

Abstract
Gedunin (1), a tetranortriterpenoid isolated from the Indian neem tree (Azadirachta indica), was recently shown to manifest anticancer activity via inhibition of the 90 kDa heat shock protein (Hsp90) folding machinery and to induce the degradation of Hsp90-dependent client proteins similar to other Hsp90 inhibitors. The mechanism of action by which gedunin induces client protein degradation remains undetermined, however, prior studies have demonstrated that it does not bind competitively versus ATP. In an effort to further probe the mechanism of action, 19 semisynthetic derivatives of gedunin were prepared and their antiproliferative activity against MCF-7 and SkBr3 breast cancer cells determined. Although no compound was found to exhibit antiproliferative activity more effective than the natural product, functionalities critical for antiproliferative activity have been identified.
Introduction
The development of resistance to chemotherapeutic agents is a major obstacle to the successful treatment of cancer.1–4 Whether because of the overexpression of efflux pumps, protein mutagenesis, or activation of alternative cell signaling pathways, resistance results from the genetic plasticity/instability of cancer cells.5,6 Cancer therapies often target a single oncogenic protein, and it is the same genetic plasticity/instability that allows cancer to select for the most adaptable mutation in order to circumvent drug effects. This type of resistance may be avoided, however, if multiple cancer signaling pathways are targeted simultaneously with a single therapeutic agent. The family of 90 kDa heat shock proteins (Hsp90a) has emerged as a drug target that mediates multiple signaling nodes as a novel strategy to combat cancer.5–7
Hsp90 is a molecular chaperone that is responsible for the conformational maturation of many nascent polypeptides, as well as denatured proteins, into their biologically active forms. To date, over 100 client proteins have been identified (see the Web page maintained by D. Picard, http://www.picard.ch/DP/downloads/Hsp90interactors.pdf) many of which are essential to oncogenic development, cancer cell survival, and the development of drug resistance.5–11 As a target for chemotherapy, Hsp90 is well validated and several Hsp90 targeting drugs are currently in clinical trials. There are, however, drawbacks associated with the current drugs that target Hsp90. For drugs based on the structure of geldanamycin (GDA), e.g., 17-allylamino-17-demethoxygeldanamycin (17-AAG) and 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17- DMAG), the major concern is related to the redox-active quinone moiety. Quinones, which are good Michael acceptors and catalysts for free-radical generation, can complicate treatment by exhibiting toxicity unrelated to Hsp90 inhibition.12 Other Hsp90 inhibitors, e.g., novobiocin, coumarin analogues, and purine analogues, target either the N- or C-terminal nucleotide binding domains, but little toxicological data have been reported to date. Since Hsp90 is known to interact with various cochaperones during the protein folding cycle, there may be additional mechanisms through which this chaperone can be modulated and thus provide an alternative to these well-established inhibitors.
Gedunin (1, Figure 1), a tetranortriterpenoid isolated from the Indian neem tree (Azadirachta indica), has demonstrated the ability to exhibit antimalarial, insecticidal, and most recently anticancer activity.13–15 The antitumor activity of gedunin was explored through the use of the connectivity map.16 Lamb et al. found, via high connectivity scores with GDA, 17-AAG, and 17-DMAG, that gedunin exhibited its antiproliferative activity through Hsp90 modulation. In a subsequent article, however, gedunin was determined to interact with Hsp90 via a mechanism that does not involve competitive inhibition of ATP,17 and therefore, the interaction of gedunin with Hsp90 is distinct from that of GDA and related analogues. While gedunin induces Hsp90-dependent client protein degradation similar to other Hsp90 inhibitors, the natural product was unable to displace GDA in a fluorescence polarization assay with purified Hsp90 at concentrations up to 100 μM. As a structurally and mechanistically distinct regulator of the Hsp90 protein folding machinery, gedunin represents a lead compound that may possess unique Hsp90 modulatory attributes.
Figure 1.

Chemical structures of GDA, gedunin (1), and novobiocin.
To date, no structure–activity relationships have been established between Hsp90 and gedunin. The ease of acquiring gedunin and its 7-desacetyl derivative from neem oil and the functionality present on the natural products make them well-suited for semisynthetic derivatization for the development of more efficacious compounds. In this article, 19 analogues of gedunin were prepared through semisynthetic procedures aimed at elucidating the role of the α,β-unsaturated ketone, exploring the sensitivity of the ketone binding pocket to steric bulk, and the effects of various substituents at the 7-position. All analogues were evaluated for antiproliferative activity against MCF-7 and SKBr-3 breast cancer cell lines, and a few compounds were subsequently analyzed by Western blot analysis of Hsp90-dependent client proteins. The results from these studies are reported herein.
Chemistry
Commercially available neem oil presents an affordable resource for a rich supply of gedunin. 1 kg of neem oil from Neem King was extracted with methanol. The methanol extracts were combined and washed with n-hexanes, and the remaining insoluble dark-brown resin was purified by flash column chromatography using a gradient of 100% hexanes to 100% ethyl acetate. Fractions containing gedunin were pooled and subjected to successive columns, each using a tighter gradient of hexanes and ethyl acetate. Gedunin-containing fractions that appeared clean by thin layer chromatography (TLC) were pooled and concentrated under reduced pressure to provide a pale-yellow resin that was crystallized from ethyl acetate to give 3 g of a white crystalline solid, which contained two products by TLC and HPLC. 1H and 13C NMR data show all peaks previously described for gedunin and desacetylgedunin.13 These two compounds were obtained in their pure forms by a final isocratic purification using n-hexanes/dichloromethane/diethyl ether in a ratio of 5:3:2.
Compounds 3a–f and 4 were synthesized from 7-desacetylgedunin (2) as outlined in Scheme 1. 7-Desacetylgedunin (2) was treated with potassium tert-butoxide and reacted with the requisite alkyl halide to afford ethers 3a–d. Standard acylation procedures were used to generate 3e. Compound 3f was produced by treatment of 7-desacetylgedunin with trichloroacetyl isocyanate followed by basic methanolic workup. Pyridinium chlorochromate oxidation of 7-desacetylgedunin provided the corresponding ketone 4.
Scheme 1a.

a Reagents and conditions: (a) alkyl iodide, KOtBu, THF, 0°C; (b) propionyl chloride, DMAP, Et3N, CH2Cl2, 0 °C; (c) trichloroacetyl isocyanate, CH2Cl2; (d) PCC, CH2Cl2.
The synthesis of compounds 6–13 are given in Schemes 2–4. Treatment of 7-desacetylgedunin with sodium hydroxide and hydrogen peroxide resulted in epoxide 5. Dihydroxylation of olefin 1 using osmium tetroxide and N-methylmorpholine N-oxide gave vicinal diol 6, which upon treatment with 2,2-dimethoxypropane and p-toluenesulfonic acid yielded the corresponding acetonide 7. In contrast, hydrogenation of the double bond was accomplished by use of palladium on carbon and hydrogen gas to afford 8. Standard Luche conditions surprisingly gave the over-reduced product, 9. After considerable investment, chemoselective reduction of the ketone was finally accomplished via Merwein–Pondorf–Verley conditions enlisting aluminum isopropoxide and isopropanol to afford 10a. The allylic alcohol of 10a was acetylated under standard conditions to give 10b. Treatment of gedunin with excess sodium borohydride without a lanthanide gave the corresponding alcohol, 11. Oxime ether derivatives of gedunin, 12a–c, were synthesized by treatment of the natural product with substituted hydroxylamines. Compound 13, the product resulting from the treatment of gedunin with hydroxylamine, is a further demonstration of unique behavior demonstrated by the lactone moiety of gedunin.
Scheme 2a.
