

Abstract
Background
Inflammatory bowel disease that begins prior to puberty frequently causes a delay in puberty resulting in losses of growth, bone mineralization, and self-esteem. A major cause of this pubertal delay is likely due in part to the effect of decreased levels of leptin on the function of the hypothalamic–pituitary–gonadal axis, though systemic inflammation is also thought to play a role.
Methods
To investigate further whether low leptin levels alone were responsible for delayed puberty in colitis, we induced colitis in 23-day-old female mice using 3% dextran sodium sulfate (DSS), resulting in 10 days of worsening colitis. These mice were compared to controls that were free-feeding and food-restricted (FR) mice that were given only enough food to keep them the same weight as the DSS group. All groups were followed for the timing of vaginal opening until 33 days old, when they were euthanized and their serum collected.
Results
DSS-treated mice exhibited later timing of vaginal opening relative to both of the other groups, as well as increased colonic inflammation by cytology and increased serum levels of interleukin (IL)-6 and tumor necrosis factor (TNF)-α. The difference in the timing of vaginal opening between the DSS and FR groups occurred despite equivalent serum levels of leptin between the groups and despite an increase in corticosterone in the FR group relative to both of the other groups.
Conclusions
We conclude that DSS colitis causes delay in puberty in sexually immature mice beyond what would be expected from decreases in weight and leptin levels.
Keywords: Colitis, Puberty, Leptin, Inflammation
Introduction
When its onset occurs prior to the onset of puberty, colitis seen as part of inflammatory bowel disease (IBD) frequently results in a delay in the timing of puberty [1, 2]. This is illustrated by one survey of adolescent girls with prepubertal onset of IBD revealing that 73% experienced menarche after their 16th birthday, compared to an average age of menarche in the general population of 12.5 years [3]. The delay in pubertal timing in IBD is important clinically because it can lead to a decrease in bone mineralization, loss of final height, and a loss of self-esteem among affected children [3-5]. The exact etiology of this pubertal delay in IBD has not yet been demonstrated.
One potential mechanism of this pubertal delay has been proposed to be a loss of body fat in IBD and resultant decrease in levels of leptin, a hormone that is released by adipocytes in proportion to the amount of stored fat [6]. Leptin is known to play a critical role in the regulation of the hypothalamic–pituitary–gonadal (HPG) axis, which is responsible for pubertal progression [7, 8]. Sufficient levels of leptin are necessary for the release of gonadotropin-releasing hormone (GnRH) from the hypothalamus, affecting the release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the pituitary, in turn affecting the release of estradiol from the ovaries [9-12]. These effects were first noted in ob/ob mice, which are unable to produce leptin and are infertile due to their subsequent lack of normal GnRH release [13-16]. However, the effect of leptin on HPG axis activity is also seen in other settings resulting in low leptin, such as abnormally low body weight [17, 18], revealing that when serum levels of leptin are low, this can lead to a decrease in HPG axis function and a delay in puberty [19-21].
Previous reports have shown that pubertal timing is delayed in rats that had colitis induced in prepubertal animals via intrarectal administration of 2,4,6-trinitrobenzenesulfonic acid (TNBS) [22]. These investigators also demonstrated that rats with colitis had a further delay in the timing of pubertal events when compared to a pair-fed group of rats that were given the same amount of food as the TNBS-treated group and that exhibited similar body weights. However, the previous researchers did not investigate whether differing levels of leptin between the colitis and pair-fed groups were responsible for the further delay in pubertal timing. Leptin levels among prepubertal animals with colitis have not previously been reported. Adult mice with colitis exhibit an initial increase in leptin levels at the onset of colitis before these levels drop related to weight loss [23].
We set out to examine whether delayed puberty in colitis could be explained solely by decreases in leptin, with a hypothesis that additional factors such as systemic inflammation are also likely to contribute to the delay in puberty. We report here results from a novel mouse model of prepubertal colitis, produced via administration of 3% dextran sodium sulfate (DSS) [24] in the drinking water of female mice starting at 23 days of age. We followed these mice for the timing of vaginal opening [22, 25]—a well-described feature of pubertal progression—as well as for serum levels of leptin and inflammatory cytokines, and colonic inflammation. We then compared the mice with DSS colitis to a group of free-feeding control mice and to food-restricted (FR) mice that were given only enough food to maintain the same weight as that seen in the DSS-treated group. We present evidence that mice with DSS colitis have a delay in puberty beyond FR mice despite equivalent levels of leptin, suggesting other factors such as inflammation are likely to play a significant role in delaying puberty in colitis.
Methods
These experiments were approved by the Animal Care and Use Committee at the University of Virginia. C57BL6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and were housed in standard wire-top cages and fed phytoestrogen-free chow. Two females and one male of childbearing age were housed together for breeding. Pups from litters of 6–10 mice were included in these experiments. Pups were weaned at 19 days of age by being removed from the mother and placed in a cage with moistened chow for 3 days before starting dry food exclusively.
