

Abstract
Cortical spreading depression is a propagating wave of depolarization that plays important roles in migraine, stroke, subarachnoid haemorrhage and brain injury. Cortical spreading depression is associated with profound vascular changes that may be a significant factor in the clinical response to cortical spreading depression events. We used a combination of optical intrinsic signal imaging, electro-physiology, potassium sensitive electrodes and spectroscopy to investigate neurovascular changes associated with cortical spreading depression in the mouse. We identified two distinct phases of altered neurovascular function, one during the propagating cortical spreading depression wave and a second much longer phase after passage of the wave. The direct current shift associated with the cortical spreading depression wave was accompanied by marked arterial constriction and desaturation of cortical haemoglobin. After recovery from the initial cortical spreading depression wave, we observed a second phase of prolonged, negative direct current shift, arterial constriction and haemoglobin desaturation, lasting at least an hour. Persistent disruption of neurovascular coupling was demonstrated by a loss of coherence between electro-physiological activity and perfusion. Extracellular potassium concentration increased during the cortical spreading depression wave, but recovered and remained at baseline after passage of the wave, consistent with different mechanisms underlying the first and second phases of neurovascular dysfunction. These findings indicate that cortical spreading depression is associated with a multiphasic alteration in neurovascular function, including a novel second direct current shift accompanied by arterial constriction and decrease in tissue oxygen supply, that is temporally and mechanistically distinct from the initial propagated cortical spreading depression wave. Vascular/metabolic uncoupling with cortical spreading depression may have important clinical consequences, and the different phases of dysfunction may represent separate therapeutic targets in the disorders where cortical spreading depression occurs.
Keywords: spreading depression, haemoglobin, neurovascular coupling, migraine, stroke
Introduction
Cortical spreading depression (CSD) is a massive spreading depolarization of neurons and glia (Leao, 1944; Somjen, 2001). CSD is thought to underlie the migraine aura, and functional imaging has shown propagated events consistent with CSD in migraine patients (Olesen et al., 1981; Hadjikhani et al., 2001). Related depolarizations occur in humans around infarcts, cortical trauma and subarachnoid haemorrhage (Mayevsky et al., 1996; Strong et al., 2002; Dohmen et al., 2008). CSD, once considered an experimental curiosity, is increasingly recognized as an element of human disease and a target for treatment.
Large changes in perfusion (blood flow, blood volume or arterial diameter) accompany the electro-cortical activity of CSD. Most prominent is a brief increase in perfusion, followed by a long-lasting hypoperfusion (Lauritzen et al., 1982; Busija et al., 2008). These changes were initially interpreted as reflecting normal neurovascular coupling, with perfusion increases to meet the needs of depolarization, and decreases afterward during the relative quiescence (‘depression’) of cortical activity. However, paradoxical reductions in perfusion, opposite to what would be expected from normal neurovascular coupling, may occur during the CSD wave in multiple species (Ayata et al., 2004; Tomita et al., 2005; Osada et al., 2006), including humans (Dreier et al., 2009). Changes in arterial carbon dioxide reactivity (Lauritzen et al., 1982, 1984; Scheckenbach et al., 2006) and in physiologically induced blood volume responses (Guiou et al., 2005; Piilgaard and Lauritzen, 2009) show that long-lasting changes in neurovascular coupling can follow the CSD wave.
The CSD perfusion response varies with the physiological state of the organism: decreasing oxygenation or blood pressure, increasing extracellular potassium ion (K+) concentration, or inhibiting nitric oxide synthesis can enhance CSD-associated perfusion decreases (Dreier et al., 1998; Sukhotinsky et al., 2008). Peri-infarct depolarizations, which are electro-physiologically indistinguishable from CSD (Czéh et al., 1993) show a more prominent constrictive vascular response than CSD in normal tissue (Shin et al., 2006; Strong et al., 2007). The perfusion response is also variable across species; mice appear to have a more pronounced hypoperfusion and less prominent hyperaemia than other animals (Ayata et al., 2004; Brennan et al., 2007). Finally, the perfusion response varies with the number of CSD events. In animals undergoing repetitive CSD, the perfusion response is attenuated, despite similar cortical depolarization (Brennan et al., 2007). These data suggest that the coupling of neural activity to blood flow during CSD functions on a physiological gradient or ‘sliding scale’ and that the conditions in which CSD occurs may thus have profound effects on the tissue outcome.
CSD is clearly an event outside the bounds of normal physiology, but the extent to which it is pathological has been debated (Nedergaard and Hansen, 1988; Gorji, 2001; Somjen, 2001, 2006). Recent evidence suggests that under certain circumstances it can be harmful. CSD has been directly recorded from humans with brain injury and stroke (Mayevsky et al., 1996; Strong et al., 2002; Dohmen et al., 2008) and in animal models, CSD around infarct is correlated with decreased perfusion and increased stroke size (Mies et al., 1993; Strong et al., 2007). It is also possible that a deleterious effect of CSD could be involved in the higher rate of stroke in patients with migraine with aura (Kruit et al., 2004; Etminan et al., 2005; Scher et al., 2009; Schürks et al., 2009).
In this study, we use optical intrinsic signal (OIS) imaging, optical spectroscopy, electro-physiology and K+ sensitive electrodes to investigate both short and longer term physiological changes during CSD in mouse. We show a significant biphasic change in direct current (DC) field potential, perfusion and haemoglobin saturation: an initial phase occurring during the CSD wave and a second prolonged phase that begins after recovery from the first phase. Though they involve similar changes—negative DC shift, arterial constriction, haemoglobin desaturation and altered neurovascular coupling—we show that these phases of CSD-associated disruption are mechanistically distinct.
Methods
Experimental preparation
Male and female C57Bl/6J mice (45 total mice, weight 22–35 g) and one male Sprague–Dawley rat (weight 350 g) were used for experiments, in accordance with the University of California, Los Angeles Animal Research Committee Guidelines. Temperature was maintained at 37.0 ± 0.7°C with a rectal temperature probe and homoeothermic blanket. Anaesthesia was induced with isoflurane (5%) in a 2:1 nitrogen:oxygen mixture and adjusted (1.0–1.6%) to maintain a respiratory rate of ∼80–120, heart rate of ∼440–530 beats/min and field potential burst-suppression with an interburst interval of 2–7 s. In selected experiments, heart rate, pulse oxygenation and respiratory rate were monitored using a pulse oximeter (three animals with Nonin 8600 V, Nonin Inc., Minneapolis, MN, USA and four animals with MouseOx, Starr Life Sciences Corp., Oakmont, PA, USA). Blood pressure was also monitored in seven experiments by cannulating the femoral artery (Blood Pressure Display Unit, Stoelting, Wood Dale, IL, USA). Arterial blood gas samples were not taken due to the duration of the experiments. In two animals each, anaesthesia was changed to a combination of low dose isoflurane (0.6–0.8%) and chlorprothixene (3.3 mg/kg); or urethane (0.75 g/kg).
Mice were placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA, USA). The skull was exposed and a rectangular section of the parietal bone (1 mm from the sagittal suture, temporal ridge, lambdoidal suture and coronal suture; Fig. 1A) was thinned to transparency. Burrholes were placed (i) 0.5 mm, antero-medial to lambda; (ii) 0.5 mm from the temporal ridge mid-way between bregma and lambda; and (iii) 0.5 mm anterior to the occipital suture. A glass recording electrode (0.5 MΩ resistance) filled with artificial CSF (in mM: 125 NaCl, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 25 NaHCO3, 11 glucose; all chemicals from Sigma-Aldrich, St Louis, MO, USA) was inserted into the first burrhole and advanced 550 μm into the cortex. In the second burrhole, either a bipolar tungsten microelectrode (80 kΩ resistance, 300 μm tip diameters, spacing 200 μm) was placed and advanced 550 μm, or a 34 gauge fused silica micropipette filled with 1 M KCl and attached to a pneumatic pico-pump (PV830, World Precision Instruments, Sarasota, FL, USA) was placed at the surface of the cortex. A silver–silver chloride ground wire was inserted into the cerebellum in the third burrhole. A thin film of silicone oil covered the skull to reduce specular artefacts and preserve bone transparency.
Figure 1.

Collection and analysis of OIS data. (A) Preparation schematic. Broad spectrum white LED light illuminated the thin skull mouse preparation. Reflected light was split between the camera and spectroscope. (B) Determination of haemoglobin content by the Beer–Lambert law. Second derivatives of measured absorbences were fit as a linear combination of empirically derived oxyhaemoglobin (HbO2) and deoxyhaemoglobin (Hb−) second-derivative absorbance standards. Conventional absorbences are on top; second derivative traces are on bottom. Haemoglobin saturation, and total haemoglobin were estimated from this fit. Since the haemoglobin spectrum displays high curvature in the region between 530–610 nm, fitting the second derivative absorbance allowed us to minimize the contribution due to background absorbers.
CSD induction
The animal rested under anaesthesia for at least 1 h after electrode placement. To ensure a constant anaesthetic environment, depth was adjusted to maintain burst-suppression activity (2–7 s interburst interval) prior to each CSD induction. For tetanic stimulation, a stimulus isolation unit (Model 2100, A-M Systems, Carlsborg, WA, USA) generated a bipolar stimulus train at 200 Hz, with a 200 μs pulse width, for 50 s. Stimulus began at 50 μA and was increased every 100 s in fixed steps to a maximum of 1.6 mA until CSD was elicited. For KCl stimulation, increasing volumes of 1 M KCl were ejected with progressively increasing pressure (4–40 psi, 0.11–1.1 μl), at 25 s intervals, until CSD was achieved. Each animal rested for 30–90 min preceding each of three CSD inductions. At the conclusion of each experiment, the animal was euthanized with anaesthetic overdose and nitrogen asphyxia.
Middle cerebral artery infarct preparation
In two animals, after thin skull preparation, the skin and muscles covering the temporal bone were dissected. The suprasylvian branch of the middle cerebral artery leading into the parietal bone window was cauterized through a burrhole and the animal was allowed to rest for 1 h before imaging.
Craniotomy preparation
Anaesthetic induction and skull exposure were performed as above. In four animals, a suspended preparation rather than a stereotaxic frame was used; the skull was affixed with cyanoacrylate glue to a circular frame and suspended from this frame in order to eliminate pressure from ear or zygoma bars. A cisternal puncture was performed and a layer of cyanoacrylate glue was placed over the exposed skull to minimize movement along suture planes. A dental cement well was built up around the craniotomy region. The craniotomy was performed with steel or diamond burrs (Meisinger USA, Centennial, CO, USA) under irrigation with chilled saline. The well was either filled with artificial CSF or 3% agarose (Sigma-Aldrich) dissolved in artificial CSF. The preparation was sealed with a coverslip. Pipettes for induction of CSD and recording electrodes were carefully advanced under the coverslip. Rat craniotomy was similar but did not require suspended preparations; for details see Brennan et al. (2007). In two animals, the contralateral hindpaw was stimulated (50 Hz square wave pulse trains through bipolar needle electrodes in the proximal and distal walking pads, pulse duration 1 ms, train duration 1 s, amplitude 0.2–0.4 mA) and evoked maps were recorded under 617 nm LED light (10 trials, baseline subtracted and divided, trials averaged).