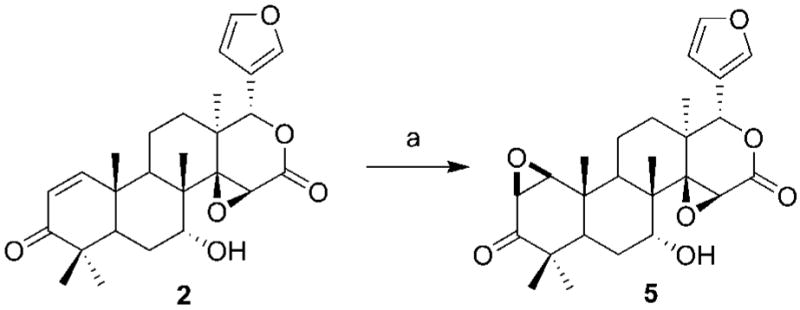
a Reagents and conditions: (a) NaOH, H2O2, acetone.
Scheme 4a.

a Reagents and conditions: (a) hydroxylamine hydrochloride, pyridine, 70 °C.
Results and Discussion
Without confirmation of the site to which gedunin binds Hsp90, we set out to synthesize 19 derivatives of the natural product in order to determine the importance of specific functional groups on the tetracyclic core, as well as to probe the chemical space surrounding these ring systems. Gedunin (1, Figure 1) presents a variety of chemical surfaces, each of which offers the potential of chemo- or regioselective modification. The red numbers in Figure 1 indicate functional groups focused upon by these studies. We sought to investigate steric limitations and electronic properties of substituents at the 7-position and the α,β-unsaturated ketone moiety (carbons 1, 2, and 3). Because the goal of this project was to generate preliminary structure–activity relationships, products of non-stereospecific reactions were not separated into their diastereomerically pure forms.
Compounds 1, 2, 3a–f, 4–9, 10a,b, 11, 12a–c, and 13 were evaluated for antiproliferative activity against MCF-7 and SKBr3 cells alongside geldanamycin, which is a previously reported Hsp90 inhibitor that binds to the N-terminal ATP-binding pocket. The antiproliferative activities for these compounds are listed in Table 1. While many of the prepared compounds retain activity, none of the derivatives are more active than the natural product, however, some important preliminary structure–activity relationships have been identified.
Table 1.
IC50 Values with Standard Deviations of Gedunin Analogues in MCF-7 and SKBr3 Breast Cancer Cell Linesa
| compd | MCF-7 | SKBr3 |
|---|---|---|
| 1 | 8.84 ± 0.03 | 3.22 ± 0.6 |
| 2 | 28.96 ± 4.48 | 21.09 ± 5.95 |
| 3a | 38.88 ± 0.36 | 12.99 ± 0.85 |
| 3b | 14.79 ± 0.87 | 12.06 ± 1.27 |
| 3c | 37.27 ± 2.20 | 18.14 ± 0.99 |
| 3d | >100 | >100 |
| 3e | >100 | >100 |
| 3f | 9.79 ± 0.69 | 7.05 ± 1.00 |
| 4 | 10.90 ± 0.41 | 4.49 ± 0.98 |
| 5 | >100 | >100 |
| 6 | >100 | >100 |
| 7 | >100 | >100 |
| 8 | >100 | >100 |
| 9 | 47.72 ± 1.59 | 82.38 ± 17.62 |
| 10a | 54.31 ± 6.78 | 47.70 ± 1.61 |
| 10b | >100 | >100 |
| 11 | >100 | >100 |
| 12a | >100 | >100 |
| 12b | >100 | >100 |
| 12c | >100 | >100 |
| 13 | 33.17 ± 2.18 | 26.49 ± 0.93 |
IC50 values are determined in μM from nonlinear dose–response curve using GraphPad Prism based on data from standard antiproliferative assay using MTS/PMS as a visualizing reagent. Each experiment was repeated three times in triplicate.
Substituents at the 7-position are placed in an environment that appears to be sensitive to the effects of steric bulk that deviates from the native ligand. From compounds 3a through 3e, a pronounced decrease in antiproliferative activity resulted in a decrease or increase of the size of the substituent from an ethyl ether (3b) to either a methyl ether or an n-propyl ether (3a and 3c) and complete loss in activity was produced by incorporation of the benzyl ether, as evidenced by compound 3d. A complete loss in activity was also noted upon incorporation of the propionate ester as in 3e. The n-propyl ether in 3c may retain activity because of its less rigid nature compared to the propionate ester 3e. Also, preliminary molecular modeling results suggest that the carbonyl oxygen present on the propionate is tilted out of alignment with the carbonyl oxygen of the natural product, suggesting that this moiety, when present, is important for binding.
From the initial data set, it also appears that the binding site for substituents at the 7-position is not sensitive to a change in the electronic nature of the substituent. Compound 3f, which incorporates a carbamate as a nonbulky hydrogen bond acceptor/donor, exhibited an IC50 value almost identical to that of the natural product. The most active analogue found in the 7-position series is compound 4, the 7-oxo derivative, further demonstrating the tolerance of this binding site to small, hydrogen bond acceptors rather than small hydrophobic moieties as in 3a. These results support two reasonable conclusions in regard to the effect of differing substituents at the 7-position: (1) the binding site complementary to substituents at this position may provide a hydrogen bond donor that resides in a shallow pocket. This may explain why hydrogen bond acceptors exhibit the most potent antiproliferative activities, while decreasing or increasing steric bulk from that possessed by the native ligand without the presence of a hydrogen bond acceptor decreases activity. (2) Substituents placed at the 7-position of the molecule are positioned in an area critical to the overall conformation of the molecule, thereby exhibiting significant impact on binding. This latter observation explains why even small changes in the size of substituents, e.g., the additional methyl group of 3e compared to 1, cause a decrease in antiproliferative activity, which is supported by preliminary molecular modeling (data not shown).
The antiproliferative activity of the α,β-unsaturated ketone series also provides key information regarding the importance of this functional group. Compounds lacking the 1,2-olefin of gedunin manifest IC50 values greater than 100 μM (i.e., 6–8 and 11). At first inspection, these data might suggest that the α,β-unsaturated ketone is acting as a Michael acceptor; however, antiproliferative activity of related compounds in this series suggest otherwise. For example, compound 9, which contains the reduced ketone and lactone moieties, retains antiproliferative activity. Although 9 exhibits activity 5- to 10-fold less potent than the natural product, evidence suggests that the α,β-unsaturated ketone is not acting as a Michael acceptor, as a far less potent compound would be expected. Support of this reasoning is substantiated with compound 10a, which resulted from chemoselective reduction of the α,β-unsaturated ketone. 10a manifests antiproliferative activitiy 5 times less potent than gedunin in both MCF-7 and SKBr3 cells. Compound 12a also retains some activity and is only 3–4 times less potent than the natural product and incorporates an electron-rich oxime, which serves as a poor Michael acceptor.
Compounds 10b, 12a–c, and 13 also provide valuable insight into the binding site of the α,β-unsaturated ketone. This binding pocket appears sensitive to steric bulk and/or intolerant to hydrophobic moieties. Compound 10b, the acetylated version of 10a, exhibits no activity. Compound 12a, the O-methyloxime, also manifests no antiproliferative activity. This is interesting because aside from the additional projection of steric bulk and the increase in hydrophobicity from the methyl group on the oxime ether, 12a appears to include the requirements for successful binding at other positions of the molecule, including the α,β-olefin, a hydrogen bond acceptor at the 3-position, and the appropriately fitting substituent at the 7-position. Neither 12b nor 12c exhibited antiproliferative activity which also parallels this trend.
In order to demonstrate the retention of Hsp90 inhibition despite structural manipulation, the most active compounds in this series were evaluated to determine their ability to induce Hsp90-dependent client protein degradation. 1, 3f, and 4 were subjected to Western blot analysis for the detection of two known Hsp90 client proteins, Raf and HER2, and the results are presented in Figure 2. Both compounds 3f and 4 demonstrate client protein degradation similar to 1 and GDA, which further supports their activity stems from modulation of the Hsp90 molecular chaperone.
Figure 2.

Western blot analysis of compounds 1, 3f, and 4 in SKBr3 breast cancer cells after 24 h of incubation at given concentrations.