Starting at day-of-life (DOL) 23, female mice were evenly divided based on starting weight into three groups: control, DSS colitis, and FR. DSS colitis mice had 3% DSS (mol wt 36 100–45 500; TdB Consultancy, Uppsala, Sweden) placed in the water supply from DOL 23–30 before DSS treatment was discontinued and animals were allowed to experience continued colitis from DOL 30–33. Control mice and FR mice received deionized water alone throughout the course of the experiment.
Both control and DSS mice were given phytoestrogen-free food ad libitum throughout the experiment. FR mice were given only enough food to maintain their weight at the same level as that in the DSS-treated group. This food administration for the FR group was done on an empirical basis each day for each mouse in the FR group, whose daily weight as a percent of baseline was compared to the DSS mean.
Vaginal opening was determined by daily inspection, using surgical loupes, and recorded as the day on which the vaginal orifice transitioned from tightly closed to patent [22, 25-29].
Animals were euthanized on DOL 33 via injection of standard mouse cocktail followed by cardiac puncture. Serum was stored at −80°C until the time of testing. Separate groups of control, DSS, and FR mice treated as described above were used to measure intermediate serum levels of leptin and cytokines. Serum samples were drawn from these mice on DOL 27 and DOL 30 and the mice were euthanized on DOL 30.
At the time of euthanizing, colons were removed and fixed in Bouin’s reagent prior to embedding in paraffin, sectioning, and staining. Slides from individual animals were read by a blinded pathologist (James Mize, MD, Anatomic and Clinical Pathology, Fairfax, VA) and scored for acute inflammatory index and chronic inflammatory index [30, 31].
Serum measurements
Serum cytokines were tested in the University of Virginia Digestive Health Research Center Core using a Beadlyte multiplex system (Upstate Cell Signaling Solutions, Temecula, CA, USA).
Serum hormone measurements were tested in the University of Virginia Center for Research in Reproduction. Estradiol measurement was performed using an Active Estradiol Coated Tube RIA (Beckman Coulter, Fullerton, CA, USA).
Luteinizing hormone (LH) was measured in serum by a modified supersensitive two-site sandwich immunoassay using monoclonal antibodies MAB1 (no. 581B7) against bovine LH (no. 5303: Medix, Kauniainen, Finland) against the human LH-beta subunit as described previously [32, 33]. The tracer antibody (no. 518B7 kindly provided by Dr. Janet Roser [34], Department of Animal Science, University of California, Davis), was iodinated by the chloramine T method and purified on Sephadex G-50 columns. The capture antibody (no. 5303) was biotinylated and immobilized on avidin-coated polystyrene beads (7 mm; Nichols Institute, San Juan Capistrano, CA, USA). Mouse LH reference preparation, provided by Dr. A.F. Parlow and the National Hormone and Peptide program was used as standard.
Follicle-stimulating hormone (FSH) measurements were determined by RIA using reagents provided by Dr. A.F. Parlow and the National Hormone and Peptide Program and procedures validated previously [35]. Mouse FSH reference preparation was used for assay standards and mouse FSH antiserum (guinea pig) AFP-1760191 diluted to a final concentration of 1:400 000 was used as primary antibody. The secondary antibody was purchased from Equitech-Bio (Kerrville, TX, USA) and was diluted to a final concentration of 1:25.
Corticosterone measurement was performed using a Corticosterone Coat-a-Count kit (Siemens Healthcare Diagnostics, Los Angeles, CA, USA).
Serum leptin measurement was performed using an enzyme-linked immunosorbent assay (ELISA) (Chrystal Chem, Downers Grove, IL, USA).
Statistics
Statistics were calculated using Prizm (GraphPad software, LaJolla, CA, USA) spreadsheet software and SAS (Cary, NC, USA). Comparisons of mean values between the three groups were performed using analysis of variance (ANOVA) with Bonferroni post-test comparison of all groups. Kaplan–Meier curves of timing of vaginal opening were analyzed via log-rank test [36].
Results
Weight gain, linear growth, and food intake
Prepubertal female mice receiving 3% DSS from DOL 23 to 30 exhibited a decrease in weight relative to controls starting at DOL 31 and continuing until mice were euthanized at DOL 33 (p < 0.01 for DOL 31 and p < 0.001 for DOL 32–33) (Fig. 1a). This decrease in weight relative to controls was matched in the FR group.
Fig. 1.
