

Abstract
Objective
Ibuprofen has been shown to reduce cerebral ischemic injury such as may occur after deep hypothermic circulatory arrest. We investigated whether ibuprofen has direct protective effects against excitotoxic neuronal injury as may be seen after cerebral ischemia using a cell culture model.
Methods
Mixed cortical cultures containing neuronal and glial cells were prepared from fetal mice at 13–15d gestation, plated on a layer of confluent astrocytes from 1–3d postnatal pups. Near-pure neuronal cultures containing <5% astrocytes were obtained from same gestational stage mice. Slow triggered excitotoxic injury was induced at 37°C by 24 hour exposure to 12.5μM N-methyl-D-aspartate (NMDA) or 50μM kainate. Neuronal death was quantified by release of lactate dehydrogenase from damaged cells. Data were analyzed using one-way ANOVA with Tukey post-hoc multiple comparisons.
Results
In mixed cultures, ibuprofen concentrations of 25, 50 and 100 μg/ml all significantly reduced NMDA induced neuronal cell death from 74.5% to 56.1%, 38.7% and 12.3%, respectively, revealing a strong dose-response (P < .001). In near-pure cultures, ibuprofen at a concentration of 25 μg/ml failed to protect neurons, indicating that the neuroprotective effects of ibuprofen require interaction with glial cells. Furthermore ibuprofen at 100μg/ml was also not protective against neuronal cell death induced by kainate exitotoxicity in near-pure culture but was effective in mixed cultures.
Conclusion
Ibuprofen provides neuroprotection through glial cells against excitotoxic neuronal injury due to glutamatergic excitotoxicity after cerebral ischemia as demonstrated by reduced neuronal cell death in mixed cell cultures. Further studies are needed to evaluate the potential of ibuprofen to reduce neurological injury in patients suffering an hypoxic/ischemic insult.
Introduction
Low flow cardiopulmonary bypass (CPB) and deep hypothermic circulatory arrest (DHCA) are used to facilitate pediatric cardiac surgery although they carry a risk of ischemic brain damage. There are also deleterious effects of CPB such as inflammation, which may exacerbate ischemic brain injury. Ibuprofen is a non-steroidal anti-inflammatory drug (NSAID) and is widely used to reduce pain, fever and inflammation. The drug inhibits cyclooxygenases and activates peroxisome proliferators-activated receptors (PPARs); both of these actions result in reduced inflammation [1–3]. Ibuprofen has been shown to reduce cerebral ischemic injury and infarct size such as may occur after deep hypothermic circulatory arrest [4, 5]. Also Ibuprofen has been reported to improve cerebral blood flow after global cerebral ischemia and to restore cerebral metabolism after cerebral traumatic injury [6, 7].
It is well known that ischemia can cause neuronal cell death through the mechanism of glutamatergic excitotoxcity. Initially an ischemic insult impairs glutamate transportation at the postsynaptic level and increases extracellular glutamate levels. Consequently it results in glutamatergic excitotoxicity, which is excessive neuronal excitation mediated by excessive calcium influx. Thus neurological problems after cardiac surgery, such as may occur with circulatory arrest, embolic injury or low flow states, may be a result of excitotoxicity after cerebral ischemia.
Glutamate is the principal neurotransmitter at the majority of synapses throughout the brain and spinal cord. N-methyl-D-aspartate (NMDA), AMPA and kainate are glutamate receptor subtypes defined by their response to these agents; the NMDA pathway causes neuronal injury more rapidly than the AMPA or kainate pathway reflecting a greater ability to induce calcium influx. Rapidly triggered excitotoxicity can be separated into two components: (A) immediate neuronal cell swelling induced by the influx of extracellular Na; and (B) delayed neuronal cell degeneration induced by excessive Ca influx. AMPA, kainate and NMDA receptors all contribute to the neuronal cell swelling seen with excitotoxic injury. In contrast, NMDA receptor activation is necessary and sufficient to induce delayed neuronal cell degeneration. Although either the acute cell swelling or delayed neuronal cell degeneration can alone produce irreversible neuronal injury, under certain conditions the majority of cells can recover and survive from cell swelling. Generally most cortical neurons die through delayed neuronal degeneration [8]. The purpose of the current study was to investigate whether Ibuprofen has direct neuroprotective effects against excitotoxicic neuronal cell death induced by NMDA and kainate using a neuronal cell culture model.