Conclusions
Preliminary structure–activity relationships with gedunin, a novel Hsp90 inhibitor, have been completed. We have shown that the α,β-unsaturated ketone, while required for antiproliferative activity, does not behave as a Michael acceptor and may therefore evade toxicity associated with this type of motif. We have also shown that the antiproliferative activity of this series of compounds is reduced by a decrease or increase in steric bulk at the 7-position compared to gedunin, suggesting that the natural product contains the optimal substituents at this position. Also, it appears that activity of compounds within the 7-position series is not affected by alteration of the electronic nature.
Of further note, the most active compounds, 3f and 4, may present scaffolds that exhibit advantages over the natural product 1. The acetate moiety present in 1 is quite labile in physiological systems, as it may be rapidly cleaved by endogenous, promiscuous esterases. The desacyl derivative of gedunin has significantly lower antiproliferative activity, and this may prevent derivatives of gedunin containing the acetate moiety from producing clinical significance. Conversely, the carbamate of 3f and the ketone of 4 represent more stable functional groups that can withstand robust physiological environments. The carbamate of 3f also has the added feature of lowering the lipophilicity of the natural product, which also increases its solubility and perhaps physiological relevance.
In summary, two lead compounds have been discovered on the basis of the structure of gedunin, which have similar activity to the natural product and improved physiological properties. Further studies based on these lead structures are underway in an effort to increase the potency of these potential chemotherapeutic agents. The results from such studies will be reported in due course.
Experimental Section
General Methods
Unless otherwise indicated, all reagents were purchased from commercial suppliers and are used without further purification. All stir bars and glassware were flame-dried, and glassware was flushed with argon immediately prior to use. The 1H and 13C NMR spectra were recorded at 500 and 125 MHz, respectively, on a Bruker DRX 500 using CDCl3 as solvent, δ values in ppm (TMS as internal standard), and J (Hz) assignments of 1H resonance coupling. Column chromatography was performed with silica gel (40–63 μm particle size) from Sorbent Technologies (Atlanta, GA). Analytical HPLC was carried out on an Agilent 1100 series capillary HPLC system with diode array detection at 254 nm (compounds 5–8, 10a, and 11 were detected at 214.15 nm) on an Agilent Eclipse XDB-C18 column (4.6 mm × 150 mm, 5 μm) with isocratic elution in 70% acetonitrile and 30% H2O at a flow rate of 5.0 mL/min.
Gedunin (1)
Spectral data correspond with that previously reported. See Supporting Information.
Desacetylgedunin (2)
Spectral data correspond with that previously reported. See Supporting Information.
General Procedure A
A solution of 2 (20 mg, 0.045 mmol, 1 equiv) in anhydrous THF (450 μL) was stirred at 0 °C under argon atmosphere. Potassium tert-butoxide (45 μL, 0.09 mmol, 2 equiv) was added dropwise and the mixture stirred for 30 min before alkyl iodide (10 equiv) was added. The resulting mixture was stirred at 0°C for 1.5 h and then quenched by the addition of H2O (1 mL). The organic layer was removed, and the aqueous layer extracted with DCM (3 × 5 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated. The residue was purified via SiO2 chromatography (5:3:2 hexanes/DCM/Et2O) to yield the desired compound.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-5-methoxy-4a1,7,7,10a,12a-pentamethyl-4a1,5,6,6a,7,10a,10b,11,12,12a-decahydronaphtho[2,1-f]oxireno[2,3-d]isochromene-3,8(1H,3aH)-dione (3a)
Compound 3a was synthesized from 2 using general procedure A and iodomethane to afford 13 mg (64%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.01 (s, 3H), 1.04 (s, 3H), 1.10 (s, 3H), 1.13 (s, 3H), 1.13 (s, 3H), 1.44–1.52 (m, 2H), 1.67–1.75 (m, 2H), 1.82–1.90 (m, 2H), 2.23 (dd, J = 2.1, 13.2 Hz, 1H), 2.38 (dd, J = 5.9, 12.7 Hz, 1H), 2.85 (d, J = 2.1 Hz, 1H), 3.25 (s, 3H), 3.55 (s, 1H), 5.52 (s, 1H), 5.76 (d, J = 10.2 Hz, 1H), 6.27 (d, J = 0.6 Hz, 1H), 7.01 (d, J = 10.2, 1H), 7.30–7.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 14.1, 16.7, 17.5, 18.9, 19.2, 20.5, 25.4, 26.4, 37.3, 37.7, 39.0, 43.1, 43.2, 43.5, 54.6, 56.4, 69.1, 77.4, 78.2, 108.9, 119.7, 124.7, 140.1, 141.9, 156.8, 167.1, 203.5. HRMS (*ESI + pos) (m/z): [M + H] calcd for C27H35O6, 455.2434; found, 455.2419. HPLC tR = 10.09 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-5-Ethoxy-1-(furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-4a1,5,6,6a,7,10a,10b,11,12,12a-decahydronaphtho[2,1-f]oxireno[2,3-d]isochromene-3,8(1H,3aH)-dione (3b)
Compound 3b was synthesized from 2 using general procedure A and 1-iodoethane to afford 7 mg (33%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.00 (s, 3H), 1.03 (s, 3H), 1.08 (s, 3H), 1.12 (s, 3H), 1.15 (s, 3H), 1.15 (t, J = 12.7 Hz, 3H), 1.46–1.50 (m, 1H), 1.53–1.57 (m, 1H), 1.61–1.65 (m, 1H), 1.69–1.73 (m, 1H), 1.81–1.89 (m, 2H), 2.24 (dd, J = 2.2, 13.1 Hz, 1H), 2.26 (dd, J = 5.8, 12.8 Hz, 1H), 2.95 (d, J = 2.1 Hz, 1H), 3.14 (dq J = 7.0, 8.7 Hz, 1H), 3.55 (s, 1H), 3.57 (dq, J = 7.0, 8.7 Hz, 1H), 5.53 (s, 1H), 5.77 (d, J = 10.2 Hz, 1H), 7.02 (d, J = 10.3 Hz, 1H), 7.31–7.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 14.0, 14.5, 16.8, 17.4, 18.9, 20.0, 20.5, 25.4, 26.4, 37.3, 37.8, 39.0, 43.0, 43.2, 43.7, 56.4, 62.8, 69.2, 77.1, 77.4, 109.0, 119.7, 124.7, 140.1, 141.9, 156.8, 167.1, 203.6. HRMS (*ESI + pos) (m/z): [M + H] calcd for C28H37O6, 469.2590; found, 469.2581. HPLC tR = 12.71 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-5-propoxy-4a1,5,6,6a,7,10a,10b,11,12,12a-decahydronaphtho[2,1-f]oxireno[2,3-d]isochromene-3,8(1H,3aH)-dione (3c)