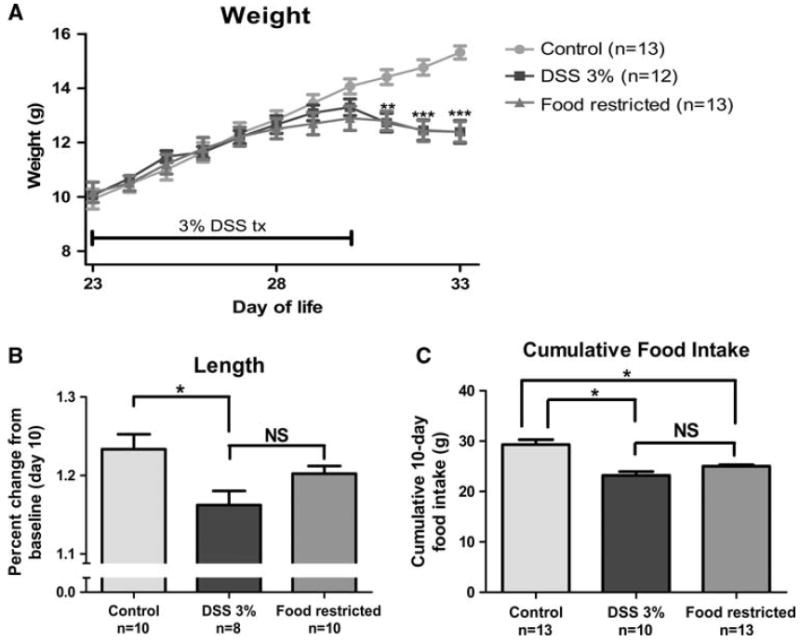
Weight, length, and food intake following colitis induction and food restriction. a Weight curve over the course of the experiment, day of life (DOL) 23–33. b Linear growth determined as follows: (length at DOL 33)/(length at DOL 23). c 10-Day cumulative food intake during experiment. *p < 0.05, **p < 0.01, ***p < 0.001, NS not significant (p > 0.05). DSS, dextran sodium sulfate; tx, treatment
Linear growth was affected for the DSS group relative to the control (p < 0.05) (Fig. 1b). Linear growth for the FR group was intermediate between that in the control and DSS groups, with no significant difference between the FR group and either of the other groups.
Food intake during the 10-day experiment was similar among the DSS and FR groups, and both groups had a decreased food intake relative to the control group (p < 0.05 for both groups) (Fig. 1c).
Colon histology
The DSS group exhibited an increase in the active inflammatory index score and the chronic inflammatory index score relative to both other groups (p < 0.001) (Fig. 2a–e).
Fig. 2.
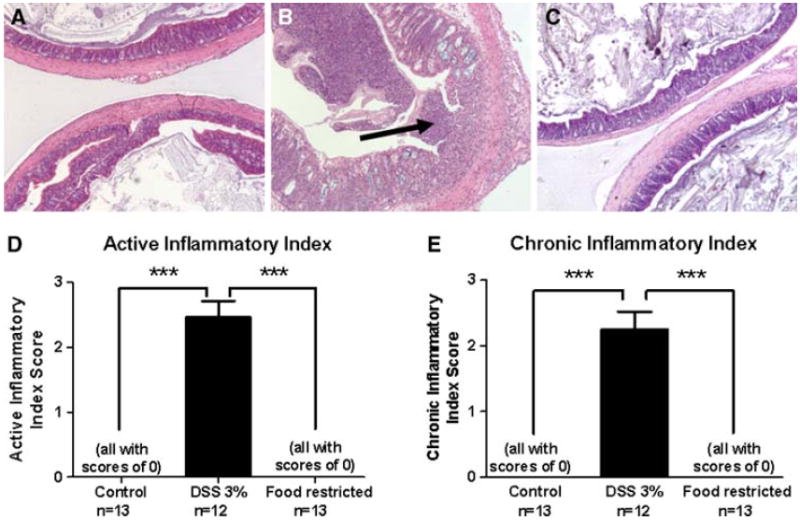
Colonic cytology, active inflammatory index, and chronic inflammatory index. Representative histological sections of colons from mice from the control. H&E, ×10 (a), DSS (b; arrow indicates ulceration of the mucosa. H&E, ×10), and food-restricted groups. H&E, 910 (c). d Active inflammatory index scores [35, 36]. e Chronic inflammatory index scores [35, 36]. ***p < 0.001
Cytokines
Starting by DOL 30, the DSS group had significant increases in the systemic inflammatory cytokines interleukin (IL)-6 (p < 0.01 vs control and p < 0.05 vs FR) (Fig. 3a) and tumor necrosis factor (TNF)-α (p < 0.05 vs control and p < 0.01 vs FR) (Fig. 3b). Absolute serum values for DOL 33 were as follows: IL-6—control 105.2 pg/mL ± 7.5, n = 12; DSS 621.9 pg/mL ± 190.7, n = 12; FR 181.9 pg/mL ± 49.1, n = 13; TNF-α—control 52.5 pg/mL ± 14.9, n = 12; DSS 102.8 pg/mL ± 30.8, n = 12; FR 53.5 pg/mL ± 18.2, n = 13.
Fig. 3.