We found that both arterial and parenchymal responses to CSD were changed by craniotomy (n = 10) in the absence of any apparent tissue or vascular injury associated with the craniotomy preparation. Arterial constriction was either significantly attenuated or absent, and the parenchymal response was characterized by much reduced hypoperfusion and more prominent hyperperfusion, quite similar to that seen in rat craniotomy preparations (Fig. 2A and B). Although CSD-associated arterial constriction was not always eliminated (4/10 experiments), when it occurred it only occurred with the first CSD (this is contrast to what is observed in the thin skull preparation, in which multiple CSD events can be elicited without loss of constriction) (Brennan et al., 2007; unpublished data). Despite the disruption of CSD-associated arterial changes, we were able to obtain evoked forepaw and hindpaw OIS maps with this preparation (Fig. 2D). While craniotomy markedly reduced the hypoperfusion associated with the CSD wave, there was no apparent effect on the second phase of sustained hypoperfusion that occurred after the CSD wave (n = 3/3 long-term craniotomy experiments) (Fig. 2C). We concluded that our craniotomy preparations were affecting CSD-associated surface vessel reactivity and thus all further experiments were conducted using thin skull preparations.
Figure 2.

Craniotomy alters vascular characteristics of CSD. (A) Images show changes in OIS reflectance under white light in mouse craniotomy preparation during passage of a CSD wave. OIS signal increases (blood volume decreases) in regions outside of the craniotomy window but decreases (blood volume increases) in region of exposed cortex. White labels are time in seconds. (B) OIS traces of (1) mouse region of interest outside craniotomy, (2) mouse region of interest inside craniotomy and rat region of interest inside craniotomy (images not shown). In the same mouse preparation, OIS traces show significant differences based on whether the cortex is exposed or not. Interestingly, the mouse craniotomy trace, though longer in duration, has a similar shape to rat craniotomy trace. Signal changes were too small outside rat craniotomy to allow measurement. Y-axis: OIS change in reflectance from baseline. (C) Contrasting arterial diameter changes in thin skull (above) and craniotomy (below) preparations in a separate experiment. Arterial constriction is attenuated in craniotomy region, but not outside it. (D) Hypoperfusion following passage of the CSD wave is preserved in craniotomy preparations even when the hypoperfusion during the wave is lost. This suggests that the mechanisms of hypoperfusion during the wave and following its passage are different (see Discussion section). (E) Despite attenuated CSD-associated arterial reactivity, evoked OIS maps are intact in craniotomy preparations. Panels show craniotomy preparation under 617 nm LED light before and during 50 Hz 1 s contralateral hind-paw stimulation (see Methods section for details). Light (617 nm) was used to optimize mapping, but maps under white light are similar. Note the difference in colour scale from the panels in A. CSD associated changes are at least two orders of magnitude larger than sensory mapping changes.
Potassium concentration sensor preparations
Electrodes were constructed according to standard protocols (Lux and Neher, 1973). Briefly, pulled capillary tubing (∼20 μm tip diameter) was silanized, the tip filled with K+ sensitive resin (Fluka Cocktail B, Sigma-Aldrich) and the remainder filled with 100 mM KCl, in contact with an Ag/AgCl electrode. The electrode and an artificial CSF-filled reference electrode were placed in a burrhole immediately adjacent to a region of thinned skull. Because of space constraints, field potentials were not recorded in these experiments. The K+ electrode was calibrated prior to each experiment, and a response of ≥54 mV/decade was verified, or the electrode was discarded.
OIS imaging
The cortex was illuminated with broad-spectrum white light (5500 K; 400–800 nm spectral range; Phillips Lumileds). Reflected light was collected with a 4× microscope lens (UPlanSApo, Olympus, Melville, NY, USA). A beamsplitter (half silvered glass, Edmund Optics, Barrington, NJ, USA) passed one half of the reflected light to the camera (902H2 Ultimate, Watec, Tsuruoka, Japan). The other half was directed to the spectroscope probe (Fig. 1A). The camera field of view was 3.2 mm × 2.4 mm with a pixel size of 5 μm. 8-bit images were acquired at 1–2 Hz, concurrent with electro-physiology, by a custom LabView Virtual Instrument (National Instruments, Austin, TX, USA).
Electro-physiology
Field potentials were acquired continuously, amplified with a band pass of 0–1 kHz (A-M Systems 3000), digitized (PCI 6251, National Instruments) and recorded simultaneously with optical data. Stability of the DC record over the course of our experiments was crucial, so experiments with >0.2 mV drift over 10 min of baseline recording were rejected.
Optical spectroscopy
A fibre optic probe (R400-7-UV/VIS, Ocean Optics; Dunedin, FL, USA) (225 μm diameter), parfocal with the OIS camera (Fig. 1A), was used to collect reflected light over a small spectral region of interest on the cortical surface. X–Y translators allowed movement of the region of interest to desired locations on the cortical surface. Regions of interest were located at distances 0.5–3.0 mm from the stimulating electrode. A spectrometer (USB400-UV-VIS, Ocean Optics) collected the probe output at 1 Hz with a 1 s integration time and 5 nm boxcar smoothing. One hundred percent oxygenated haemoglobin spectrum was obtained by bubbling a 0.5% by volume solution of mouse blood in pH 7.4 buffered saline with oxygen gas for 20 min. Deoxygenated haemoglobin spectrum was obtained by reducing the previous solution with sodium hydrosulphite (10 mg/ml; Sigma-Aldrich). Sodium hydrosulphite had no appreciable effect on absorbance within our wavelength window.
The emittance spectrograph of the incident light, π(λ), was sampled off a thin 20% Intralipid (Baxter Healthcare, Deerfield, IL, USA) emulsion spread over the surface of the imaging window. Reflectance, R(λ), was calculated as the proportion of the light reflected into the detector at a given wavelength λ. For in vivo preparations, reflectance was typically within the range of 15–75% for wavelength between 530 and 610 nm, placing our measurements within the range of linearity needed for application of the Beer–Lambert law. Designating reflected light intensity as I(λ), the absorbance was calculated as follows: A(λ) = −log R(λ) = log π(λ) − log I(λ).
Data analysis
Optical spectroscopy
Relative quantities of haemoglobin moieties were determined using a second-derivative form of the Beer–Lambert law:
| (1) |
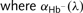 and
and 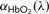 are the experimentally determined pathlength-normalized optical attenuation coefficients of unbound haemoglobin (Hb−) and oxygen-bound haemoglobin (HbO2).
are the experimentally determined pathlength-normalized optical attenuation coefficients of unbound haemoglobin (Hb−) and oxygen-bound haemoglobin (HbO2).
The second derivative method (Merrick and Pardue, 1986; Myers et al., 2005) exploits the curvature of the haemoglobin spectra to minimize contributions due to other absorbers and pathlength-independent scatter. Second derivative spectra were calculated using penalized fifth degree polynomial splines (Ripley, 2007) and fit to the model specified by equation (1) at 397 equally spaced points between 530 and 610 nm. This procedure allowed estimation of [Hb−] and [HbO2], from which haemoglobin oxygen saturation {SatO2 = [HbO2]/([Hb−] + [HbO2])}, and total haemoglobin ([Hb−] + [HbO2] = [Hbtot]) were calculated. Assuming constant haematocrit, [Hbtot] measures the total blood volume within penetrating depth of the light source. The half-width of the 95% confidence interval for SatO2 was ∼3% (parametric bootstrap).
Validation of spectroscopic measures
To determine normoxic cortical SatO2 in our thin skull preparation, we sampled spectroscopic data from 14 mice preceding burrhole and electrode placement. Mean SatO2 was 66.6% (CI: 63.9–68.5). Pre-CSD values were slightly lower—mean SatO2 was 65% (CI: 63–67). Note that tissue haemoglobin saturation is not equivalent to pulse oxygenation, which measures only arterial haemoglobin saturation; our values integrate arterial, capillary and venous haemoglobin saturation, and are in agreement with other published measures of tissue haemoglobin saturation (Benaron et al., 2004).
Superior branch middle cerebral artery infarct provided scaling for CSD-induced SatO2 changes. SatO2 dropped notably in post-infarct cortical tissue. Relative to a visible sharp border (Fig. 3A), the region closest to the cauterized vessel (>500 μm from the border) ranged in saturation from 13.8% (CI: 10.6–17.0) to 26.6% (CI: 22.5–30.8), the region surrounding the border (±500 μm) ranged from 26.6% (CI: 22.5–30.8) to 40.4% (CI: 37.7–43.1). Additional scaling was performed with nitrogen asphyxia at the end of each experiment (Fig. 3D). SatO2 reached zero upon nitrogen asphyxia in all experiments, confirming the validity of our measures. As SatO2 decreased, [Hbtot] increased sharply, revealing a physiological response to hypoxia and confirming that SatO2 and [Hbtot] (normally closely associated) were being measured independently.
Figure 3.