Compound 3c was synthesized from 2 using general procedure A and 1-iodopropane to afford 5 mg (23%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.86 (t, J = 7.4 Hz, 3H), 1.01 (s, 3H), 1.04 (s, 3H), 1.08 (s, 3H), 1.13 (s, 3H), 1.16 (s, 3H), 1.46–1.50 (m, 1H), 1.50–1.61 (m, 3H), 1.62–1.67 (m, 1H), 1.70–1.74 (m, 1H), 1.84–1.88 (m, 1H), 1.90–1.95 (m, 1H), 2.25 (dd, J = 2.2, 13.1 Hz, 1H), 2.42 (dd, J = 6.0, 12.8 Hz, 1H), 2.94 (d, J = 2.2 Hz, 1H), 3.10 (dt, J = 2.3, 6.2 Hz, 1H), 3.42 (dt, J = 1.6, 6.5 Hz, 1H), 3.55 (s, 1H), 5.52 (s, 1H), 5.77 (d, J = 10.2 Hz, 1H), 6.27–6.29 (m 1H), 7.02 (d, J = 10.2 Hz, 1H), 7.31–7.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 10.1, 14.0, 16.8, 17.5, 18.9, 19.9, 20.5, 22.3, 25.4, 26.5, 37.3, 37.8, 39.0, 43.0, 43.2, 43.7, 56.4, 68.9, 69.1, 76.8, 77.4, 108.9, 119.7, 124.7, 140.1, 141.9, 156.9, 167.0, 203.6. HRMS (*ESI + pos) (m/z): [M + H] calcd for C29H39O6, 483.2747; found, 483.2737. HPLC tR = 15.90 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-5-(Benzyloxy)-1-(furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-4a1,5,6,6a,7,10a,10b,11,12,12a-decahydronaphtho[2,1-f]oxireno[2,3-d]isochromene-3,8(1H,3aH)-dione (3d)
Compound 3d was synthesized from 2 using general procedure A and benzyl bromide and catalytic tetrabutylammonium iodide to afford 14 mg (59%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.02 (s, 3H), 1.04 (s, 3H), 1.07 (s, 3H), 1.12 (s, 3H), 1.14 (s, 3H), 1.42–1.46 (m, 1H), 1.59–1.68 (m, 2H), 1.68–1.72 (m, 1H), 1.80–1.85 (m, 1H), 2.02 (dt, J = 3, 14.7 Hz, 1H), 2.32 (dd, J = 2.2, 13.1 Hz), 2.41 (dd, J = 5.9, 12.7 Hz, 1H), 3.14 (d, J = 2.2 Hz, 1H), 3.67 (s, 1H), 4.25 (d, J = 10.2 Hz, 1H), 4.45 (d, J = 10.2 Hz, 1H), 5.51 (s, 1H), 5.75 (d, J = 10.2 Hz, 1H), 6.25 (d, J = 1.0 Hz, 1H), 6.99 (d, J = 10.2 Hz, 1H), 7.24–7.30 (m, 2H), 7.35–7.21 (m, 5H). 13C NMR (125 MHz, CDCl3): δ 14.0, 16.9, 17.6, 19.1, 19.9, 20.5, 25.5, 26.6, 37.3, 37.7, 39.1, 43.2, 43.3, 43.7, 56.3, 69.0, 69.6, 76.4, 77.3, 109.0, 119.6, 124.7, 127.1, 127.5, 127.5, 127.7, 127.7, 135.8, 140.1, 141.9, 156.9, 166.9, 203.4. HRMS (*ESI + pos) (m/z): [M + H] calcd for C33H39O6, 531.2747; found, 531.2762. HPLC tR = 16.08 min. Purity = 97.8%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-3,8-dioxo-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11,12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Propionate (3e)
A solution of 2 (20 mg, 0.045 mmol, 1 equiv), dimethylaminopyridine (catalytic), and Et3N (35 μL, 0.248 mmol, 5.5 equiv) in 450 μL of anhydrous DCM was stirred under argon atmosphere at 0 °C until completely dissolved. Propionyl chloride (20 μL, 0.225 mmol, 5 equiv) was added dropwise to the solution and then stirred for 5 h before the reaction was quenched by addition of H2O (1 mL). The organic layer was removed and the aqueous layer extracted with DCM (3 × 5 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated. The resulting yellow oil was purified via SiO2 chromatography (4:3:3 hexanes/DCM/Et2O) to yield 17 mg (76%) 3e as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.98 (s, 3H), 1.00 (t, J = 7.6 Hz, 3H), 1.00 (s, 3H), 1.12 (s, 3H), 1.17 (s, 3H), 1.36 (s, 3H), 1.66–1.97 (m, 4H), 2.09 (dd, J = 2.4, 13.4 Hz, 1H), 2.20–2.38 (m, 1H), 2.30 (dq, J = 2.6, 7.7 Hz, 1H), 2.40–2.46 (m, 2H), 2.42 (dq, J = 2.6, 7.7 Hz, 1H), 3.48 (s, 1H), 4.52 (m, 1H), 5.52 (s, 1H), 5.79 (d, J = 10.2 Hz, 1H), 6.26 (m, 1H), 7.02 (d, J = 10.2 Hz, 1H), 7.34 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 10.5, 14.1, 15.2, 17.7, 18.9, 19.9, 21.3, 23.3, 25.3, 27.3, 38.9, 39.6, 40.2, 42.8, 44.1, 46.1, 57.6, 69.4, 72.7, 78.0, 110.0, 114.0, 120.5, 126.0, 141.2, 143.1, 157.0, 165.1, 166.9, 204.1. HRMS (*ESI + pos) (m/z): [M + H] calcd for C29H37O7, 497.2539; found, 497.2542. HPLC tR = 13.66 min. Purity = 97.1%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-3,8-dioxo-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11,12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Carbamate (3f)
A solution of 2 (20 mg, 0.045 mmol, 1 equiv) in 450 μL of anhydrous DCM under argon atmosphere was stirred at 0 °C. Trichloroacetyl isocyanate (10.7 μL, 0.09 mmol, 2 equiv) was added dropwise, and the solution was allowed to warm to room temperature and stirred for 3 h, at which point the reaction was quenched by the addition of H2O (1 mL). The organic layer was collected and the aqueous layer extracted with DCM (3 × 5 mL). The combined organic extracts were concentrated and then dissolved in 1 mL of 0.1 M K2CO3 in MeOH, and the mixture was stirred for 1.5 h at ambient temperature. The reaction was quenched by the addition of H2O (1 mL). The reaction mixture was extracted with DCM (3 × 5 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated. The resulting colorless oil was purified via SiO2 chromatography (4:3:3 hexanes/DCM/Et2O) to yield 21 mg (97%) 3f as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.01 (s, 3H), 1.03 (s, 3H), 1.09 (s, 3H), 1.16 (s, 6H), 1.49–1.53 (m, 1H), 1.65–1.68 (m, 1H), 1.778–1.81 (m, 2H), 1.85–1.93 (m, 2H), 2.15 (dd, J = 2.5, 12.7 Hz, 1H), 2.37 (dd, J = 6.1, 12.7 Hz, 1H), 3.66 (s, 1H), 4.45 (s, 1H), 4.67 (br s, 2H), 5.54 (s, 1H), 5.79 (d, J = 10.2 Hz, 1H), 6.25–6.27 (m, 1H), 7.02 (d, J = 10.2 Hz, 1H), 7.32–7.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 15.0, 18.0, 18.5, 19.8, 21.3, 23.6, 26.0, 27.2, 38.6, 39.5, 40.0, 42.9, 44.1, 45.9, 56.9, 69.7, 73.5, 78.2, 109.9, 120.5, 126.0, 141.2, 143.1, 155.1, 157.1, 167.4, 204.2. HRMS (*ESI + pos) (m/z): [M + H] calcd for C27H34N1O7, 484.2335; found, 484.2320. HPLC tR = 3.59 min. Purity = 98.7%.