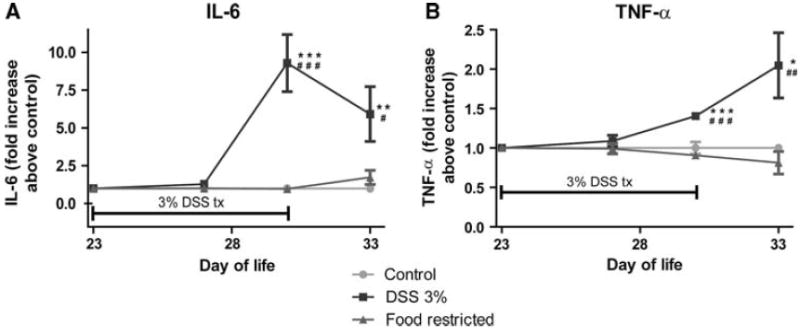
Systemic inflammation. Serum measurement of cytokines at baseline, mid-points (days of life 27 and 30), and the end of the experiment (day of life 33), expressed as -fold change above control, calculated on a per-assay basis. Mid-point values were performed in separate groups of animals under the same protocol. See text for absolute serum values. a Interleukin-6 (IL-6). b Tumor necrosis factor-α (TNF -α). *p < 0.05, **p < 0.01, NS not significant (p > 0.05)
There were no significant differences in levels of IL-1β or IL-10. Absolute serum values were as follows: IL-1β—control 144.5 pg/mL ± 22.7, n = 12; DSS 113.8 pg/mL ± 26.8, n = 12; FR 191.8 pg/mL ± 49.3, n = 13; IL-10—control 112.0 pg/mL ± 28.5, n = 9, DSS 171.4 pg/mL ± 52.9, n = 11; FR 311.4 pg/mL ± 49.9, n = 11).
Vaginal opening
The DSS group exhibited a delay in vaginal opening relative to both the control group and the FR group, as analyzed both by Kaplan–Meier curve plotting of the percentage of animals without vaginal opening (Fig. 4b, p < 0.001 relative to both groups) and by mean age of vaginal opening (Fig. 4a, p < 0.05 relative to both groups).
Fig. 4.
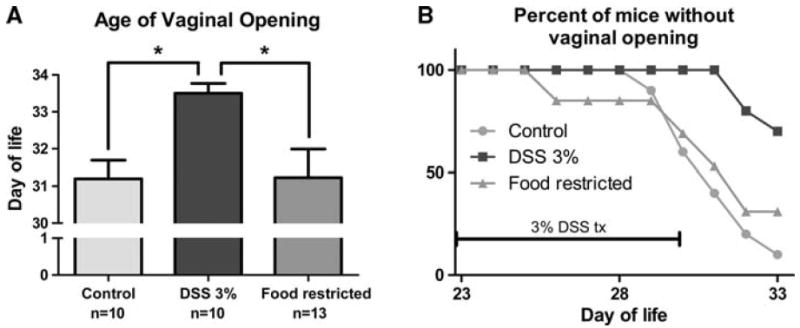
Timing of vaginal opening. a Age of vaginal opening as assessed by daily examination. Animals that had not experienced vaginal opening by the end of the experiment (day of life 33) were assigned a value of 34 days old. b Kaplan–Meier curve showing the proportion of mice without vaginal opening over time. DSS vs control and food-restricted p < 0.05 as determined by logrank test. *p < 0.05
Hormone levels
The prepubertal and peripubertal mice used in this protocol exhibited estradiol levels that were almost entirely at or below the limit of detection for the ultra-sensitive assay used, including in control animals. There were no significant differences in estradiol between the groups (Table 1). There were also no significant differences in levels of LH and FSH between the DSS group and either the control or FR groups (Table 1).
Table 1.
Assessment of hypothalamic–pituitary–gonadal axis
| Estradiol (pg/mL, limit of detection of assay 5.0 pg/mL) | LH (pg/mL, limit of detection of assay 0.01 ng/mL) | FSH (pg/mL, limit of detection of assay 2.0 ng/mL) | |
|---|---|---|---|
| Control | 6.37 ± 1.00, n = 12 | 0.349 ± 0.167, n = 15 | 6.31 ± 0.70, n = 11 |
| DSS colitis | 5.62 ± 0.63, n = 12 | 0.068 ± 0.018, n = 12 | 6.36 ± 0.53, n = 12 |
| Food-restricted | 5.55 ± 0.35, n = 13 | 0.248 ± 0.141, n = 13 | 7.46 ± 0.55, n = 12 |
Serum levels of estradiol, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) at the end of the experiment (day of life 33). Values shown are means ± standard error, with total number of samples. For calculating means, samples that yielded values below the limit of detection were given a value at that limit. None of the values listed were significantly different from each other (p > 0.05)
DSS dextran sodium sulfate
Leptin levels increased from baseline in all groups initially (p < 0.05), but were not different from baseline starting at DOL 30 for both the DSS and FR groups (Fig. 5a). By DOL 30, leptin levels were decreased in both the DSS and FR groups relative to controls. There were no differences in leptin levels between the DSS and FR groups at any time point. On DOL 33, corticosterone levels were increased in the FR group relative to either of the other groups (Fig. 5b, p < 0.01). There were no significant differences in corticosterone levels between the DSS and control groups.
Fig. 5.