CSD causes haemoglobin saturation (SatO2) to drop to levels seen in ischaemic cortex. (A) Measuring SatO2 in superior branch middle cerebral artery infarct. Spectral regions of interest (circles) shown dispersed throughout middle cerebral artery infarct preparation window, with superior division middle cerebral artery cauterized just outside the imaging window (bottom centre). A sharp border is visible in OIS images (dotted line). SatO2 was lower than 27% in the region >500 μm proximal to the OIS border (closest to the cauterized middle cerebral artery; solid white circles) and was <40% within 500 μm of the border (dotted circles). Saturation was >40% (but not necessarily normal) in regions of interest >500 μm distal to the border. (B and C) Comparing SatO2 in CSD to SatO2 in middle cerebral artery infarct. Scatterplot shows that following middle cerebral artery infarct, SatO2 depends linearly upon distance from ischaemic core. Desaturation in regions closest to the infarct locus is the most severe. Boxplots of observed minimum SatO2 values in CSD are overlaid on scatterplots of middle cerebral artery infarct SatO2, to gauge the severity of the CSD related desaturation relative to middle cerebral artery infarct related desaturation. (B) Results from long-term recordings where the animal was given 90 min to recover completely between CSD inductions. Note that both the first desaturation, associated with the CSD wave (1) and the second desaturation, after passage of the wave (2), were in an ischaemic range. (C) CSD-associated desaturations for repetitive CSD separated by 30 min intervals (incomplete recovery; see Fig. 7). The saturation drop was smaller with subsequent CSDs, though still in an ischaemic range. This was not the case with complete recovery, where no difference in saturation drop between CSD episodes was seen. (D) Upon nitrogen asphyxia, SatO2 dropped to zero while [Hbtot] increased, showing that SatO2 and [Hbtot], which generally move in the same direction, were effectively distinguished by our spectroscopic techniques. (E) Magnitude of changes in the pial artery diameter (PAD), [Hbtot] and SatO2 (rectified normalized traces averaged over 50 s) for each of three time periods: pre-CSD baseline (BL), DC shift (DC) and end of experiment (END). During DC shift, arterial diameter and SatO2 (both unlikely to be affected by cell swelling—see Methods section) undergo overall changes of ∼10% whereas [Hbtot] change (likely to be affected by cell swelling) is significantly attenuated at ∼2%. This shows that relative spectroscopic measures such as SatO2 may be more reliable indicators of perfusion than absolute measures like [Hbtot] during cortical events involving significant tissue swelling. Vertical scale is in arbitrary units. (F) [Hbtot] gives insight into optical pathlength changes caused by tissue swelling. The ratio of [Hbtot] to arterial diameter gives an approximation of mean optical pathlength. This ratio peaks during the CSD wave and is slightly attenuated after passage of the wave, before returning to baseline. Change in pathlength is indicative of change in refractive properties of the tissue, including changes in cellular volume and thus light scatter. The duration of these changes agrees with invasive measures of tissue swelling (see Methods). Dotted lines show 95% confidence intervals. Vertical scale is in arbitrary units.
CSD is associated with significant cellular swelling, which can alter spectroscopic measures by changing the pathlength of light through tissue (Kohl et al., 1998). In contrast to SatO2, a ratio measure that intrinsically normalizes for pathlength changes, spectroscopic blood volume ([Hbtot]) is susceptible to these changes (Fig. 3E). Though this makes for inaccurate moiety measurements, it also provides an indicator of tissue swelling if it can be referenced to a measure unaffected by such changes. Pial arteries provide such a measure because they rest on the cortical surface. The ratio of [Hbtot] to pial artery diameter gives an approximation of mean optical pathlength which corresponds to tissue swelling. While this ratio was stable at baseline before CSD, the onset of CSD coincided with a marked increase (Fig. 3F). The duration of these changes corresponded to reported durations of tissue swelling during CSD using more invasive methods (Hansen and Olsen, 1980; Mazel et al., 2002; Takano et al., 2007), as well as different spectroscopic methods (Kohl et al., 1998).
Arterial diameter measurements
ImageJ (NIH; Bethesda, MD, USA) was used for all image analysis. To measure arterial diameter, a square 225 × 225 μm section of the OIS imaging field was chosen according to the following criteria: (i) the section contained only one artery of at least a 5 pixel diameter and (ii) the section was within 1 mm of the centre of the spectroscopic region of interest. If no section of the image satisfied these conditions, the experiment was rejected for analysis. Vessel discrimination was performed by applying the IsoData threshold algorithm (Velasco, 1980). This technique gave a binary map distinguishing the artery from background tissue and enabled a pixel count of cross-sectional arterial area, proportional to arterial diameter. Threshold vessel diameters were in close agreement with manual vessel diameter measurements (±2 pixels on average).
Time series analysis
For OIS analysis, the mean pixel value (OIS reflectance) was calculated within a square surrounding our spectroscopic region of interest. CSD can be characterized in terms of its two distinct phases: the first during the spreading CSD wave and the second following its passage. Phase duration was determined using cross-validated cubic spline-smoothed traces (Ripley, 2007). In general, the time series were characterized by a sharp deflection, followed by a much longer second deflection before final recovery. First derivatives were used to identify local extrema (minima/maxima). First phase amplitude (associated with passage of the CSD wave) was measured between the first and second extremum and duration was measured between the first and third extremum.
For measurement of long-term changes, a physiological measure was defined as having returned to baseline if its spline-interpolated mean returned to the baseline median and remained within baseline interquartile range. To account for unobserved recovery, the Kaplan–Meier estimator (Armitage et al., 2002) was used to estimate the empirical cumulative distribution function of the time elapsed until return to baseline.
To quantify recovery of oscillatory activity after CSD, wideband OIS and field potential data were high pass filtered at 0.5 Hz and rectified (Igor Pro 6, Wavemetrics, Oswego, OR, USA). Onset of EEG and OIS activity after CSD was defined as the time point after which at least 3 bursts within a 50 s period exceeded the root mean square (RMS) value of a 50 s pre-CSD baseline.
Response kinetics for OIS and long-term changes in all measures were modelled as first order linear time–invariate systems (Liptak, 2003), with time constant (τ) defined as the time taken to rise to 63% or fall to 37% of maximum.
Wavelet coherence analysis for neurovascular coupling
Neurovascular coupling was determined based on the degree by which oscillations in EEG activity corresponded with similar responses in the OIS signal. The wavelet transform coherence (Torrence and Compo, 1998) finds regions in time–frequency space where two signals are correlated. The Wavelet Coherence Toolkit for MATLAB (Grinsted et al., 2004) was used to calculate the continuous wavelet transform, cross-wavelet transform and wavelet transform coherence.
In addition to the time–frequency representation of wavelet coherence, a frequency-averaged coherence was calculated. Fisher transformed (Fisher, 1915) coherence values were weighted by the continuous wavelet transform spectral power. This approach is similar to other approaches in literature (Porges et al., 1980) and measures how well the OIS signal responds to EEG activity across all frequencies.
Statistical inference
We present bias-adjusted estimates using non-parametric bootstraps with 10 000 replicates. Confidence intervals for correlations were computed using the Fisher transformation. All confidence intervals for these estimates are given as 95% bootstrap bias-corrected and accelerated (BCa) intervals (Efron and Tibshirani, 1986) unless otherwise stated. Multiple group-wise comparisons and repeated measurement comparisons were performed within a generalized linear regression framework using generalized linear mixed effects modelling (Pinheiro and Bates, 1999). Subject variability was controlled for by treating it as a random effect (Laird and Ware, 1982). Post hoc pairwise multiple comparisons were completed using Tukey’s honestly significant differences test while holding family wise type-I error to under α = 0.05. Inferences on repeated measures over continuous time were made by using bootstrap repeated measures spline regression (Tibshirani and Knight, 1999). To determine continuous confidence bands, 1200 bootstrap resamplings of subjects were used, controlling for subject wise effects. All statistical inference was performed using the R Language and Environment for Statistical Computing (http://www.r-project.org).
Results
Physiological variables are not affected by CSD
Continuous monitoring of blood pressure, heart rate, respiration rate and pulse oximetry revealed spontaneous fluctuations within a physiological range. CSD however had no noticeable effect on these variables, showing that the haemodynamic changes observed in cortex did not have systemic correlates (Table 1).
Table 1.
CSD has no effect on heart rate, respiratory rate, pulse oximetry and blood pressure
| Before CSD | During CSD | After CSD | |
|---|---|---|---|
| Heart rate (b.p.m.) | 440–533 | 475–515 | 440–522 |
| Respiratory rate (breaths/min) | 81–114 | 86–117 | 77–117 |
| Pulse oximetry (% saturation) | 97.9–99.0 | 97.8–99.1 | 97.8–99.1 |
| Mean arterial pressure (mmHg) | 69.9–99.8 | 70.2–99.5 | 70.6–99.1 |
Biphasic neurovascular response to CSD
Following CSD induction, we observed changes in all measures—direct and alternating current field potential, cortical haemoglobin saturation, spectroscopic blood volume [Hbtot], pial arterial diameter and OIS—over two distinct phases (Fig. 4A). The first centred around the acute spreading changes of CSD and lasted at most a few minutes (Fig. 4A and Table 2). The second occurred after a brief recovery following the passage of the wave, and was many times longer (lasting at least 1 h for all measures).
Figure 4.

Biphasic tissue response to CSD. (A) Physiological measurements over a long time scale following CSD induction by tetanic stimulation in a representative experiment. Note the distinct biphasic response in each measure. Additional phases [most prominent on the pial artery diameter (PAD) trace and driven by arterial constriction and dilation] could sometimes be seen superimposed on the long second phase (arrow on pial artery diameter trace; Supplementary Fig. S1). Small arrows above OIS trace indicate thresholding steps, which cause decreases in OIS signal due to cortical activation. Similar changes can be seen on other traces. Large arrow under OIS trace indicates when CSD wave starts. Field potential is in millivolts, cortical haemoglobin saturation (SatO2) in percent. [Hbtot], pial arterial diameter and OIS reflectance are normalized to pre-CSD baseline. Inset of 20 s samples of field potential at selected time points to show: (1) baseline burst-suppression, (2) return of spontaneous activity after DC shift, (3) return of irregular bursting activity with intervals of quiescence and (4) burst-suppression at recovered baseline. Horizontal scale bar = 5 s; vertical scale bar = 0.5 mV. (B) Empirical distribution function [1-Survival(t)] plots for total CSD recovery time. Recovery time is total time (in min) elapsed from the beginning of the DC shift—including both phases of neurovascular dysfunction. To account for experiments where recovery was not observed, the Kaplan–Meier estimator was used. Dashed dark lines show boundaries for 95% confidence intervals of the empirical cumulative distribution function. Dashed grey lines intersect the expected median recovery times.
Table 2.
Amplitude and duration of CSD-associated measures
| First phase (CSD wave) | Second phase | |||
|---|---|---|---|---|
| Amplitude (95% CI) | Duration (95% CI) | Amplitude (95% CI) | Duration (lower 95% CL) | |
| DC shift | −18.5 mV (−19.7 to −16.9) | 68 s (51–90) | −6.52 mV (−7.16 to −5.88) | 73 min (62) |
| SatO2 | 53.2% (46.2–60.9) | 97 s (77–127) | 59.5% (55.5–62.8) | 67 min (55) |
| [Hbtot] | 82.0%a (79.6–84.7) | 108 s (93–123) | 66.3% (64.2–68.8) | 75 min (75) |
| Arterial diameter | 50.2% (38.9–60.3) | 84 s (70–108) | 64.2% (60.4–68.7) | 69 min (48) |
| OIS reflectance | 137% (131–144) | 108 s (100–122) | 140% (136–145) | 68 min (64) |
a[Hbtot] value is likely to be affected by CSD-associated cellular swelling (see Methods section, Fig. 3E and F).
n = 7 animals (21 CSD events). OIS mean pixel intensity maximum, pial arterial diameter minimum, haemoglobin saturation (SatO2) and spectroscopic blood volume ([Hbtot]) minimum are reported as percentage of pre-CSD baseline. Second phase durations are defined as the median times to persistent recovery from CSD induction, determined by the Kaplan–Meier Estimator, with first phase duration subtracted; lower tail 95% confidence limit is given.