(1S,3aS,41R,4a1R,10aS,12aS)-1-(Furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-6,6a,7,10a,10b,11,12,12a-octahydronaphtho[2,1-f]oxireno[2,3-d]isochromene-3,5,8(1H,3aH,4a1H)-trione (4)
A solution of 2 (20 mg, 0.045 mmol, 1 equiv) in 450 μL of anhydrous DCM was stirred at ambient temperature. Pyridinium chlorochromate (29.1 mg, 0.135 mmol, 3 equiv) was added in one portion, and the reaction mixture was stirred overnight before the solution was filtered through a plug of silica using diethyl ether as eluent. The colorless solution was dried (Na2SO4), filtered, and concentrated. The resulting colorless oil was purified via SiO2 chromatography (5:3:2 hexanes/DCM/Et2O) to yield 19 mg (96%) of 4 as a colorless solid. 1H NMR (125 MHz, CDCl3): δ 1.07 (s, 3H), 1.07 (s, 3H), 1.09 (s, 3H), 1.15 (s, 3H), 1.29 (s, 3H), 1.41 (dt, J = 3.9, 9.8 Hz, 1H), 1.71–1.77 (m, 2H), 1.91–1.95 (m, 1H), 2.10–2.17 (m, 2H), 2.34 (dd, J = 3.2, 14 Hz, 1H), 2.86 (t, J = 14.4 Hz, 1H), 3.80 (s, 1H), 5.4 (s, 1H), 5.85 (d, J = 10.2 Hz, 1H), 6.29 (dd, J = 0.7, 1.8 Hz, 1H), 7.03 (d, J = 10.2 Hz, 1H), 7.32–7–34 (m, 1H), 7.34–7.36 (m, 1H). 13C NMR (500 MHz, CDCl3): δ 16.2, 16.4, 18.8, 19.6, 19.9, 25.7, 31.2, 35.7, 36.7, 35.7, 36.7, 38.6, 44.2, 46.6, 52.4, 52.6, 53.5, 64.5, 77.0, 108.8, 119.2, 125.4, 140.0, 142.1, 154.9, 165.8, 202.2, 207.1. HRMS (*ESI + pos) (m/z): [M + H] calcd for C26H31O6,439.2121; found, 439.2129. HPLC tR = 5.45 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-5-hydroxy-4a1,7,7,10a,12a-pentamethyl-4a1,5,6,6a,7,10a,10b,11,12,12a-decahydronaphtho[2,1-f][9,10]dioxireno[2,3-d]isochromene-3,8(1H,3aH)-dione (5)
A solution of 2 (20 mg, 0.045 mmol, 1 equiv) in 450 μL of acetone was stirred at 0 °C. A solution of 8% aqueous sodium hydroxide (85 μL) and a solution of 30% aqueous hydrogen peroxide (52 μL) were added. The reaction mixture was allowed to warm to room temperature. After the mixture was stirred overnight, 2 N hydrochloric acid (1 mL) was added. The reaction mixture was extracted with DCM (3 × 5 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated. The resulting colorless oil was purified via SiO2 chromatography (5: 3:2 hexanes/DCM/Et2O) to yield 14 mg (68%) of 5 as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.92 (s, 3H), 0.96 (s, 3H), 1.00 (s, 3H), 1.04 (s, 3H), 1.22 (s, 3H), 1.45–1.52 (m, 2H), 1.63 (dd, J = 4.1, 8.1 Hz, 1H), 1.71–1.77 (m, 2H), 1.92–1.96 (m, 1H), 2.64 (dd, J = 2.6, 13.7 Hz, 1H), 2.72 (dd, J = 6.6, 12.7 Hz, 1H), 3.32 (d, J = 4.6 Hz, 1H), 3.46 (s, 1H), 3.48 (d, J = 4.5 Hz, 1H), 3.85 (s, 1H), 5.53 (s, 1H), 6.29 (s, 1H), 7.33 (s, 1H), 7.32–7.34 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 14.5, 14.8, 16.4, 17.7, 19.8, 25.0, 26.0, 26.5, 35.9, 36.1, 37.4, 38.2, 42.2, 43.2, 55.6, 57.0, 62.1, 68.5, 68.9, 77.4, 109.0, 119.6, 140.2, 141.9, 167.2, 210.7. HRMS (*ESI + pos) (m/z): [M + H] calcd for C26H33O7, 457.2226; found, 457.2220. HPLC tR = 6.40 min. Purity = 98.6%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-9,10-dihydroxy-4a1,7,7,10a,12a-pentamethyl-3,8-dioxohexadecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (6)
A solution of 1 (100 mg, 0.21 mmol, 1 equiv) in 1.78 mL of fully degassed acetone was stirred under argon atmosphere at 0 °C. A 4% solution of osmium tetroxide in water (72 μL, 0.011 mmol, 0.05 equiv) and a 1 M solution of N-methylmorpholine N-oxide in deionized water (315 μL, 0.315 mmol, 1.5 equiv) were added. After the mixture was stirred overnight, saturated aqueous sodium sulfite was added (4 mL) and the reaction mixture was stirred for 1 h at room temperature. The reaction mixture was extracted 4 times with ethyl acetate. The combined organics were dried (Na2SO4), filtered, and concentrated. The resulting black oil was purified via SiO2 chromatography (7:6:6 hexanes/DCM/Et2O) to yield 97 mg (89%) of 6 as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.99 (s, 3H), 1.02 (s, 3H), 1.07 (s, 3H), 1.18 (s, 3H), 1.19 (s, 3H), 1.48–1.51 (m, 1H), 1.60–1.63 (m, 2H), 1.79–1.82 (m, 2H), 2.03 (s, 3H), 2.16 (dd, J = 4.1, 11.5 Hz, 1H), 2.48 (s, 1H), 2.96 (dd, J = 6.4, 12.1 Hz, 1H), 3.42 (s, 1H), 3.86 (d, J = 2.3 Hz, 1H), 3.89 (d, J = 2.8 Hz, 1H), 4.44 (br s, 1H), 4.60–4.62 (m, 1H), 5.54 (s, 1H), 6.25–6.27 (m, 1H), 7.32–7.34 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 13.5, 15.3, 15.7, 17.5, 19.9, 20.1, 22.0, 22.8, 24.6, 35.7, 37.9, 40.2, 40.8, 41.0, 45.5, 55.7, 69.0, 70.2, 72.5, 75.9, 77.5, 109.0, 119.6, 140.1, 141.9, 166.6, 169.1, 213.0. HRMS (*ESI + pos) (m/z): [M + H] calcd for C28H37O9, 517.2438; found, 517.2421. HPLC tR = 4.71 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-9,10-dihydroxy-4a1,7,7,10,10,10a,12a-heptamethyl-3,8-dioxohexadecahydronaphtho-9,11-dioxol[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (7)
A solution of 6 (10.5 mg, 0.02 mmol, 1 equiv) in 100 μL of acetone was stirred at room temperature. 2,2-Dimethoxypropane (11 μL, 0.088 mmol, 4.4 equiv) was added followed by the addition of catalytic p-toluenesulfonic acid. After the mixture was stirred for 3 h, solvent was blown dry and residue was taken up in 300 μL of DCM and washed with sodium bicarbonate and brine. The organic layer was dried (Na2SO4), filtered, and concentrated. The resulting colorless oil was purified via SiO2 chromatography (5:3:2 hexanes/DCM/Et2O) to yield 10 mg (90%) of 7 as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.98 (s, 3H), 1.02 (s, 3H), 1.13 (s, 3H), 1.22 (s, 3H), 1.23 (s, 3H), 1.48 (s, 3H), 1.48–1.51 (m, 1H), 1.50 (s, 3H), 1.56–1.63 (m, 3H), 1.71–1.73 (m, 1H), 1.83 (dt, J = 3.7, 14.9 Hz, 1H), 2.05 (s, 3H), 2.42 (dd, J = 3.6, 13.7 Hz, 1H), 2.95 (dd, J = 6.2, 12.4 Hz, 1H), 3.48 (s, 1H), 4.05 (d, J = 7.5 Hz, 1H), 4.31 (d, J = 7.5 Hz, 1H), 4.45 (dd, J = 1.9, 3.4, 1H), 5.55 (s, 1H), 6.27 (d, J = 1.0 Hz, 1H), 7.32–7.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 13.8, 14.5, 15.7, 16.9, 20.1, 20.4, 22.5, 22.9, 24.5, 24.9, 28.4, 32.6, 36.7, 37.8, 39.5, 40.4, 44.0, 52.4, 55.7, 69.1, 72.3, 77.4, 81.1, 109.0, 109.4, 119.6, 140.1, 141.9, 166.7, 168.9, 210.9. HRMS (*ESI + pos) (m/z): [M + Na] calcd for C31H40O9Na, 579.2570; found, 579.2548. HPLC tR = 14.17 min. Purity = 96.1%.