Methods
Near-Pure Neuronal Cultures
Near-pure neuronal cultures containing less than 5% astrocytes were obtained from fetal mice at 13–15d gestation (Taconic, Rockville, MD). Dissociated cortical cells in plating medium of media stock (MS) (Dulbecco’s modified Eagle’s medium with 25 mM glucose; SIGMA, St. Louis, MO) supplemented with 5% fetal bovine serum (GIBCO, Carlsbad, CA), 5% horse serum (GIBCO, Carlsbad, CA) and 2 mM glutamine (SIGMA, St. Louis, MI) were plated in 24-well plates coated with poly-D-lysine (0.1 mg/dl; Invitrogen, Carlsbad, CA) and laminin (0.02 mg/ml; Invitrogen, Carlsbad, CA). After 3 days in vitro (DIV), non-neuronal cell division was halted by exposure to 10 μM cytosine arabinoside (Ara-C; SIGMA, St. Louis, MO). There was no further exchange of the media except adding DMEM for evaporation. After 12 DIV, cultures did not need the presence of serum to survive. They were shifted to a maintenance medium identical to plating medium but lacking serum. All cultures were kept at 37°C in a humidified 5% CO2 incubator. Cultures were used after 13–14 DIV for excitotoxic injury [9].
Glial cultures
Glial cell cultures were prepared from 1–3 day old postnatal mice (1–3 days after birth from Taconic, Rockville, MD). Dissociated cortical cells were plated in 24-well plates previously coated with poly-D-lysine (0.1 mg/dl) using a plating medium of MS supplemented with 10% horse serum, 10% fetal bovine serum and 2 mM glutamine. Cultures were kept at 37°C in a humidified 5% CO2-containing atmosphere until they reached confluency, 7–14 DIV. Confluent cultures were then used as a support for mixed cultures [9].
Mixed Cortical Cultures
Mixed cortical cultures containing both neurons and astrocytes were prepared from fetal mice at 13–15 d gestation. Dissociated cortical cells were plated in 24 wells on a layer of confluent astrocytes, using plating medium supplemented with 5% horse serum, 5% fetal bovine serum and 2 mM glutamine. After 7 DIV, non-neuronal cell division was halted by a 2–3 days exposure to 10 μM Ara-C. Subsequent partial medium replacement was performed twice per week, and after 12 DIV, cultures were shifted to a maintenance medium identical to plating medium though lacking serum since neurons survive without it. Experiments were performed on cortical cultures after 13–14 DIV [9].
Slowly triggered excitotoxicity
Since lower levels of glutamate exposure induce delayed neuronal cell death in most cortical neurons, slowly triggered excitotoxicity was induced at 37°C by 24 hr exposure to 12.5 μM NMDA or 50 μM kainate as an excitotoxin in MS supplemented with 10 μM glycine. These concentrations do not influence glial cells. 10 μM MK-801 was always added concurrently with kainate to block secondary NMDA receptor activation [9]. Ibuprofen was co-applied at three different concentrations, 25, 50 or 100 μg/ml, with the excitotoxin and left for 24 hours in the bathing medium. A clinically effective concentration of ibuprofen is between 25 and 100 μg/ml since the inhibitory effect of ibuprofen on neutrophil activation and migration generally occurs at concentrations over 50 μg/ml [10].
Assessment of Neuronal Cell Death
The % neuronal cell death was calculated by lactate dehydrogenase (LDH) assay which is a standard method that is very widely applied for quantitation of cell death in neuronal cell culture[8, 9, 11]. Briefly, neuronal death was confirmed qualitatively by examining cultures under phase-contrast microscopy and was quantified by measurement of LDH release from damaged cells into the bathing medium 24 hours after the onset of excitotoxin exposure. Only damaged cells (dead neurons) release LDH. LDH was measured by ELISA kit (Promega, Madison, WI). The % neuronal cell death was calculated by LDH levels in the bathing medium. The LDH level corresponding to complete neuronal death (without glial death) was determined in sister cultures exposed to 100 μM NMDA. Background LDH levels were determined in sister cultures with sham wash as a control and subtracted from experimental values to yield the signal specific for experimentally induced injury [11].