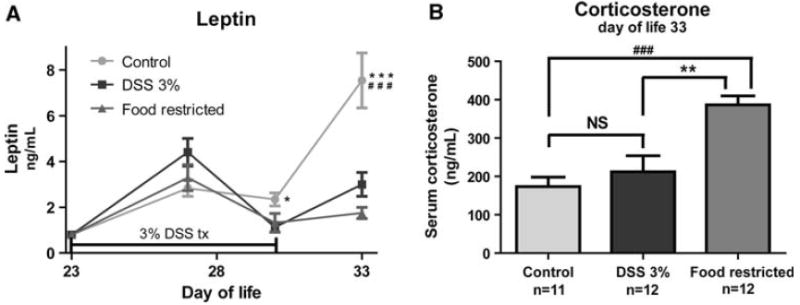
Serum levels of leptin and corticosterone. a Serum levels of leptin at baseline, mid-points (days of life 27 and 30), and the end of the experiment (day of life 33). Mid-point values were performed in separate groups of animals under the same protocol, with equal food-restricted and DSS animal weights. b Serum levels of corticosterone at the end of the experiment (day of life 33). *p < 0.05 vs DSS; **p < 0.01 vs DSS; ***p < 0.001 vs. DSS; ###p < 0.001 vs food-restricted, NS not significant (p > 0.05)
Discussion
We have developed a model of DSS colitis in sexually immature female mice that approximates the insidious onset of IBD in children with respect to a gradual onset of decreasing weight gain and increasing systemic inflammation. We have used this model to demonstrate delayed vaginal opening among mice with DSS colitis vs an FR group with similar body weight. Moreover, the DSS colitis group and FR groups had similar serum levels of leptin at three time points measured during the 10-day treatment course, indicating that the difference in timing of puberty between these groups was not due to the DSS mice having a lower level of leptin. To our knowledge these experiments are the first evaluation of leptin levels in peripubertal mice with colitis and demonstrate that in the later stages of colitis these animals do not exhibit differences in leptin levels beyond what would be expected from restricted body weight alone.
These findings are significant because of the well-documented effects that low leptin can have on the function of the HPG axis and the timing of puberty. These effects are seen among ob/ob mice that are infertile because of their absence of leptin [13, 14]. Clinically the importance of leptin for puberty is seen among young women with low body mass due to intense exercise or anorexia nervosa [17, 18]. These young women have low levels of leptin and exhibit delayed puberty similar to that seen among children with IBD [20, 21]. Studies have demonstrated that among postpubertal women with anorexia, increases in leptin levels following weight gain appear necessary prior to the resumption of menses [17, 37], and treatment with exogenous leptin in the absence of weight gain can induce normal menses [38]. Prepubertal girls with IBD also exhibit low levels of leptin similar to weight-matched controls [39], and while no studies have been performed linking low leptin levels to delayed puberty in colitis, leptin is felt to be a major contributor to pubertal delay in IBD [40].
It was certainly conceivable that leptin levels in advanced colitis could be lower than those in weight-matched controls. Though leptin production is initially stimulated by inflammation in both humans [41] and rodent models of colitis [23], these levels decrease with time. We have demonstrated that the decrease in leptin with continued colitis does not drop leptin levels below those of weight-matched controls as a cause of the further delay in puberty seen during colitis. Also, while the DSS colitis group exhibited a modest trend toward increased leptin levels during the early stages of colitis (p = 0.1 vs controls), this increase in leptin clearly did not induce an earlier timing of puberty, further demonstrating a restraint on puberty by other factors.
Other potential contributors to delayed puberty in the setting of colitis include decreased food intake, increased stress response, and systemic inflammation, which will be considered individually below. Regarding the effect of food intake, we empirically restricted food intake among the FR group without specifically aiming to give the same amount of food to the FR group as was consumed by the DSS group. Nevertheless, the DSS and FR groups were found to have similar food intake, indicating that the majority of the cause of poor weight gain for the DSS group was due to a decrease in food intake. This finding parallels that seen in the prior experiment of TNBS colitis among prepubertal rats, in which the investigators pair-fed their weight-restricted group [22]. In these experiments, then, the two groups of animals ate exactly the same amount of food and were also found to be of nearly identical weights. Taken together, these experiments suggest that the major cause of weight loss in colitis is a decrease in food intake and not poor food absorption or increased basal metabolic rate. Given that the DSS and FR groups did not exhibit a difference in food intake, altered nutrition does not account for the difference in timing of their pubertal onset.
Another potential cause of delayed puberty in colitis is the stress involved in chronic illness. To assess for whether stimulation of the hypothalamic–pituitary–adrenal (HPA) axis was the main explanation of pubertal delay in our mice, we measured levels of corticosterone, the major adrenal corticosteroid in mice. While corticosterone levels have been shown to be initially elevated in colitis, they moderate over time [42], potentially due to the long-term nature of stress in the setting of colitis. This phenomenon has been described in patients in the intensive care unit in whom the HPA axis is paradoxically downregulated after prolonged stress [43]. Additionally, other investigators have reported corticosterone levels that were similar between mice with DSS colitis and controls [44]. Consistent with these findings, we observed similar levels of corticosterone between mice with DSS colitis and controls. By contrast, the FR animals exhibited a pronounced increase in levels of corticosterone, consistent with prior studies of stress among starved animals [42]. Taken together, we conclude that activation of the HPA axis resulting in corticosterone stimulation does not appear to be a major influence on the timing of puberty in this setting, since both unstressed controls and stressed FR animals exhibited an advanced pubertal timing relative to DSS animals.
Finally, another factor potentially related to the delay in puberty is related to the systemic inflammation observed during colitis. Mice with DSS colitis exhibited increases in both serum IL-6 and TNF-α relative to both other groups, starting by day 7 of colitis treatment. This raises a potential cause of pubertal delay, given that peripheral inflammation has been shown to have a negative effect on HPG axis activation in other settings [45]. Nevertheless, other factors may also predominate and further experiments are necessary.