(1S,3aS,41R,4a1S,5R,10aR,12aS)-1-(Furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-3,8-dioxohexadecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (8)
A solution of 1 (20 mg, 0.041 mmol, 1 equiv) and 10% Pd/C (2 mg) in 1 mL of methanol was stirred at room temperature under an atmosphere of argon. The reaction flask was purged with H2 repeatedly. The fully H2 purged flask was stirred at room temperature for 5 h, after which the solution was run through a plug of silica using ether as eluent. The organic filtrate was dried (Na2SO4), filtered, and concentrated. The resulting colorless oil was purified via SiO2 chromatography (5:3:2 hexanes/DCM/Et2O) to yield 7 mg (35%) of 8 as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.96 (s, 3H), 1.00 (s, 3H), 1.05 (s, 3H), 1.17 (s, 3H), 1.42 (s, 3H), 1.48–1.53 (m, 2H), 1.63–1.71 (m, 4H), 1.77–1.81 (m, 1H), 1.81–1.83 (m, 1H), 1.84–1.86 (m, 1H), 2.24 (dd, J = 6.9, 12.1 Hz, 1H), 2.38–2.40 (m, 1H), 2.50–2.52 (m, 1H), 3.45 (s, 1H), 4.44–4.47 (m, 1H), 5.54 (s, 1H), 6.25–6.26 (m, 1H), 7.31–7.33 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 14.0, 14.7, 16.4, 17.0, 19.9, 20.1, 22.7, 24.8, 25.0, 32.7, 36.3, 37.8, 37.9, 40.9, 43.0, 45.6, 46.7, 55.7, 68.78, 72.7, 77.3, 108.9, 119.5, 140.1, 142.0, 166.6, 169.0, 214.9. HRMS (*ESI + pos) (m/z): [M + H] calcd for C28H37O7, 485.2539; found, 485.2549. HPLC tR = 6.35 min. Purity = 97.6%.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-1-(Furan-3-yl)-3,8-dihydroxy-4a1,7,7,10a,12a-pentamethyl-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11,12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (9)
A solution of 1 (500 mg, 1.04 mmol, 1 equiv) in 4 mL of a 2:1 mixture of methanol and chloroform was stirred under argon atmosphere at 0 °C. Cerium trichloride hexahydrate (740 mg, 2.08, 2 equiv) was added followed by the slow addition of sodium borohydride (39.33 mg, 1.04 mmol, 1 equiv). The reaction mixture was stirred for 3 min and quenched by the addition of a solution of 1% acetic acid in water. The reaction mixture was extracted 3 times with DCM. The combined organic layers were dried (Na2SO4), filtered, and concentrated. The resulting colorless crystals were dissolved in minimal DCM and purified via SiO2 chromatography (6:7:7 hexanes/DCM/Et2O) to yield 473 mg (93%) of 9 as a colorless solid. Major diastereomer: 1H NMR (500 MHz, CDCl3) δ 0.74 (s, 3H), 0.83 (s, 3H), 0.95 (s, 3H), 1.02 (s, 3H), 1.16 (s, 3H), 1.45–1.80 (m, 7H), 2.05 (s, 3H), 2.30 (dd, J = 5.4, 12.9 Hz, 1H), 3.00–3.10 (m, 2H), 3.21 (d, J = 1.8 Hz, 1H), 3.84 (bs, 1H), 4.56–4.60 (m, 1H), 4.92 (s, 1H), 5.05 (d, J = 10.2 Hz, 1H), 5.28 (d, J = 10.2, 1H), 5.80 (m, 1H), 6.20 (m, 1H), 7.23 (m, 1H), 7.27 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 14.6, 16.1, 17.2, 18.5, 18.7, 20.3, 22.2, 25.6, 26.5, 35.6, 35.7, 338.4, 40.9, 41.9, 45.0, 57.5, 68.5, 71.4, 73.4, 73.4, 76.2, 86.8, 109.2, 121.9, 125.1, 136.3, 125.1, 136.3, 139.6, 141.3, 168.8. HRMS (*ESI + pos) (m/z): [M + Na] calcd for C28H38O7Na, 509.2515; found, 509.2500. HPLC tR = 6.12 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-1-(Furan-3-yl)-8-hydroxy-4a1,7,7,10a,12a-pentamethyl-3-oxo-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11, 12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (10a)
A solution of 1 (500 mg, 1.04 mmol, 1 equiv) in 1.3 mL of toluene was stirred at ambient temperature under argon atmosphere. Isopropyl alcohol (687 mg, 11.44 mmol, 11 equiv) and aluminum triisopropoxide (127.5 mg, 0.624, 0.6 equiv) were added, and the reaction mixture was heated at 70 °C for 20 h. After being allowed to cool to ambient temperature, the reaction mixture was quenched by the addition of 1 N HCl (4 mL) and ethyl acetate (4 mL) and stirred for 1.5 h. The organic layer was washed with water and concentrated. The resulting colorless oil was purified via SiO2 chromatography (3:1:1 hexanes/DCM/Et2O) to yield 284 mg (56%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.75 (s, 3H), 0.83 (s, 3H), 1.02 (s, 3H), 1.04 (s, 3H), 1.16 (s, 3H), 1.44–1.48 (m, 1H), 1.57–1.61 (m, 1H), 1.59–1.63 (m, 1H), 1.66–1.69 (m, 2H), 1.82–1.86 (m, 1H), 1.90 (dd, J = 2.6, 11.7 Hz, 1H), 2.05 (s, 3H), 2.29 (dd, J = 6.4, 12.6 Hz, 1H), 3.42 (s, 1H), 3.84 (d, J = 8.3, 1H), 4.45 (bs, 1H), 5.29 (d, J = 10.5 Hz, 1H), 5.53 (s, 1H), 5.78 (d, J = 10.6 Hz, 1H), 6.26 (s, 1H), 7.33 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 15.4, 17.3, 17.5, 18.5, 19.6, 21.2, 22.6, 26.0, 27.4, 36.5, 38.8, 39.7, 42.0, 42.7, 45.8, 56.8, 65.9, 69.9, 73.9, 78.4, 109.9, 120.6, 126.5, 136.7, 141.2, 143.0, 167.8, 170.0. HRMS (*ESI + pos) (m/z): [M + H] calcd for C28H37O7, 485.2539; found, 485.2540. HPLC tR = 4.31 min. Purity = 98.1%.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-1-(Furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-3-oxo-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11,12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromene-5,8-diyl Diacetate (10b)
A solution of 10a (10 mg, 0.0206 mmol, 1 equiv), dimethylaminopyridine (catalytic), and Et3N (15 μL, 0.103 mmol, 5 equiv) in anhydrous THF (250 μL) was stirred under argon atmosphere at 0 °C. Acetyl chloride (8 μL, 0.103 mmol, 5 equiv) was added dropwise, and the reaction mixture was stirred overnight while allowing to warm to ambient temperature. The reaction mixture was quenched by the addition of water (500 μL). The organic layer was collected, and the aqueous layer was washed with DCM (3 × 5 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated. The resulting yellow oil was purified via SiO2 chromatography (5:3:2 hexanes/DCM/Et2O) to yield 9 mg (83%) of 10b as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 0.74 (s, 3H), 0.83 (s, 3H), 1.02 (s, 3H), 1.07 (s, 3H), 1.15 (s, 3H), 1.46–1.50 (m, 1H), 1.59–1.61 (m, 1H), 1.66–1.72 (m, 2H), 1.81 (m, 2H), 1.90 (d, J = 15.0 Hz, 1H), 2.03 (s, 3H), 2.05 (s, 3H), 2.31 (dd, J = 6.4, 12.7 Hz, 1H), 3.42 (s, 1H), 4.45 (bs, 1H), 5.07 (d, J = 1.8 Hz, 1H), 5.17 (dd, J = 1.4, 9.7 Hz, 1H), 5.52 (s, 1H), 5.82 (d, J = 10.5 Hz, 1H), 6.26 (s, 1H), 7.33 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 14.4, 16.4, 17.3, 17.4, 18.4, 20.2, 21.4, 25.0, 26.4, 34.5, 37.7, 38.7, 41.0, 41.6, 44.8, 55.7, 68.8, 72.7, 77.4, 77.7, 108.9, 119.5, 121.8, 136.6, 140.1, 142.0, 166.6, 169.0, 170.4. HRMS (*ESI + pos) (m/z): [M + H] calcd for C30H39O8, 527.2645; found, 527.2654. HPLC tR = 16.63 min. Purity = 96.2%.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-1-(Furan-3-yl)-3,8-dihydroxy-4a1,7,7,10a,12a-pentamethylhexadecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (11)
A solution of 1 (20 mg, 0.041 mmol, 1 equiv) in 450 μL of ethanol was stirred at ambient temperature under argon atmosphere. Sodium borohydride (10 mg, 0.25 mmol, 6 equiv) was added in one portion, and the reaction mixture was stirred for 45 min. The reaction mixture was quenched by the addition of 1% aqueous acetic acid. The reaction mixture was extracted 3 times with chloroform. The combined organic layers were dried (Na2SO4), filtered, and concentrated. The resulting colorless oil was purified via SiO2 chromatography (4:3:3 hexanes/DCM/Et2O) to yield 17 mg (85%) 11 as a colorless solid. Major diasteromer: 1H NMR (500 MHz, CDCl3) δ 0.69 (s, 3H), 0.81 (s, 3H), 0.85 (s, 3H), 0.94 (s, 3H), 1.18 (s, 3H), 1.15–1.36 (m, 2H), 1.45–1.75 (m, 9H), 2.05 (s, 3H), 2.10–2.21 (m, 1H), 2.96 (d, J = 10.4, 1H), 3.17–3.20 (m, 1H), 3.22 (s, 1H), 4.56–4.59 (m, 1H), 4.92 (s, 1H), 5.06 (d, J = 10.4, 1H), 6.21 (m, 1H), 7.22–7.26 (m, 1H), 7.26–7.28 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 14.2, 16.8, 17.2, 18.5, 20.4, 22.3, 25.4, 25.7, 26.5, 26.7, 26.5, 26.7, 35.7, 36.8, 37.3, 37.4, 43.3, 45.0, 47.0, 52.4, 57.8, 72.2, 73.8, 77.5, 86.8, 109.3, 122.4, 139.6, 141.3, 168.9. HRMS (*ESI + pos) (m/z): [M + Na] calcd for C28H40O7Na, 511.2672; found, 511.2681. HPLC tR = 4.23 min. Purity = 98.3%.