To estimate temporal alignment of changes, we placed the spectroscopic region of interest directly over the field potential electrode (n = 6 animals, 15 CSDs). To the limits of our temporal resolution, constrained by spectroscopy to 1 Hz, spectroscopic, OIS and field potential changes were concurrent (Fig. 5A). Arterial constriction also appeared to coincide with CSD onset, but because arteries were sampled outside the spectral region of interest this could not be confirmed.
Figure 5.

Haemoglobin desaturation during CSD wave is due to coincident arterial constriction and cortical depolarization. (A) Field potential (FP), SatO2, [Hbtot], pial artery diameter (PAD) and OIS reflectance changes during the CSD wave. The size of the haemoglobin desaturation is likely to be due to the simultaneous occurrence of a metabolically expensive depolarization with a paradoxical reduction in blood supply. Field potential is shift from baseline in mV. OIS, [Hbtot] and arterial diameter are normalized to pre-CSD baseline. SatO2 is percent oxyhaemoglobin. Labelled time points correspond to images in panel B. (B) Raw OIS images (above) and binarized images used for measurement of arterial diameter (below): (1) immediately before CSD, (2) at maximal constriction during the CSD wave and (3) at maximal dilation. Circle: spectroscopic and intrinsic signal region of interest. Square: section of surface artery selected for analysis. (C) Raw tissue absorbance at time points indicated in (A). Experimentally derived absorbance traces for oxyhaemoglobin (solid) and deoxyhaemoglobin (dotted) are included for reference. Note that absorbance is dominated by oxyhaemoglobin at baseline and during the brief recovery after passage of the CSD wave, but is closer to the pure deoxyhaemoglobin spectrum during passage of the wave.
In order to verify that the biphasic changes we observed were not due to anaesthetic depth or type of anaesthesia, we used either an isoflurane/chlorprothixene combination (n = 2 animals) or urethane anaesthesia (n = 2 animals). Both resulted in markedly different EEG records (continuous activity rather than burst-suppression) as compared with the standard isoflurane regime. Similar biphasic changes in DC field potential, spectroscopic measurements and OIS changes were observed with isoflurane/chlorprothixene and urethane, despite different EEG activity (Supplementary Fig. S2).
Neurovascular response during the CSD wave
There was a rapid and significant desaturation of cortical haemoglobin coincident with the DC shift of the propagating CSD wave (Figs 4 and 5, Table 2). This desaturation was sizable, involving a halving of pre-CSD baseline values [pre-CSD baseline SatO2 65% (CI: 63–67); minimum SatO2 35% (CI: 30–39); n = 21 CSDs]. Putting the desaturation into physiological perspective, SatO2 during passage of the CSD wave dropped to levels seen after middle cerebral artery infarct (infarct was performed in separate experiments) (Fig. 3B and C). In 14 of 21 trials, SatO2 dropped to <40% (equivalent to levels within 500 µm of the infarct border in stroke experiments) and the time spent below this threshold ranged from 2 to 86 s (interquartile range: 7.5–37.25 s). In half of these trials (7/14), SatO2 dropped to <27% (equivalent to levels >500 µm inside the infarct border). Time spent under this threshold ranged from 1 to 39 s (interquartile range: 6.5–25 s). The similarity in magnitude to middle cerebral artery infarct suggests that the desaturation associated with the CSD wave is physiologically significant.
The desaturation was not artefactual to stimulation. There was no significant difference between desaturation in CSD elicited by KCl ejection (n = 12) or tetanic electrical stimulation (n = 31). Desaturation could be observed over the entire exposed region and we detected no relationship between depth of desaturation and distance from the CSD stimulus (r = 0.07; CI: −0.28 to +0.38). Finally, if the animal was allowed to recover completely, there was no significant difference in desaturation in the same animal across multiple CSD inductions (this changed with incomplete CSD recovery; see below).
Significant vascular and electro-physiological changes accompanied the haemoglobin desaturation (Figs 4A and 5). The amplitude of desaturation during the CSD wave was positively associated with the amplitudes of changes in haemodynamic variables: arterial constriction (r = 0.33; CI: 0.01–0.58), [Hbtot] decrease (r = 0.69; CI: 0.40–0.83) and OIS increase (r = 0.69; CI: 0.45–0.84). The amplitude of desaturation was also positively associated with the duration (r = 0.43; CI: 0.14–0.64), but not the amplitude of the DC shift (r = 0.25; CI: −0.03 to +0.49). Overall, both vascular supply (arterial diameter, [Hbtot], OIS reflectance) and metabolic demand (approximated by DC shift) are likely to contribute to haemoglobin desaturation during the CSD wave.
Neurovascular response after the CSD wave
Continuous recording allowed investigation of DC field potential both during and after an individual CSD event. After recovery from the classic CSD-associated DC shift, there was a second sustained DC shift (Fig. 4A, Table 2 and Supplementary Fig. S2). Although smaller than the initial DC shift, this second change was sizeable (∼6 mV) and significantly greater than any non-CSD changes we have encountered (n > 100 mice; data not shown).
The second phase of sustained DC shift was accompanied by a second phase of reduction in SatO2. Although the SatO2 drop with the CSD wave was significantly larger (average difference in saturation 4.0; lower CI: 0.1), SatO2 in the second phase also dropped to ischaemic levels: all 14 trials that dropped below SatO2 40% during the CSD wave also dropped below this level in the second phase. Moreover, SatO2 was depressed far longer in the second phase, with time under SatO2 40%, ranging from 15 s to 31 min (interquartile range: 40 s to 12.5 min) (Fig. 4 and Table 2).
The magnitude of the second SatO2 drop was significantly correlated with the magnitude of SatO2 drop during the CSD wave (r = 0.78; CI: 0.44–0.93). The minimum SatO2 during each of these phases was also correlated (r = 0.65; CI: 0.22–0.87). This suggests that second phase saturation changes might represent a response to the propagated wave of CSD.
Arterial diameter and DC field potential changes in general paralleled SatO2 (Fig. 4 and Table 2), with smaller but long-lasting changes in both variables compared to the CSD wave. The only exception was [Hbtot], where the decrease was greater in the second phase than the first. However, this was likely artefactual, due to optical pathlength changes caused by tissue swelling during passage of the CSD wave (Fig. 3E and F, Table 2, Methods section). There was also some variability in the arterial response. Immediately after passage of the CSD wave, arterial diameter often underwent a small constriction and dilation (Fig. 4 and Supplementary Fig. S1) before the long-lasting second constriction. However these changes did not appear to affect the local perfusion state of the tissue as OIS, SatO2 and [Hbtot] remained relatively unchanged.
The second DC shift appeared to have different kinetics from second phase haemodynamic changes (Fig. 5A). Duration of DC field potential changes was similar to arterial diameter, [Hbtot], SatO2, and OIS duration (Fig. 4A and Table 2). However minimum levels were achieved much faster (Fig. 4A): mean time constant (τ) of the second DC shift was 65 s (CI: 34–163 s; n = 9), compared with: 7 min (CI: 3–12 min), 4 min (CI: 2–8 min), 5 min (CI: 3–7 min) and 3.4 min (CI: 2.5–5.5 min), for arterial diameter, [Hbtot], SatO2 and OIS, respectively.
Neurovascular uncoupling following passage of the CSD wave
Return of EEG oscillations precedes return of OIS oscillations
The baseline EEG record (alternating current field potential; high passed at 0.5 Hz) consisted of burst-suppression activity with a mean burst frequency of 0.14 Hz (CI: 0.12–0.16 Hz; n = 9). This activity was attenuated during the first DC shift, as is classically the case for EEG during CSD (Leao, 1944). However, it returned to baseline level more quickly than other measures (Fig. 6A and B). Bursting EEG activity began an average of 99 s (CI: 50.2–186.3 s) after the end of the first DC shift, and reached equal amplitude to baseline bursts 7 min (CI: 5–13 min) after the end of the first DC shift. In contrast, oscillations in whole field OIS reflectance (which normally correlates with EEG during burst-suppression), did not return to baseline amplitude for 17 min (CI: 15–19 min) after the end of the first DC shift (Fig. 6A). During the second phase of neurovascular dysfunction, burst frequency significantly increased to an average of 0.25 Hz (CI: 0.22–0.27 Hz; n = 9) at the second DC nadir (typically 18–24 min after the end of the first DC shift). Then it gradually decreased, reaching an average of 0.18 Hz (CI: 0.16–0.21 Hz), but not regaining its original frequency by the end of the recording period.
Figure 6.

Neurovascular uncoupling after passage of the CSD wave. (A) Electro-physiological activity returns well before the haemodynamic signal and recovery begins only after the haemodynamic response is re-established. Four representative long-term experiments (coloured) and average trace (black). Upper traces show OIS reflectance, lower traces show haemoglobin saturation, beginning after the end of the first DC shift. H-shaped bars mark the time period when significant EEG activity (defined as discharges at 1–2 root mean square of baseline discharge amplitude) returns, and straight bars mark the time period when OIS oscillations (1–2 root mean square) return, for each experiment. In the period after the first DC shift, bursting activity returns well before oscillations in OIS signal. Note that the second desaturation peaks after significant EEG bursting activity returned, and recovery of saturation occurs only after significant oscillations return in the OIS (and saturation) signal. (B) OIS oscillatory response (orange; corresponding to orange trace in A) to EEG bursts (black) shows changes in neurovascular coupling. OIS activity was impaired 7 min after CSD (bursts of this amplitude and duration normally are associated with an OIS response, but are not here), showed some recovery at 24 min, and largely returned by 34 min. Vertical scale bar: 2% OIS change, 1.2 mV field potential. The small regular upward deflections during suppression periods (between bursts) are respiratory artefact. (C) EEG-power-weighted coherence across all long-term experiments (n = 8 animals, 17 CSDs). Dashed line represents the 95% confidence boundary. Please see Methods section and Supplementary Fig. S3 for how coherence was calculated.
Uncoupling of EEG and OIS activity
The beginning of the second SatO2 drop coincided with the resumption of bursting EEG activity, vasoconstriction, decreased blood volume and increased OIS (Fig. 6A). This pattern was similar to that seen during the CSD wave, with a paradoxical decrease in perfusion in the face of metabolic demand.