Whatever the cause of this pubertal delay among the DSS mice relative to controls or FR mice, the effect appears to have been at the level of the hypothalamus and/or pituitary and not at the level of the ovary. This is seen in that mice with DSS colitis had a delay of vaginal opening, which has been validated as a characteristic driven by minor changes in estrogen during development [22, 26-29]. We were unable to demonstrate these changes in estradiol production via serum levels because of the low levels of estradiol needed to produce vaginal opening. In our experiments even the control mice had levels of estradiol near the limit of detection in the assay, reflecting the early nature of this testing in the course of their pubertal progression. However, in previous investigations using TNBS colitis in rats, investigators found lower levels of estradiol among the rats with colitis vs controls, in addition to a delay in vaginal opening [22]. Estrogen production is driven by the pituitary gonadotropins LH and FSH, and in the absence of normal ovarian function, LH and FSH levels increase due to a lack of feedback inhibition. However, in the absence of elevated LH and FSH, low levels of estrogen are due to a decrease in activation of gonadotropins [46]. This is the process that both we and the prior investigators noted, since LH and FSH in the DSS-treated mice were similar to the levels in the control group even in the face of apparently low estradiol levels. Should estrogen production at the level of the ovary have been the main site of the inflammatory effect, we would have expected LH and FSH to have been elevated in the DSS-treated mice as a result of negative feedback, as has been seen in similarly aged mice following oophorectomy [47]. The lack of such elevation thus suggests the effect was upstream of estrogen production, at the level of GnRH or LH/FSH regulation.
The implication of this study clinically is that therapy aimed to treat the underlying IBD may result in improvements in the timing of puberty and related endpoints beyond what would be expected from weight gain alone. Many such treatments for IBD, such as infliximab and adalimumab, target bowel and systemic inflammation. Further experiments will be needed to test for potential connections between colitis-related inflammation and HPG axis function.
Acknowledgments
We would like to thank Matt Gurka, Ph.D., for his assistance with statistical evaluation in these experiments. This work was supported by NIH HD060739-01 (to M.D.D.) and a pilot-feasibility grant (to M.D.D.) and the Immunology/Cell Isolation Core of the UVa Silvio O. Conte Digestive Health Research Center NIH P30 DKO67629. Reproductive hormone levels were measured by the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core U54-HD28934.
Contributor Information
Mark D. DeBoer, Division of Pediatric Endocrinology, Department of Pediatrics, University of Virginia, P.O. Box 800386, Charlottesville, VA 22908, USA, deboer@virginia.edu
Yongli Li, Division of Pediatric Endocrinology, Department of Pediatrics, University of Virginia, P.O. Box 800386, Charlottesville, VA 22908, USA.
Steven Cohn, Department of Medicine, University of Virginia Digestive, Health Research Center, Charlottesville, VA, USA.
References
- 1.Ballinger AB, Savage MO, Sanderson IR. Delayed puberty associated with inflammatory bowel disease. Pediatr Res. 2003;53(2):205–10. doi: 10.1203/01.PDR.0000047510.65483.C9. [DOI] [PubMed] [Google Scholar]
- 2.Brain CE, Savage MO. Growth and puberty in chronic inflammatory bowel disease. Baillieres Clin Gastroenterol. 1994;8(1):83–100. doi: 10.1016/s0950-3528(06)80020-5. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson A, Sedgwick DM. Juvenile onset inflammatory bowel disease: height and body mass index in adult life. BMJ. 1994;308(6939):1259–63. doi: 10.1136/bmj.308.6939.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Otley AR, Griffiths AM, Hale S, Kugathasan S, Pfefferkorn M, Mezoff A, et al. Health-related quality of life in the first year after a diagnosis of pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2006;12(8):684–91. doi: 10.1097/00054725-200608000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Paganelli M, Albanese C, Borrelli O, Civitelli F, Canitano N, Viola F, et al. Inflammation is the main determinant of low bone mineral density in pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(4):416–23. doi: 10.1002/ibd.20039. [DOI] [PubMed] [Google Scholar]
- 6.Simon D. Puberty in chronically diseased patients. Horm Res. 2002;57(suppl 2):53–6. doi: 10.1159/000058102. [DOI] [PubMed] [Google Scholar]