General Procedure B
A solution of 1 (20 mg, 0.041 mmol, 1 equiv) was stirred in pyridine (450 μL) at room temperature. Hydroxylamine or appropriate hydroxylamine derivative (2 equiv) was added, and the reaction mixture was stirred at 70 °C in a sealed tube overnight. The reaction mixture was diluted with toluene, and solvents were condensed in vacuo. The coevaporation procedure was repeated 2 more times. The resulting oil was dissolved in DCM and washed with saturated aqueous sodium bicarbonate (2 × 5 mL). The organic layer was collected, and the aqueous layer was re-extracted with dichloromethane (2 × 5 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated. The resulting oil was purified via SiO2 chromatography (6:2:2, hexanes/DCM/Et2O) to yield the desired oxime.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-1-(Furan-3-yl)-8-(methoxyimino)-4a1,7,7,10a,12a-pentamethyl-3-oxo-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11, 12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (12a)
Compound 12a was synthesized from 1 using general procedure B and methoxyhydroxylamine hydrochloride to afford 13 mg (62%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.05 (s, 3H), 1.06 (s, 3H), 1.07 (s, 3H), 1.08 (s, 3H), 1.16 (s, 3H), 1.46–1.50 (m, 1H), 1.63–1.65 (m, 1H), 1.67–1.71 (m, 1H), 1.77 (dd, J = 1.8, 13.4, 1H), 1.76–1.78 (m, 1H), 1.90 (dd, J = 2.2, 12.7, 1H), 1.89–1.93 (m, 1H), 2.32 (dd, J = 6.0, 12.8 Hz, 1H), 3.45 (s, 1H), 3.79 (s, 3H), 4.45–4.49 (m, 1H), 5.54 (s, 1H), 6.24–6.28 (m, 1H), 6.33 (d, J = 10.4 Hz, 1H), 6.49 (d, J = 10.4, 1H), 7.32–7.34 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 14.0, 16.7, 17.2, 18.0, 20.1, 21.9, 23.2, 25.0, 28.7, 36.3, 37.7, 38.8, 38.9, 41.5, 45.4, 55.8, 60.6, 68.9, 72.5, 77.3, 108.9, 113.3, 119.5, 140.1, 142.0, 145.4, 157.4, 166.6, 168.9. HRMS (*ESI + pos) (m/z): [M + H] calcd for C29H38N1O7, 512.2649; found, 512.2635. HPLC tR = 21.37 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-8-(Ethoxyimino)-1-(furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-3-oxo-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11, 12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (12b)
Compound 12b was synthesized from 1 using general procedure B and ethoxyhydroxylamine hydrochloride to afford 17 mg (79%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.05 (s, 3H), 1.06 (s, 3H), 1.08 (s, 3H), 1.16 (s, 3H), 1.18 (s, 3H), 1.18 (t, J = 6.9 Hz, 3H), 1.60–1.75 (m, 5H), 1.75–1.92 (m, 2H), 2.02 (s, 3H), 2.33 (dd, J = 5.9, 12.8 Hz, 1H), 3.45 (s, 1H), 4.03 (q, J = 7 Hz, 2H), 4.47 (s, 1H), 5.54 (s, 1H), 6.26 (m, 1H), 6.32 (d, J = 10.3 Hz, 1H), 6.52 (d, J = 10.3 Hz, 1H), 7.33 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 14.6, 15.0, 17.7, 18.2, 19.1, 21.1, 22.9, 24.2, 26.1, 29.6, 37.4, 38.7, 39.9, 40.0, 42.5, 46.5, 56.9, 69.4, 70.0, 73.6, 78.3, 109.9, 114.5, 120.5, 141.2, 143.0, 146.0, 158.1, 163.1, 164.0. HRMS (*ESI + pos) (m/z): [M + Na] calcd for C30H39NO7Na, 548.2624; found, 548.2616. HPLC tR = 28.90 min. Purity = 100%.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-8-(Benzyloxyimino)-1-(furan-3-yl)-4a1,7,7,10a,12a-pentamethyl-3-oxo-1,3,3a,4a1,5,6,6a,7,8,10a, 10b,11,12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (12c)
Compound 12c was synthesized from 1 using general procedure B and benzyloxyhydroxylamine hydrochloride to afford 10 mg (42%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.05 (s, 6H), 1.06 (s, 3H), 1.07 (s, 3H), 1.15 (s, 3H), 1.60–1.75 (m, 5H), 1.78–1.88 (m, 2H), 2.02 (s, 3H), 2.31 (dd, J = 6.05, 12.8 Hz, 1H), 3.45 (s, 1H), 4.46 (t, J = 1.6 Hz, 1H), 5.02 (s, 2H), 5.53 (s, 1H), 6.26 (m, 1H), 6.32 (d, J = 10.4 Hz, 1H), 6.55 (d, J = 10.4 Hz, 1H), 7.25–7.28 (m, 5H), 7.28–7.29 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 14.0, 16.7, 17.2, 18.1, 20.1, 21.9, 23.2, 24.5, 28.6, 36.4, 37.6, 38.8, 38.9, 41.4, 45.3, 55.8, 68.9, 72.5, 74.9, 77.3, 108.9, 113.5, 119.4, 126.6, 126.8, 127.2, 127.2, 127.3, 137.0, 140.1, 142.0, 145.3, 157.8, 166.6, 169.0. HRMS (*ESI + pos) (m/z): [M + H] calcd for C35H42N1O7, 588.2961; found, 588.2915. HPLC tR = 15.06 min. Purity = 96.3%.
