google Scholar]
- 133.Artemov D, Mori N, Okollie B, Bhujwalla ZM. Magn Reson Med. 2003;49:403–408. doi: 10.1002/mrm.10406. [DOI] [PubMed] [Google Scholar]
- 134.Funovics MA, Kapeller B, Hoeller C, Su HS, Kunstfeld R, Puig S, Macfelda K. Magnetic resonance imaging. 2004;22:843–850. doi: 10.1016/j.mri.2004.01.050. [DOI] [PubMed] [Google Scholar]
- 135.Yu MK, Jeong YY, Park J, Park S, Kim JW, Min JJ, Kim K, Jon S. Angew Chem Int Ed Engl. 2008;47:5362–5365. doi: 10.1002/anie.200800857. [DOI] [PubMed] [Google Scholar]
- 136.Cheresh DA, Smith JW, Cooper HM, Quaranta V. Cell. 1989;57:59–69. doi: 10.1016/0092-8674(89)90172-4. [DOI] [PubMed] [Google Scholar]
- 137.Haubner R, Gratias R, Diefenbach B, Goodman SL, Jonczyk A, Kessler H. J Am Chem Soc. 1996;118:7461–7472. [Google Scholar]
- 138.Khemtong C, Kessinger CW, Ren J, Bey EA, Yang SG, Guthi JS, Boothman DA, Sherry AD, Gao J. Cancer Res. doi: 10.1158/0008-5472.CAN-08-3231. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]