- 7.Bluher S, Mantzoros CS. Leptin in reproduction. Curr Opin Endocrinol Diabetes Obes. 2007;14(6):458–64. doi: 10.1097/MED.0b013e3282f1cfdc. [DOI] [PubMed] [Google Scholar]
- 8.Chan JL, Matarese G, Shetty GK, Raciti P, Kelesidis I, Aufiero D, et al. Differential regulation of metabolic, neuroendocrine, and immune function by leptin in humans. Proc Natl Acad Sci USA. 2006;103(22):8481–6. doi: 10.1073/pnas.0505429103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahima RS, Dushay J, Flier SN, Prabakaran D, Flier JS. Leptin accelerates the onset of puberty in normal female mice. J Clin Invest. 1997;99(3):391–5. doi: 10.1172/JCI119172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caprio M, Fabbrini E, Isidori AM, Aversa A, Fabbri A. Leptin in reproduction. Trends Endocrinol Metab. 2001;12(2):65–72. doi: 10.1016/s1043-2760(00)00352-0. [DOI] [PubMed] [Google Scholar]
- 11.Cheung CC, Thornton JE, Kuijper JL, Weigle DS, Clifton DK, Steiner RA. Leptin is a metabolic gate for the onset of puberty in the female rat. Endocrinology. 1997;138(2):855–8. doi: 10.1210/endo.138.2.5054. [DOI] [PubMed] [Google Scholar]
- 12.Luque RM, Kineman RD, Tena-Sempere M. Regulation of hypothalamic expression of KiSS-1 and GPR54 genes by metabolic factors: analyses using mouse models and a cell line. Endocrinology. 2007;148(10):4601–11. doi: 10.1210/en.2007-0500. [DOI] [PubMed] [Google Scholar]
- 13.Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12(3):318–20. doi: 10.1038/ng0396-318. [DOI] [PubMed] [Google Scholar]
- 14.Smith JT, Acohido BV, Clifton DK, Steiner RA. KiSS-1 neurones are direct targets for leptin in the ob/ob mouse. J Neuroendocrinol. 2006;18(4):298–303. doi: 10.1111/j.1365-2826.2006.01417.x. [DOI] [PubMed] [Google Scholar]
- 15.Kawa S, Kumar A, Smith JS, Becerra SP, Beard WA, Wilson SH, et al. Expression and purification of the HIV-1 reverse transcriptase using the baculovirus expression vector system. Protein Expr Purif. 1993;4(4):298–303. doi: 10.1006/prep.1993.1038. [DOI] [PubMed] [Google Scholar]
- 16.Farooqi IS. Leptin and the onset of puberty: insights from rodent and human genetics. Semin Reprod Med. 2002;20(2):139–44. doi: 10.1055/s-2002-32505. [DOI] [PubMed] [Google Scholar]
- 17.Audi L, Mantzoros CS, Vidal-Puig A, Vargas D, Gussinye M, Carrascosa A. Leptin in relation to resumption of menses in women with anorexia nervosa. Mol Psychiatry. 1998;3(6):544–7. doi: 10.1038/sj.mp.4000418. [DOI] [PubMed] [Google Scholar]
- 18.Matejek N, Weimann E, Witzel C, Molenkamp G, Schwidergall S, Bohles H. Hypoleptinaemia in patients with anorexia nervosa and in elite gymnasts with anorexia athletica. Int J Sports Med. 1999;20(7):451–6. doi: 10.1055/s-1999-8834. [DOI] [PubMed] [Google Scholar]
- 19.Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382(6588):250–2. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 20.Misra M, Aggarwal A, Miller KK, Almazan C, Worley M, Soyka LA, et al. Effects of anorexia nervosa on clinical, hematologic, biochemical, and bone density parameters in community-dwelling adolescent girls. Pediatrics. 2004;114(6):1574–83. doi: 10.1542/peds.2004-0540. [DOI] [PubMed] [Google Scholar]
- 21.Misra M, Miller KK, Almazan C, Ramaswamy K, Aggarwal A, Herzog DB, et al. Hormonal and body composition predictors of soluble leptin receptor, leptin, and free leptin index in adolescent girls with anorexia nervosa and controls and relation to insulin sensitivity. J Clin Endocrinol Metab. 2004;89(7):3486–95. doi: 10.1210/jc.2003-032251. [DOI] [PubMed] [Google Scholar]
- 22.Azooz OG, Farthing MJ, Savage MO, Ballinger AB. Delayed puberty and response to testosterone in a rat model of colitis. Am J Physiol Regul Integr Comp Physiol. 2001;281(5):R1483–91. doi: 10.1152/ajpregu.2001.281.5.R1483. [DOI] [PubMed] [Google Scholar]
- 23.Barbier M, Cherbut C, Aube AC, Blottiere HM, Galmiche JP. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut. 1998;43(6):783–90. doi: 10.1136/gut.43.6.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2(3):541–6. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 25.Nelson JF, Karelus K, Felicio LS, Johnson TE. Genetic influences on the timing of puberty in mice. Biol Reprod. 1990;42(4):649–55. doi: 10.1095/biolreprod42.4.649. [DOI] [PubMed] [Google Scholar]
- 26.Brill DS, Moenter SM. Androgen receptor antagonism and an insulin sensitizer block the advancement of vaginal opening by high-fat diet in mice. Biol Reprod. 2009;81(6):1093–8. doi: 10.1095/biolreprod.109.079301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thigpen JE, Setchell KD, Padilla-Banks E, Haseman JK, Saunders HE, Caviness GF, et al. Variations in phytoestrogen content between different mill dates of the same diet produces significant differences in the time of vaginal opening in CD-1 mice and F344 rats but not in CD Sprague-Dawley rats. Environ Health Perspect. 2007;115(12):1717–26. doi: 10.1289/ehp.10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Zhu W, Guo Z, Zhao Y, Song Z, Xiao J. Effects of maternal nuclear genome on the timing of puberty in mice offspring. J Endocrinol. 2007;193(3):405–12. doi: 10.1677/joe.1.07049. [DOI] [PubMed] [Google Scholar]