Because burst-suppression EEG is generally a pan-cortical phenomenon (Swank and Watson, 1949; Steriade et al., 1994), it reliably generates whole field changes in OIS signal, with decreased reflectance in response to each burst. We used the EEG/OIS interaction as an index of neurovascular coupling, quantifying it with wavelet coherence (Fig. 6C, Supplementary Fig. S3). The recovery of other measures (SatO2, [Hbtot], arterial diameter) closely followed the resumption of coherent EEG and OIS activity (Figs 4 and 6). As EEG burst amplitude had been at baseline levels for several minutes prior (Fig. 6A), the increasing coherence was probably brought about by the return of OIS response. This in turn suggested that the recovery depended on intact functional hyperaemia.
Once neurovascular coupling returned, it was quantitatively similar to baseline. We computed the time constant (τ) of OIS changes in response to spontaneous EEG activity before CSD, sub-CSD-threshold tetanic stimulation and spontaneous EEG bursts during the second phase after recovery of coherence. Differences in τ were minimal (mean pre-CSD spontaneous τ 2.1 s, pre-CSD induced τ 2.0 s, second phase τ 1.9 s; n = 10 experiments), suggesting that mechanisms of coupling were not deranged by CSD in the long term.
Uncoupling of DC and EEG activity
Comparing DC and EEG changes, there was no significant correlation between second DC shift duration or amplitude and time to recovery of EEG bursting. Likewise we found no significant correlation between latency or amplitude of the second DC nadir, and recovery of EEG bursts. Finally, we found no correlation between DC level (either at the second DC nadir or at the end of the experiment) and burst rate (n = 10 animals, 10 CSDs). These data suggest that different mechanisms may be involved in DC and alternating current electro-physiological activity in the wake of CSD.
Attenuation of arterial response when CSD is elicited before full recovery
Our results suggested a prolonged uncoupling of cortical supply and demand in the wake of a single CSD, with a prominent deficit in vascular response. CSD is often repetitive, even in humans (Dreier et al., 2009), and close spacing of CSD episodes can significantly alter subsequent events. For example, continuous repetitive CSD enhances dissociation between vascular and electro-cortical activity (Shibata et al., 1990; Brennan et al., 2007). We examined whether repetitive CSD, spaced 30 min apart, would elicit further dissociation between haemodynamic and electro-physiological measures.
In contrast to CSD elicited after full recovery, CSD elicited at 30 min intervals was associated with an attenuated haemodynamic response. We found significantly larger and faster desaturation in the first CSD wave when compared to the two subsequent waves (Figs 7 and 3C). The vascular response paralleled the haemoglobin desaturation: vessel constriction amplitude and rate of constriction were more pronounced in the first wave than in subsequent waves (Fig. 7). In contrast, there was no significant difference in the amplitude or kinetics of the first (CSD wave-associated) DC shift. Though we intentionally did not observe full recovery in these experiments, we saw the beginnings of second phase dysfunction in DC and EEG field potential, spectroscopic measures, pial artery diameter and OIS in each experiment. These data strongly suggest that distinct mechanisms mediate the response of surface vessels and underlying cortex. They also confirm that the vascular response is more susceptible to derangement than the parenchymal response.
Figure 7.

Reduced haemodynamic response but preserved electro-physiological response when CSD is elicited at short intervals. (A) Haemoglobin saturation, arterial diameter and DC field potential for three CSD waves elicited at 30 min intervals (i.e. before complete recovery from the prior event). The magnitude and rate of desaturation and vascular constriction decreases with subsequent CSDs, but the size and kinetics of the DC shift do not change. SatO2 values are shown in percent saturation. Pial artery diameter measurements are normalized to pre-CSD baseline. Note that baseline saturation is similar in all three experiments, while in Fig. 4 saturation 30 min after CSD is significantly lower than baseline. This is because the tetanic electrical stimulation used to induce CSD causes a blood volume, arterial diameter and saturation increase over the whole field. The fact that the saturation response is attenuated despite this ‘normalization’ to pre-CSD baseline values points to a specific change in vascular reactivity. (B) Tukey box and whisker plots showing trends in magnitude (top row) and rate (bottom row) of the change of haemoglobin saturation, pial artery diameter and DC field potential across each experiment. While there was a decrement in the size and speed of haemoglobin desaturation and arterial constriction across each subsequent CSD, there was no such change in DC shift measures; n = 20 animals, 60 CSDs. Tukey 95% family-wise contrasts: magnitude of desaturation was 8.44 (†CI: 4.60–12.29) greater in first CSD versus second, 4.89 (†CI: 1.03–8.74) greater in second versus third; rate of desaturation was 0.37/s (†CI: 0.02–0.72/s) greater in first versus second, 0.18/sec (†CI: 0.17–0.53/s) greater is second versus third. Arterial constriction was 13% greater in first versus second (†CI: 3.26–23.23%), 2.99% (CI: −6.99 to 12.98%) greater in second versus third; rate of constriction was 1.44%/s (†CI: 0.37–2.42%/s) greater in first CSD versus second, 0.22%/s (CI: −0.86 to 1.29%/s) greater in second versus third. †Denotes statistically significant difference in means.
Extracellular potassium is not involved in the vascular response after passage of the CSD wave
We measured extracellular K+ concentration in response to CSD (n = 5 animals) to evaluate its effect on arterial diameter (Fig. 8). We observed a mean pre-CSD baseline extracellular K+ concentration of 5.89 mM (CI: 4.82–7.38 mM) which increased to a mean of 32.1 mM (CI: 26.1–37.2 mM) at peak, coincident with the first DC shift. After passage of the CSD wave, extracellular K+ concentration returned to a baseline value of 6.22 mM (CI: 4.9–8.1 mM). With repetitive CSD, the duration of extracellular K+ concentration elevation increased (Fig. 8B). Comparing the kinetics of extracellular K+ concentration to arterial diameter, extracellular K+ concentration time to peak was longer, at an average of 67 s (CI: 56–84 s) to reach peak, compared to 13.8 s (CI: 8–35 s) for arterial constriction. Duration of extracellular K+ concentration elevation was 7 min (CI: 6–9 min), compared to 43.5 s (CI: 39.6–47.6 s) for arterial constriction. Arteries constricted monotonically with the onset of the CSD wave—there was no dilation in response to lower levels of extracellular K+ concentration elevation, and arteries ended constriction and began to re-dilate well before extracellular K+ concentration returned to baseline. Finally, there was no extracellular K+ concentration change accompanying the arterial constriction that followed the passage of the CSD wave (Fig. 8C).
Figure 8.

Extracellular K+ does not affect perfusion after passage of the CSD wave. (A) Extracellular K+ concentration [K+] increases to over 30 mM while surface vessels constrict and OIS signal increases during the CSD wave. (B) After passage of the CSD wave, extracellular K+ concentration levels returns to baseline even though other measures are disturbed, showing that extracellular K+ concentration is not involved in the second phase of CSD-associated neurovascular derangement. (C) During CSD waves with incomplete recovery, the rise time of extracellular K+ concentration increases, as does the time to recover to baseline values.
Discussion
Biphasic DC shift associated with CSD
Following recovery from the classic DC shift associated with the CSD wave, our field potential recordings reveal a significant and long-lasting second DC shift in cortical potential, whose duration was similar to the haemodynamic changes (Fig. 4 and Table 2). To our knowledge a long-term DC shift of this nature has not been reported.
The incidence, amplitude, duration and kinetics of the second DC shift were highly replicable—we observed it on every long-term recording (n = 14) and observed its beginning in every recording (n = 43). Interestingly, though its duration was similar to that of haemodynamic changes, its onset was significantly faster (Fig. 4).
Several physiological processes can cause DC shifts, among them hyper- and hypoventilation, hypo- and hypercapnoea, administration of anaesthetic agents, and disruption of the blood brain barrier (Nita et al., 2004). However, respiratory rate was unaffected by CSD (Table 1) and no changes to anaesthetic parameters were made during recordings. Moreover, hypoventilation, isoflurane anaesthesia and blood brain barrier disruption all cause positive, not negative, DC shifts and of much smaller amplitude (usually <1 mV) than we observed (Nita et al., 2004). The possibility of electrode compromise is made unlikely by the fact that the majority of DC recordings returned to baseline potential, and that further recording of DC shifts and EEG activity was unimpeded.
Large extracellular negative DC shifts are associated with neuronal depolarization, both in vitro and in vivo, during spreading depression but also during seizure activity and stroke (Marshall, 1959; Wadman et al., 1992; Somjen, 2001; Canals et al., 2005). They are also associated with glial depolarization; in fact, Sugaya et al. (1975) found that the DC shift of CSD correlated better with astrocytic than neuronal potential. Without current source density analysis or intracellular recordings, we could not conclusively determine the origin of the second DC shift. We find it reasonable to propose that our electrodes detected a cortical (neuronal, glial or vascular) depolarization during the second DC shift, given that (i) other sources of DC shift were ruled out and (ii) the same electrode, in the same location, detected negative CSD wave-associated DC shifts (known to involve cortical depolarization) both before and after the second DC shift. A complementary explanation, based on the findings of Makarova et al. (2008) is that increases in extracellular resistivity, likely to be due to tissue swelling, play a role in the second DC shift. A tissue resistivity increase correlates temporally with the known duration of tissue swelling in CSD and contributes to the large size of the first DC shift. However, though our spectroscopic measures agree with other measures of tissue swelling in the literature, they do not show pathlength changes consistent with swelling after passage of the CSD wave (there are mild but significant decreases in pathlength after the wave-associated increase) (Fig. 3F). This does not rule out either swelling or resistance changes as mechanisms for the DC shift, as our technique might lack the resolution to detect subtle changes.
Biphasic disruption of neurovascular function associated with CSD
We observed two large desaturations of cortical haemoglobin, the first during the CSD wave and the second, after a brief recovery, in its aftermath. Both desaturations were within an ischaemic range, and appeared to be due to a paradoxical combination of increased tissue demand and reduced arterial supply, opposite to what would be expected from conventional neurovascular coupling. Despite some similarities, the first and second phases of haemoglobin desaturation clearly involve distinct mechanisms, as evidenced by their different relations to the CSD wave, different spatio-temporal characteristics, different relation to extracellular K+ concentration and differential sensitivity to craniotomy.