(1S,3aS,41R,4a1S,5R,10aS,12aS)-1-(Furan-3-yl)-3,8-bis(hydroxyimino)-4a1,7,7,10a,12a-pentamethyl-1,3,3a,4a1,5,6,6a,7,8,10a,10b,11, 12,12a-tetradecahydronaphtho[2,1-f]oxireno[2,3-d]isochromen-5-yl Acetate (13)
Compound 13 was synthesized from 1 using general procedure B and hydroxylamine hydrochloride to afford 7 mg (34%) as a colorless solid. 1H NMR (500 MHz, CDCl3): δ 1.06 (s, 3H), 1.07 (s, 3H), 1.07 (s, 6H), 1.18 (s, 3H), 1.38–1.40 (m, 1H), 1.59 (m, 1H), 1.72 (m, 2H), 1.82 (dd, J = Hz, 1H), 1.89 (m, 2H), 2.03 (s, 3H), 2.34 (dd, J = 6.1, 12.8 Hz, 1H), 3.48 (s, 1H), 4.50 (m, 1H), 5.37 (s, 1H), 6.31 (m, 1H), 6.41 (d, J = 10.4 Hz, 1H), 6.60 (d, J = 10.4 Hz, 1H), 6.66 (br s, 1H), 7.34 (m, 1H), 7.38 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 14.1, 16.8, 17.0, 18.1, 20.2, 22.0, 23.1, 25.0, 28.6, 29.3, 36.4, 37.8, 39.0, 41.2, 45.3, 55.1, 66.5, 72.5, 76.5, 109.0, 112.4, 120.0, 140.3, 141.9, 146.1, 149.2, 158.8, 169.0. HRMS (*ESI + pos) (m/z): [M + H] calcd for C28H37N2O7, 513.2595; found, 513.2509. HPLC tR = 4.26 min. Purity = 100%.
Antiproliferative Assay
MCF-7 and SKBr3 cells were maintained in a 1:1 mixture of Advanced DMEM/F12 (Gibco) supplemented with nonessential amino acids, L-glutamine (2 mM), streptomycin (500 g/mL), penicillin (100 units/mL), and 10% FBS. Cells were grown to confluence in a humidified atmosphere (37°C, 5% CO2), seeded (2000/well, 100 μL) in 96-well plates, and allowed to attach overnight. Compound or GDA at varying concentrations in DMSO (1% DMSO final concentration) was added, and cells were returned to the incubator for 72 h. At 72 h, the number of viable cells was determined using an MTS/PMS cell proliferation kit (Promega) per the manufacturer’s instructions. Cells incubated in 1% DMSO were used as 100% proliferation, and values were adjusted accordingly. IC50 values were calculated from separate experiments performed in triplicate using GraphPad Prism.
Western Blot Assay
SKBr3 cells were maintained in a 1:1 mixture of Advanced DMEM/F12 (Gibco) supplemented with nonessential amino acids, L-glutamine (2 mM), streptomycin (500 g/mL), penicillin (100 units/mL), and 10% FBS. Cells were grown to confluence in a humidified atmosphere (37 °C, 5% CO2), seeded (1 000 000 per dish, 5 mL) in sterile culture dishes, and allowed to attach overnight. Compounds or GDA at varying concentrations in DMSO (1% DMSO final concentration) was added, and cells were returned to the incubator for 24 h. Cells were washed once with cold phosphate buffered saline (pH 7.0) and lysed by scraping in TMNS (50 mM Tris-HCl, pH 7.5, 20 mM Na2MoO4, 0.1% NP-40, 150 mM NaCl) supplemented with 20 μg/mL aprotinin, 20 μg/mL leupeptin, and 1 mM phenylmethanesulfonyl fluoride. Cell lysate was clarified by centrifugation at 14 000 rpm at 4 °C for 15 min, and protein concentration was determined by using the BCA method (Pierce, Rockford, IL). An amount of 20 μg of total protein from cell lysates was separated by 4–20% gradient SDS–PAGE (Bio-Rad, Hercules, CA). Western blotting for ErbB2 was performed as described previously. Blotting for actin was used to verify equal loading of lanes. Antibodies for R-tubulin and HER2 were from Calbiochem (La Jolla, CA). Antibodies for actin, HER2, and Raf were from Calbiochem (La Jolla, CA).
Supplementary Material
Scheme 3a.

a Reagents and conditions: (a) NMO, OsO4, acetone, H2O, 0 °C; (b) 1,2-dimethoxypropane, pTsOH, acetone; (c) 10% Pd/C, MeOH; (d) NaBH4, CeCl3, MeOH/CHCl3; (e) iPrOH, Al(OiPr)3, toluene, 70 °C; (f) acyl chloride, DMAP, Et3N, CH2Cl2; (g) NaBH4, EtOH; (h) alkoxyamine hydrochloride, pyridine, 70 °C.
Acknowledgments
This work described was supported by the University of Kansas. The authors also thank support from the American Foundation for Pharmaceutical Education (G.E.L.B. and M.D.S.) and the Madison and Lila Self Graduate Fellowship Program (G.E.L.B.).
Footnotes
Abbreviations: 17-AAG, 17-allylamino-17-demethoxygeldanamycin; ATP, adenosine triphosphate; 17-DMAG, 17-dimethylaminoethlamino-17-demethoxygeldanamycin; DCM, dichloromethane; GDA, geldanamycin; HER2, human estrogen receptor 2; HPLC, high performance liquid chromatography; Hsp90, 90 kDa heat shock protein; MCF-7, human breast adenocarcinoma cell line; MTS/PMS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, innersalt/phenazine methosulfate; Raf, mitogen-activated protein kinase kinase kinase; SKBr3,Sloan-Ketteringbreastcancercellline;TLC, thinlayerchromatography.
Supporting Information Available: 1H NMR, 13C NMR, and HPLC results of compounds 1–13. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Borst P, Jonkers J, Roftenberg S. What makes tumors multidrug resistant? Cell Cycle. 2007;6:2782–2787. doi: 10.4161/cc.6.22.4936. [DOI] [PubMed] [Google Scholar]
- 2.Nibili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- 3.Duesberg P, Li R, Sachs R, Fabarius A, Upender M, Hehlmann R. Cancer drug resistance: the central role of the karyotype. Drug Resist Updates. 2007;10:51–58. doi: 10.1016/j.drup.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Nooter K, Stoter G. Molecular mechanisms of multidrug resistance in cancer chemotherapy. Pathol, Res Pract. 1996;192:768–780. doi: 10.1016/S0344-0338(96)80099-9. [DOI] [PubMed] [Google Scholar]
- 5.Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–530. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 6.Xiao L, Lu X, Ruden DM. Effectiveness of Hsp90 inhibitors as anti-cancer drugs. Mini-Rev Med Chem. 2006;6:1137–1143. doi: 10.2174/138955706778560166. [DOI] [PubMed] [Google Scholar]
- 7.Blagg BSJ, Kerr TD. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med Res Rev. 2005;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 8.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer theapeutics. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 9.Maloney A, Workman P. Hsp90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 10.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by Hsp90/Hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 11.Pratt WB, Galigniana MD, Harrell JM, Defranco DB. Role of Hsp90 and the Hsp90-binding immunophilins in signaling protein movement. Cell. 2004;16:857–872. doi: 10.1016/j.cellsig.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 13.Khalid SA, Duddeck H, Gonzalez-Sierra M. Isolation and characterization of an antimalarial agent of the neem tree Azadirachta indica. J Nat Prod. 1989;52:922–927. doi: 10.1021/np50065a002. [DOI] [PubMed] [Google Scholar]
- 14.Nathan SS, Kalaivani K, Chung PG, Murugan K. Effect of neem limonoids on lactate dehydrogenase (LDH) of the rice leafholder, Cnaphalocrocis medinalis (Guenee) (Insecta: Lepidoptera: Pyralidae) Chemosphere. 2006;62:1388–1393. doi: 10.1016/j.chemosphere.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Uddin SJ, Nahar L, Shilpi JA, Shoeb M, Borkowski T, Gibbons S, Middleton M, Byres M, Sarker SD. Gedunin, a limonoid from Xylocarpus granatum, inhibits the growth of CaCo-2 colon cancer cell line in vitro. Phytother Res. 2007:757–761. doi: 10.1002/ptr.2159. [DOI] [PubMed] [Google Scholar]
- 16.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 17.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR. Gene expression signature-based chemical genomic prediction identifies a novel class of Hsp90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.