- 29.Nathan BM, Hodges CA, Supelak PJ, Burrage LC, Nadeau JH, Palmert MR. A quantitative trait locus on chromosome 6 regulates the onset of puberty in mice. Endocrinology. 2006;147(11):5132–8. doi: 10.1210/en.2006-0745. [DOI] [PubMed] [Google Scholar]
- 30.Burns RC, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Ley K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology. 2001;121(6):1428–36. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 31.Marini M, Bamias G, Rivera-Nieves J, Moskaluk CA, Hoang SB, Ross WG, et al. TNF-alpha neutralization ameliorates the severity of murine Crohn’s-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc Natl Acad Sci USA. 2003;100(14):8366–71. doi: 10.1073/pnas.1432897100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fallest PC, Trader GL, Darrow JM, Shupnik MA. Regulation of rat luteinizing hormone beta gene expression in transgenic mice by steroids and a gonadotropin-releasing hormone antagonist. Biol Reprod. 1995;53(1):103–9. doi: 10.1095/biolreprod53.1.103. [DOI] [PubMed] [Google Scholar]
- 33.Haavisto AM, Pettersson K, Bergendahl M, Perheentupa A, Roser JF, Huhtaniemi I. A supersensitive immunofluorometric assay for rat luteinizing hormone. Endocrinology. 1993;132(4):1687–91. doi: 10.1210/endo.132.4.8462469. [DOI] [PubMed] [Google Scholar]
- 34.Matteri RL, Roser JF, Baldwin DM, Lipovetsky V, Papkoff H. Characterization of a monoclonal antibody which detects luteinizing hormone from diverse mammalian species. Domest Anim Endocrinol. 1987;4(3):157–65. doi: 10.1016/0739-7240(87)90011-7. [DOI] [PubMed] [Google Scholar]
- 35.Gay VL, Midgley AR, Jr, Niswender GD. Patterns of gonadotrophin secretion associated with ovulation. Fed Proc. 1970;29(6):1880–7. [PubMed] [Google Scholar]
- 36.Bland JM, Altman DG. The logrank test. BMJ. 2004;328(7447):1073. doi: 10.1136/bmj.328.7447.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swenne I. Weight requirements for return of menstruations in teenage girls with eating disorders, weight loss and secondary amenorrhoea. Acta Paediatr. 2004;93(11):1449–55. doi: 10.1080/08035250410033303. [DOI] [PubMed] [Google Scholar]
- 38.Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351(10):987–97. doi: 10.1056/NEJMoa040388. [DOI] [PubMed] [Google Scholar]
- 39.Hoppin AG, Kaplan LM, Zurakowski D, Leichtner AM, Bousvaros A. Serum leptin in children and young adults with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1998;26(5):500–5. doi: 10.1097/00005176-199805000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Ballinger A. Divergency of leptin response in intestinal inflammation. Gut. 1999;44(5):588–9. doi: 10.1136/gut.44.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zumbach MS, Boehme MW, Wahl P, Stremmel W, Ziegler R, Nawroth PP. Tumor necrosis factor increases serum leptin levels in humans. J Clin Endocrinol Metab. 1997;82(12):4080–2. doi: 10.1210/jcem.82.12.4408. [DOI] [PubMed] [Google Scholar]
- 42.Kojima K, Naruse Y, Iijima N, Wakabayashi N, Mitsufuji S, Ibata Y, et al. HPA-axis responses during experimental colitis in the rat. Am J Physiol Regul Integr Comp Physiol. 2002;282(5):R1348–55. doi: 10.1152/ajpregu.00260.2001. [DOI] [PubMed] [Google Scholar]
- 43.Menon K, Clarson C. Adrenal function in pediatric critical illness. Pediatr Crit Care Med. 2002;3(2):112–6. doi: 10.1097/00130478-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 44.Reber SO, Obermeier F, Straub RH, Falk W, Neumann ID. Chronic intermittent psychosocial stress (social defeat/overcrowding) in mice increases the severity of an acute DSS-induced colitis and impairs regeneration. Endocrinology. 2006;147(10):4968–76. doi: 10.1210/en.2006-0347. [DOI] [PubMed] [Google Scholar]
- 45.Nappi RE, Rivest S. Effect of immune and metabolic challenges on the luteinizing hormone-releasing hormone neuronal system in cycling female rats: an evaluation at the transcriptional level. Endocrinology. 1997;138(4):1374–84. doi: 10.1210/endo.138.4.5044. [DOI] [PubMed] [Google Scholar]
- 46.Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357(9):863–73. doi: 10.1056/NEJMoa066494. [DOI] [PubMed] [Google Scholar]
- 47.Darney KJ, Jr, Goldman JM, Vandenbergh JG. Neuroendocrine responses to social regulation of puberty in the female house mouse. Neuroendocrinology. 1992;55(4):434–43. doi: 10.1159/000126155. [DOI] [PubMed] [Google Scholar]