Arterial constriction (van Harreveld and Stamm, 1952; Osada et al., 2006; Brennan et al., 2007; Chuquet et al., 2007), blood flow or volume decrease (Fabricius et al., 1995; Sonn et al., 1996; Reuter et al., 1998; Ba et al., 2002; Tomita et al., 2005) and hypoxia or haemoglobin desaturation (Marshall, 1959; Mayevsky et al., 1980; LaManna et al., 1989; Haselgrove et al., 1990; Takano et al., 2007; Piilgaard and Lauritzen, 2009) all occur in multiple species, including humans (Dreier et al., 2009), during CSD. The CSD-associated arterial constriction in mouse is larger and more consistent than in other species (Ayata et al., 2004; Brennan et al., 2007), but tissue hypoxia has also been reported with CSD even in species where there is prominent arterial dilation or hyperperfusion (Marshall, 1959; Mayevsky et al., 1980; LaManna et al., 1989; Haselgrove et al., 1990; Piilgaard and Lauritzen, 2009), probably because metabolic demand outstrips vascular supply. Moreover, the transition from a dilatory to a constrictive response to the CSD wave has been demonstrated, under conditions of metabolic challenge, in animals that normally have a predominantly dilatory response (Dreier et al., 1998; Sukhotinsky et al., 2008). Manipulations that favour this response in experimental animals—elevations in extracellular K+, depletion of nitric oxide, hypotension, ischaemia and hypoxia (Duckrow, 1993; Dreier et al., 1998; Shin et al., 2006; Sukhotinsky et al., 2008)—are all consistent with either increases in metabolic demand or reduction in vascular supply of metabolic substrates. In this regard, the mouse may have a decreased metabolic/perfusion reserve compared to larger animals. However it should be emphasized that neurovascular coupling is likely to function on a gradient in all species and can actively contribute to pathology when deranged. Moreover, all species studied thus far can undergo constrictive neurovascular coupling during the CSD wave.
While the response to the CSD wave in mouse may or may not be different from other species, long-lasting hypoperfusion following the wave is a consistent finding in all model systems (Lauritzen et al., 1982; Busija et al., 2008) and in humans after CSD-like events (Olesen et al., 1981; Lauritzen et al., 1983, 1984). We observed a sustained vasoconstriction and hypoperfusion which began after a brief recovery from the initial wave of CSD. The vascular changes occur in the face of a second sustained DC shift and recovery of spontaneous electro-cortical activity, and are associated with a second nadir in haemoglobin saturation, consistent with a prolonged derangement of neurovascular coupling. As with the CSD wave itself, the result is paradoxically reduced perfusion in the setting of increased parenchymal demand.
Our finding of persistent haemoglobin desaturation after the CSD wave complements the recent work of Piilgaard and Lauritzen (2009), who report a long-lasting decrease in pO2, accompanied reduced blood flow and increased cerebral metabolic rate of O2, following the wave. That the authors found these changes in rat, a species with a different response to the acute CSD wave than mouse, is strong evidence that significant, metabolically relevant changes in perfusion response following CSD are generalized. Moreover, their finding of increased cerebral metabolic rate of O2 after passage of the CSD wave is consistent with an increase in cortical metabolic demand that is not met by vascular supply.
We also found that arterial response could vary in the face of a consistent electro-physiological response. When repetitive CSD was elicited at intervals shorter than required for complete recovery, the rate and amplitude of arterial constriction during the CSD wave decreased, but no such decrement occurred in DC shift parameters (Fig. 7). This confirms that the vascular and parenchymal compartments are separable in CSD (Brennan et al., 2007) and further suggests that the ‘weak link’ in CSD-associated neurovascular uncoupling lies in the vascular response. Given the smaller size of desaturation with equally sized DC shifts, it also shows conclusively that arterial constriction contributes to CSD-associated desaturation in mouse.
Another indication of the potential variability of the vascular response was the finding that apparently atraumatic craniotomies resulted in substantial changes to the vascular and parenchymal CSD signal in mouse (Fig. 2). Our findings are consistent with those of Ayata and Moskowitz (2006), who reported arterial dilation rather than constriction in a closed craniotomy preparation of N-methyl-D-aspartic acid-induced mouse CSD. While we cannot rule out the possibility of subtle trauma or unrecorded CSD in our preparations, our results suggest that alterations in the perivascular environment caused by craniotomy can result in markedly different vascular responses. The finding of a ‘normal’ mouse neurovascular response outside the craniotomy in the same experiments that showed altered craniotomy response (Fig. 2A and B), provides strong evidence that the changes we observed were not due to generalized dysfunction like that generated by unrecorded CSD.
The craniotomy preparation also provides important evidence that the first and second neurovascular derangements are mechanistically distinct. Long-term craniotomy recordings showed intact hypoperfusion after passage of the CSD wave, even when the perfusion response associated with the wave itself was reversed (Fig. 2C). The finding that evoked OIS maps are apparently normal in craniotomy (Fig. 2D) suggests that the parenchymal dilation response is intact, at least to physiological stimuli. This difference in parenchymal and surface vessel response may be due to the different innervation of pial and penetrating arteries (Cipolla et al., 2004).
Finally, our craniotomy findings may resolve a discrepancy in the literature regarding the vascular response to CSD. Takano et al. (2007) measured subsurface arterial response using two-photon microscopy, and unexpectedly found that parenchymal arterioles dilated mildly rather than constricted during the CSD wave in mouse. We also saw evidence of parenchymal vessel dilation during the CSD wave in craniotomy (decrease in OIS signal consistent with increased blood volume). It is possible that the craniotomy necessary to perform two-photon imaging caused a change in the parenchymal arterial response to the CSD wave, such that a normally constrictive event became dilatory. Taken together with our finding of apparently normal dilation during cortical mapping, these data suggest an impairment of parenchymal as well as surface artery constriction during the CSD wave in craniotomy. Future studies of vascular reactivity in different species would benefit from a comparison of thin skull versus craniotomy methods, not only to determine whether differences occur, but also to investigate possible mechanisms (mechanical, vasoactive mediators) contributing to the difference.
Potassium concentration and mechanisms of arterial behaviour
Extracellular K+ levels closely follow the initial DC shift of CSD (Brinley et al., 1960; Vyskocil et al., 1972; Hansen et al., 1980). K+ is also highly vasoactive (Golding et al., 2000; Horiuchi et al., 2002), causing constriction via L-type voltage-gated calcium channels (Nelson et al., 1990) and dilation through inwardly rectifying K+ channels (Knot et al., 1996), in addition to modulation through indirect mechanisms (Windmuller et al., 2005). It could thus account for the large changes in arterial diameter both during and following CSD, perhaps by overwhelming normal mechanisms of neurovascular coupling (Ayata et al., 2004). We found that cortical surface arteries underwent a monophasic constriction and return to baseline during the period of extracellular K+ concentration elevation, which corresponded with the CSD wave. Thus our data are consistent with increasing extracellular K+ concentration as a possible driver of arterial constriction. However, the recovery from the initial constriction appears to be mediated by other mechanisms. Moreover, in contrast to the CSD wave, we were able to rule out any role of extracellular K+ concentration elevations in the sustained arterial changes in the second phase, as extracellular K+ concentration had returned to normal levels before the second desaturation began (Fig. 8). This highlights the fact that the two phases of vascular change with CSD have different mechanisms of action.
Spontaneous vasomotion, OIS responses to evoked neuronal activity, and arterial responses to pH, extracellular K+ concentration, adenosine, papaverine, bradykinin and hypercapnoea are all attenuated for at least an hour after CSD in different species (Wahl et al., 1987; Piper et al., 1991; Lacombe et al., 1992; Florence et al., 1994; Seitz et al., 2004; Guiou et al., 2005; Scheckenbach et al., 2006). The depletion or exhaustion of a vasodilator is one possible mechanism for this attenuated response. Nitric oxide depletion occurs in the wake of the CSD wave and decreased cerebrovascular reactivity after the wave can be rescued by treatment with nitric oxide donors or cyclic GMP agonists (Fabricius et al., 1995; Scheckenbach et al., 2006). It is also possible that the increased release of a vasoconstrictor contributes to the disruption. Shibata et al. (1992) found that long-lasting arterial constriction after CSD in rabbits was inhibited by indomethacin, suggesting a role for prostanoids in this constriction.
We found that there was delayed clearance of extracellular K+ on repetitive CSD with incomplete recovery (Fig. 8C). This may be due to the ischaemic levels of hypoperfusion we encountered: Hansen et al. (1981) observed reduced extracellular K+ clearance in post-ischaemic rat brain. Impaired astrocyte function may also play a role: astrocytic aquaporin mutations (Padmawar et al., 2005) and selective poisoning of astrocytes (Lian and Stringer, 2004) reduce extracellular K+ clearance. As astrocytic K+ transport depends on intact membrane potential (Kucheryavykh et al., 2007), the second DC shift we observed might be a source of astrocytic dysfunction.
Relation of second DC shift to haemodynamic and electro-physiological measures
A long-lasting cortical depolarization could have effects on vascular reactivity: cerebral vessels can respond directly to depolarization, as well as to mediators released (or depleted) during depolarization (Bai et al., 2004). We ruled out a role for K+, but to our knowledge the effect of persistent DC shift on other mechanisms has not been investigated.
We hypothesized that the EEG and DC traces would bear some relation to each other, with a presumed depolarization favouring higher frequency EEG bursts and a longer time to burst recovery. However, we found no significant correlations in these variables, suggesting that the DC and EEG portions of the field potential might not be mechanistically linked in the wake of CSD. This could be due to a predominant glial, rather than neuronal, contribution to the DC shift (Sugaya et al., 1975). Glial swelling might also contribute to cortical resistance changes (Makarova et al., 2008), which could result in DC changes uncorrelated with EEG. Further investigation of glial roles in the post-CSD state is warranted.
Whatever the mechanism of the second DC shift, its occurrence coincided with changes in neuronal activity. Early in the course of the second depolarization, EEG activity was disorganized and often paroxysmal in appearance (Fig. 4 and Supplementary Fig. S2). We could not conclusively demonstrate spike-wave discharges or other clearly epileptic phenomena, but the appearance of the traces was suggestive of increased excitability. Seizure-like activity has been noted in the setting of CSD since the time of Leao (1944) and van Harreveld (1953), and recently Fabricius et al. (2008) have reported association of seizures and CSD in injured human brain, so an increase in cortical excitability after passage of the CSD wave is tenable. The idea of an increase in excitability following CSD is supported by the work of Krüger et al. (1996) and Piilgaard and Lauritzen (2009), who showed a reduction in presumed GABAergic activity in the wake of CSD. Even after other measures recovered, EEG activity was characterized by burst-suppression at a higher frequency than prior to CSD (Fig. 4), again suggestive of a possible increase in excitability.
Physiological and clinical significance
Our results have potential clinical significance at multiple levels. We show that there are two separate phases of neurovascular dysfunction associated with CSD that involve distinct mechanisms. Consideration of the physiological consequences of CSD must include not only the initial propagated wave, but also the sustained changes that occur after the wave. Given both the first and second DC shift and desaturation, our results support studies in humans indicating that CSD may not be a benign phenomenon, especially if the cortex is already compromised. The sustained derangement of neurovascular coupling following CSD may represent a distinct therapeutic target in migraine, stroke, subarachnoid haemorrhage, brain trauma and other disorders where CSD occurs.
Funding
The National Institutes of Health (T32 GM008185 to J.C.C., R01 MH52083 to A.W.T., K08 NS059072 to K.C.B.); Larry L. Hillblom Foundation (to A.C.C. and K.C.B.).
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations
- CSD
cortical spreading depression
- DC
direct current
- [Hbtot]
total haemoglobin
- K+
potassium ion
- OIS
optical intrinsic signal
- SatO2
haemoglobin oxygen saturation
References
- Armitage P, Berry G, Matthews J. Blackwell Science Malden, MA; 2002. Statistical methods in medical research. [Google Scholar]
- Ayata C, Shin H, Salomone S, Ozdemir-Gursoy Y, Boas D, Dunn AK, et al. Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab. 2004;24:1172–82. doi: 10.1097/01.WCB.0000137057.92786.F3. [DOI] [PubMed] [Google Scholar]
- Ayata C, Moskowitz MA. Cortical spreading depression confounds concentration-dependent pial arteriolar dilation during N-methyl-D-aspartate superfusion. Am J Physiol Heart Circ Physiol. 2006;290:H1837–41. doi: 10.1152/ajpheart.01102.2005. [DOI] [PubMed] [Google Scholar]
- Ba A, Guiou M, Pouratian N, Muthialu A, Rex D, Cannestra A, et al. Multiwavelength optical intrinsic signal imaging of cortical spreading depression. J Neurophysiol. 2002;88:2726–35. doi: 10.1152/jn.00729.2001. [DOI] [PubMed] [Google Scholar]
- Bai N, Moien-Afshari F, Washio H, Min A, Laher I. Pharmacology of the mouse-isolated cerebral artery. Vasc Pharmacol. 2004;41:97–106. doi: 10.1016/j.vph.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Benaron DA, Parachikov IH, Friedland S, Soetikno R, Brock-Utne J, van der Starre PJA, et al. Continuous, noninvasive, and localized microvascular tissue oximetry using visible light spectroscopy. Anesthesiology. 2004;100:1469–75. doi: 10.1097/00000542-200406000-00019. [DOI] [PubMed] [Google Scholar]
- Brennan KC, Beltrán-Parrazal L, López-Valdés HE, Theriot J, Toga AW, Charles AC. Distinct vascular conduction with cortical spreading depression. J Neurophysiol. 2007;97:4143–51. doi: 10.1152/jn.00028.2007. [DOI] [PubMed] [Google Scholar]
- Brinley FJ, Kandel ER, Marshall WH. Potassium outflux from rabbit cortex during spreading depression. J Neurophysiol. 1960;23:246–56. doi: 10.1152/jn.1960.23.3.246. [DOI] [PubMed] [Google Scholar]
- Busija DW, Bari F, Domoki F, Horiguchi T, Shimizu K. Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol. 2008;86:379–95. doi: 10.1016/j.pneurobio.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals S, Makarova I, López-Aguado L, Largo C, Ibarz JM, Herreras O. Longitudinal depolarization gradients along the somatodendritic axis of CA1 pyramidal cells: a novel feature of spreading depression. J Neurophysiol. 2005;94:943–51. doi: 10.1152/jn.01145.2004. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Hollender L, Nimchinsky E. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci. 2007;27:4036–44. doi: 10.1523/JNEUROSCI.0721-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ, Li R, Vitullo L. Perivascular innervation of penetrating brain parenchymal arterioles. J Cardiovasc Pharmacol. 2004;44:1–8. doi: 10.1097/00005344-200407000-00001. [DOI] [PubMed] [Google Scholar]
- Czéh G, Aitken PG, Somjen GG. Membrane currents in CA1 pyramidal cells during spreading depression (SD) and SD-like hypoxic depolarization. Brain Res. 1993;632:195–208. doi: 10.1016/0006-8993(93)91154-k. [DOI] [PubMed] [Google Scholar]
- Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus R, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63:720–8. doi: 10.1002/ana.21390. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132:1866–81. doi: 10.1093/brain/awp102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier J, Korner K, Ebert N, Gorner A, Rubin I, Back T, et al. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab. 1998;18:978–90. doi: 10.1097/00004647-199809000-00007. [DOI] [PubMed] [Google Scholar]
- Duckrow R. A brief hypoperfusion precedes spreading depression if nitric oxide synthesis is inhibited. Brain Res. 1993;618:190–5. doi: 10.1016/0006-8993(93)91265-t. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani R. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci. 1986;1:54–77. [Google Scholar]
- Etminan M, Takkouche B, Isorna F, Samii A. Risk of ischaemic stroke in people with migraine: systematic review and meta-analysis of observational studies. Br Med J. 2005;330:63–6. doi: 10.1136/bmj.38302.504063.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabricius M, Akgoren N, Lauritzen M. Arginine-nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. Am J Physiol Heart Circ Physiol. 1995;269:23–9. doi: 10.1152/ajpheart.1995.269.1.H23. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, Willumsen L, Dreier J, Bhatia R, Boutelle M, et al. Association of seizures with cortical spreading depression and peri-infarct depolarisations in the acutely injured human brain. Clin Neurophysiol. 2008;119:1973–84. doi: 10.1016/j.clinph.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. Frequency distribution of the values of the correlation coefficients in samples from and indefinitely large population. Biometrika. 1915;10:507–18. [Google Scholar]
- Florence G, Bonvento G, Charbonne R, Seylaz J. Spreading depression reversibly impairs autoregulation of cortical blood flow. Am J Physiol Regul Integr Comp Physiol. 1994;266:R1136–40. doi: 10.1152/ajpregu.1994.266.4.R1136. [DOI] [PubMed] [Google Scholar]
- Golding EM, Steenberg ML, Johnson TD, Bryan RM. The effects of potassium on the rat middle cerebral artery. Brain Res. 2000;880:159–66. doi: 10.1016/s0006-8993(00)02793-1. [DOI] [PubMed] [Google Scholar]
- Gorji A. Spreading depression: a review of the clinical relevance. Brain Res Rev. 2001;38:33–60. doi: 10.1016/s0165-0173(01)00081-9. [DOI] [PubMed] [Google Scholar]
- Grinsted A, Moore JC, Jevrejeva S. Application of the cross wavelet transform and wavelet coherence to geophysical time series. Nonlinear Proc Geophys. 2004;11:561–6. [Google Scholar]
- Guiou M, Sheth S, Nemoto M, Walker M, Pouratian N, Ba A, et al. Cortical spreading depression produces long-term disruption of activity-related changes in cerebral blood volume and neurovascular coupling. J Biomed Opt. 2005;10:11004. doi: 10.1117/1.1852556. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA. 2001;98:4687–92. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ, Gjedde A, Siemkowicz E. Extracellular potassium and blood flow in the post-ischemic rat brain. Pflügers Arch. 1981;389:1–7. doi: 10.1007/BF00587921. [DOI] [PubMed] [Google Scholar]
- Hansen A, Olsen C. Brain extracellular space during spreading depression and ischemia. Acta Physiol Scand. 1980;108:355–65. doi: 10.1111/j.1748-1716.1980.tb06544.x. [DOI] [PubMed] [Google Scholar]
- Hansen A, Quistorff B, Gjedde A. Relationship between local changes in cortical blood flow and extracellular K+ during spreading depression. Acta Physiol Scand. 1980;109:1–6. doi: 10.1111/j.1748-1716.1980.tb06557.x. [DOI] [PubMed] [Google Scholar]
- Haselgrove JC, Bashford CL, Barlow CH, Quistorff B, Chance B, Mayevsky A. Time resolved 3-dimensional recording of redox ratio during spreading depression in gerbil brain. Brain Res. 1990;506:109–14. doi: 10.1016/0006-8993(90)91205-u. [DOI] [PubMed] [Google Scholar]
- Horiuchi T, Dietrich H, Hongo K, Dacey R. Mechanism of extracellular K+-induced local and conducted responses in cerebral penetrating arterioles. Stroke. 2002;33:2692–9. doi: 10.1161/01.str.0000034791.52151.6b. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492 (Pt 2):419–30. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl M, Lindauer U, Dirnagl U, Villringer A. Separation of changes in light scattering and chromophore concentrations during cortical spreading depression in rats. Opt Lett. 1998;23:555–7. doi: 10.1364/ol.23.000555. [DOI] [PubMed] [Google Scholar]
- Kruit M, van Buchem M, Hofman P, Bakkers J, Terwindt G, Ferrari M, et al. Migraine as a risk factor for subclinical brain lesions. JAMA. 2004;291:427–34. doi: 10.1001/jama.291.4.427. [DOI] [PubMed] [Google Scholar]
- Krüger H, Luhmann HJ, Heinemann U. Repetitive spreading depression causes selective suppression of GABAergic function. Neuroreport. 1996;7:2733–6. doi: 10.1097/00001756-199611040-00065. [DOI] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, et al. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–81. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- Lacombe P, Sercombe R, Correze J, Springhetti V, Seylaz J. Spreading depression induces prolonged reduction of cortical blood flow reactivity in the rat. Exp Neurol. 1992;117:278–86. doi: 10.1016/0014-4886(92)90137-f. [DOI] [PubMed] [Google Scholar]
- Laird N, Ware J. Random-effects models for longitudinal data. Biometrics. 1982;38:963–74. [PubMed] [Google Scholar]
- LaManna J, McCracken K, Patil M, Prohaska O. Stimulus-activated changes in brain tissue temperature in the anesthetized rat. Metab Brain Dis. 1989;4:225–37. doi: 10.1007/BF00999769. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Long-lasting reduction of cortical blood flow of the brain after spreading depression with preserved autoregulation and impaired CO2 response. J Cereb Blood Flow Metab. 1984;4:546–54. doi: 10.1038/jcbfm.1984.79. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Balslev Jorgensen M, Diemer N, Gjedde A, Hansen A. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann Neurol. 1982;12:469–74. doi: 10.1002/ana.410120510. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Olsen T, Lassen N, Paulson O. Changes in regional cerebral blood flow during the course of classic migraine attacks. Ann Neurol. 1983;13:633–41. doi: 10.1002/ana.410130609. [DOI] [PubMed] [Google Scholar]
- Leao A. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–90. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Lian X, Stringer J. Astrocytes contribute to regulation of extracellular calcium and potassium in the rat cerebral cortex during spreading depression. Brain Res. 2004;1012:177–84. doi: 10.1016/j.brainres.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Liptak BG. CRC Press: Boca Raton, FL; 2003. Instrument Engineers' Handbook: Process control and optimization. [Google Scholar]
- Lux HD, Neher E. The equilibration time course of (K +) 0 in cat cortex. Exp Brain Res. 1973;17:190–205. doi: 10.1007/BF00235028. [DOI] [PubMed] [Google Scholar]
- Makarova J, Gómez-Galán M, Herreras O. Variations in tissue resistivity and in the extension of activated neuron domains shape the voltage signal during spreading depression in the CA1 in vivo. Eur J Neurosci. 2008;27:444–56. doi: 10.1111/j.1460-9568.2008.06022.x. [DOI] [PubMed] [Google Scholar]
- Marshall W. Spreading cortical depression of Leao. Physiol Rev. 1959;39:239–79. doi: 10.1152/physrev.1959.39.2.239. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Lebourdais S, Chance B. The interrelation between brain PO2 and NADH oxidation-reduction state in the Gerbil. J Neurosci Res. 1980;5:173–82. doi: 10.1002/jnr.490050302. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Doron A, Manor T, Meilin S, Nili Zarchin, Ouaknine G. Cortical spreading depression recorded from the human brain using a multiparametric monitoring system. Brain Res. 1996;740:268–74. doi: 10.1016/s0006-8993(96)00874-8. [DOI] [PubMed] [Google Scholar]
- Mazel T, Richter F, Vargova L, Sykova E. Changes in extracellular space volume and geometry induced by cortical spreading depression in immature and adult rats. Physiol Res. 2002;51:85–94. [PubMed] [Google Scholar]
- Merrick MF, Pardue HL. Evaluation of absorption and first- and second-derivative spectra for simultaneous quantification of bilirubin and hemoglobin. Clin Chem. 1986;32:598–602. [PubMed] [Google Scholar]
- Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport. 1993;4:709–11. doi: 10.1097/00001756-199306000-00027. [DOI] [PubMed] [Google Scholar]
- Myers D, Anderson L, Seifert R, Ortner J, Cooper C, Beilman G, et al. Noninvasive method for measuring local hemoglobin oxygen saturation in tissue using wide gap second derivative near-infrared spectroscopy. J Biomed Opt. 2005;10:034017. doi: 10.1117/1.1925250. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain. Brain Res. 1988;449:395–8. doi: 10.1016/0006-8993(88)91062-1. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Nita D, Vanhatalo S, Lafortune F, Voipio J, Kaila K, Amzica F. Nonneuronal origin of CO2-related DC EEG shifts: an in vivo study in the cat. J Neurophysiol. 2004;92:1011–22. doi: 10.1152/jn.00110.2004. [DOI] [PubMed] [Google Scholar]
- Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rcbf in classic migraine. Ann Neurol. 1981;9:344–52. doi: 10.1002/ana.410090406. [DOI] [PubMed] [Google Scholar]
- Osada T, Tomita M, Suzuki N. Spindle-shaped constriction and propagated dilation of arterioles during cortical spreading depression. Neuroreport. 2006;17:1365. doi: 10.1097/01.wnr.0000227992.00175.fa. [DOI] [PubMed] [Google Scholar]
- Padmawar P, Yao X, Bloch O, Manley G, Verkman A. K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat Meth. 2005;2:825–7. doi: 10.1038/nmeth801. [DOI] [PubMed] [Google Scholar]
- Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab. 2009;29:1517–27. doi: 10.1038/jcbfm.2009.73. [DOI] [PubMed] [Google Scholar]
- Pinheiro JC, Bates DM. Madison: Bell Labs, Lucent Technologies and University of Wisconsin; 1999. Lme and nlme: mixed-effects methods and classes for S and S-plus. [Google Scholar]
- Piper RD, Lambert GA, Duckworth JW. Cortical blood flow changes during spreading depression in cats. Am J Physiol Heart Circ Physiol. 1991;261:96–102. doi: 10.1152/ajpheart.1991.261.1.H96. [DOI] [PubMed] [Google Scholar]
- Porges S, Bohrer R, Cheung M, Drasgow F, McCabe P, Keren G. New time-series statistic for detecting rhythmic co-occurrence in the frequency domain: the weighted coherence and its application to psychophysiological research. Psychol Bull. 1980;88:580–87. [PubMed] [Google Scholar]
- Reuter U, Weber J, Gold L, Arnold G, Wolf T, Dreier J, et al. Perivascular nerves contribute to cortical spreading depression-associated hyperemia in rats. Am J Physiol Heart Circ Physiol. 1998;274:H1979–87. doi: 10.1152/ajpheart.1998.274.6.H1979. [DOI] [PubMed] [Google Scholar]
- Ripley B. pspline: penalized smoothing splines. R package version 1.0–12. 2007 [Google Scholar]
- Scheckenbach KEL, Dreier JP, Dirnagl U, Lindauer U. Impaired cerebrovascular reactivity after cortical spreading depression in rats: restoration by nitric oxide or cGMP. Exp Neurol. 2006;202:449–55. doi: 10.1016/j.expneurol.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Scher A, Gudmundsson L, Sigurdsson S, Ghambaryan A, Aspelund T, Eiriksdottir G, et al. Migraine headache in middle age and late-life brain infarcts. JAMA. 2009;301:2563. doi: 10.1001/jama.2009.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürks M, Rist PM, Bigal ME, Buring JE, Lipton RB, Kurth T. Migraine and cardiovascular disease: systematic review and meta-analysis. Br Med J. 2009;339:b3914. doi: 10.1136/bmj.b3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz I, Dirnagl U, Lindauer U. Impaired vascular reactivity of isolated rat middle cerebral artery after cortical spreading depression in vivo. J Cereb Blood Flow Metab. 2004;24:526–30. doi: 10.1097/00004647-200405000-00006. [DOI] [PubMed] [Google Scholar]
- Shibata M, Leffler CW, Busija DW. Cerebral hemodynamics during cortical spreading depression in rabbits. Brain Res. 1990;530:267. doi: 10.1016/0006-8993(90)91294-q. [DOI] [PubMed] [Google Scholar]
- Shibata M, Leffler C, Busija D. Pial arteriolar constriction following cortical spreading depression is mediated by prostanoids. Brain Res. 1992;572:190–7. doi: 10.1016/0006-8993(92)90469-p. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb. Blood Flow Metab. 2006;26:1018–30. doi: 10.1038/sj.jcbfm.9600252. [DOI] [PubMed] [Google Scholar]
- Somjen G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–96. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Is spreading depression bad for you? Focus on “repetitive normoxic spreading depression-like events result in cell damage in juvenile hippocampal slice cultures”. J Neurophysiol. 2006;95:16–7. doi: 10.1152/jn.01008.2005. [DOI] [PubMed] [Google Scholar]
- Sonn J, Dekel N, Kadusi R, Mayevsky A. Responses to cortical spreading depression of the normoxic and ischemic brain. Israel J Med Sci. 1996;32:34. [Google Scholar]
- Steriade M, Amzica F, Contreras D. Cortical and thalamic cellular correlates of electroencephalographic burst-suppression. Electroencephalogr Clin Neurophysiol. 1994;90:1–16. doi: 10.1016/0013-4694(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Anderson PJ, Watts HR, Virley DJ, Lloyd A, Irving EA, et al. Peri-infarct depolarizations lead to loss of perfusion in ischaemic gyrencephalic cerebral cortex. Brain. 2007;130:995. doi: 10.1093/brain/awl392. [DOI] [PubMed] [Google Scholar]
- Strong A, Fabricius M, Boutelle M, Hibbins S, Hopwood S, Jones R, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738–43. doi: 10.1161/01.str.0000043073.69602.09. [DOI] [PubMed] [Google Scholar]
- Sugaya E, Takato M, Noda Y. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J Neurophysiol. 1975;38:822–41. doi: 10.1152/jn.1975.38.4.822. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Dilekoz E, Moskowitz M, Ayata C. Hypoxia and hypotension transform the blood flow response to cortical spreading depression from hyperemia into hypoperfusion in the rat. J Cereb Blood Flow Metab. 2008;28:1369. doi: 10.1038/jcbfm.2008.35. [DOI] [PubMed] [Google Scholar]
- Swank R, Watson C. Effects of barbiturates and ether on spontaneous electrical activity of dog brain. J Neurophysiol. 1949;12:137–60. doi: 10.1152/jn.1949.12.2.137. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian G, Peng W, Lou N, Lovatt D, Hansen A, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- Tibshirani R, Knight K. Model search by bootstrap “Bumping”. J Comput Graph Stat. 1999:671–86. [Google Scholar]
- Tomita M, Schiszler I, Tomita Y, Tanahashi N, Takeda H, Osada T, et al. Initial oligemia with capillary flow stop followed by hyperemia during K+-induced cortical spreading depression in rats. J Cereb Blood Flow Metab. 2005;25:742–7. doi: 10.1038/sj.jcbfm.9600074. [DOI] [PubMed] [Google Scholar]
- Torrence C, Compo G. A practical guide to wavelet analysis. Bull Am Meteorol Soc. 1998;79:61–78. [Google Scholar]
- Velasco RD. Thresholding using the ISODATA clustering algorithm. IEEE Trans Sys Man Cybernet. 1980;10:771. [Google Scholar]
- van Harreveld A, Stamm JS. Cerebral asphyxiation and spreading cortical depression. Am J Physiol. 1953;173:171–5. doi: 10.1152/ajplegacy.1953.173.1.171. [DOI] [PubMed] [Google Scholar]
- van Harreveld A, Stamm J. Vascular concomitants of cortical spreading depression. J Neurophysiol. 1952;16:487–96. doi: 10.1152/jn.1952.15.6.487. [DOI] [PubMed] [Google Scholar]
- Vyskocil F, Kritz N, Bures J. Potassium-selective microelectrodes used for measuring the extracellular brain potassium during spreading depression and anoxic depolarization in rats. Brain Res. 1972;39:255–9. doi: 10.1016/0006-8993(72)90802-5. [DOI] [PubMed] [Google Scholar]
- Wadman WJ, Juta AJ, Kamphuis W, Somjen GG. Current source density of sustained potential shifts associated with electrographic seizures and with spreading depression in rat hippocampus. Brain Res. 1992;570:85–91. doi: 10.1016/0006-8993(92)90567-s. [DOI] [PubMed] [Google Scholar]
- Wahl M, Lauritzen M, Schilling L. Change of cerebrovascular reactivity after cortical spreading depression in cats and rats. Brain Res. 1987;411:72–80. doi: 10.1016/0006-8993(87)90682-2. [DOI] [PubMed] [Google Scholar]
- Windmuller O, Lindauer U, Foddis M, Einhaupl K, Dirnagl U, Heinemann U, et al. Ion changes in spreading ischaemia induce rat middle cerebral artery constriction in the absence of NO. Brain. 2005;128:2042. doi: 10.1093/brain/awh545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.