Statistical Analysis
The primary outcome measure for evaluating efficacy was % neuronal cell death calculated by LDH levels. Results are expressed as mean ± standard error. When n = 12 is indicated, this value corresponds to 12 different well pools derived from three different dissections. Analysis of these data included factorial one-way analysis of variance (ANOVA) with the Tukey post-hoc method for multiple group comparisons to protect against Type I (false positive) results [12]. The SPSS software package was used for statistical analysis (version 15.1, SPSS Inc., Chicago, IL). Two-tailed values of P < .05 were considered statistically significant, with Tukey correction as appropriate. Power analysis indicated that the sample sizes provided 80% power to detect a significant difference in neuronal cell death of 15% or more between treatment groups assuming a standard deviation of 5–10% (version 7.0, nQuery Advisor, Statistical Solutions, Saugus, MA).
Results
Sham wash and addition of Ibuprofen alone at the concentration of 100 μg/ml did not influence neuronal cells in either mixed or pure neuronal culture (Figure 1a, b). Excitotoxicity was induced by exposure to 12.5 μM NMDA or 50 μM Kainate for 24 hours and demonstrated an acute swelling of neuronal cell bodies followed by widespread necrotic neuronal degeneration resulting in disrupted neurons and segmentalized neurites in both near-pure and mixed cultures (Figure 1c). Ibuprofen at a concentration of 100 μg/ml was co-applied in the medium with excitotoxin (NMDA). Ibuprofen was able to preserve neuronal cells including neurites in both cultures (Figure 1d). Percent neuronal cell death induced by exposure to 12.5 μM NMDA revealed about 70% neuronal cell death by LDH assay measuring LDH levels in bathing medium in both cultures. Ibuprofen at a concentration of 25, 50 and 100 μg/ml reduced % neuronal cell death significantly from 74.5±3.8% to 56.1±4.0%, 38.7±2.8% and 12.3±1.4% in mixed cultures, respectively, revealing a strong dose-response (P < .001). However low concentration Ibuprofen failed to protect neurons in near-pure cultures (Figure 2a, b). % neuronal cell death induced by exposure to 50 μM kainate revealed about 30% neuronal cell death. Ibuprofen also reduced % neuronal cell death induced by exposure to 50 μM kainate from 36.1±3.5% to 20.0±3.1% in mixed cultures but failed in near-pure cultures (Figure 3a, b). These results suggest that Ibuprofen may reduce glutamatergic excitotoxic neuronal cell death induced by NMDA or Kainate, resulting from ischemia, although it may require interaction with glial cells.
Figure 1.




(A): Sham wash (Mixed cortical cultures), (B) : Ibuprofen (100 μg/ml) alone, (C) : Mixed cortical cultures were exposed to 12.5 μM NMDA for 24 hours and demonstrated an acute swelling of neuronal cell bodies followed by a widespread necrotic neuronal degeneration resulting in disrupted neurons and segmentalized neuritis, (D) : Ibuprofen at a concentration of 100 μg/ml was co-applied in the medium with excitotoxin (NMDA). Ibuprofen preserved neuronal cells and protected neurites. Original magnification is X200. Scale bars indicated 100 μm.
NMDA : N-methyl-D-aspartate, Ib : Ibuprofen.
Figure 2.
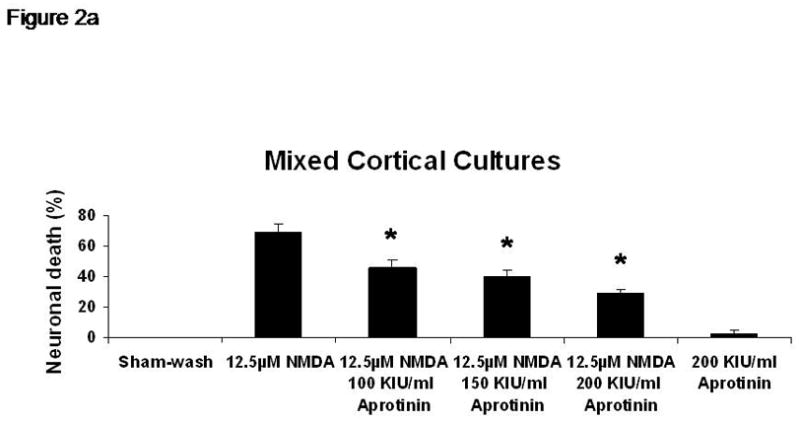
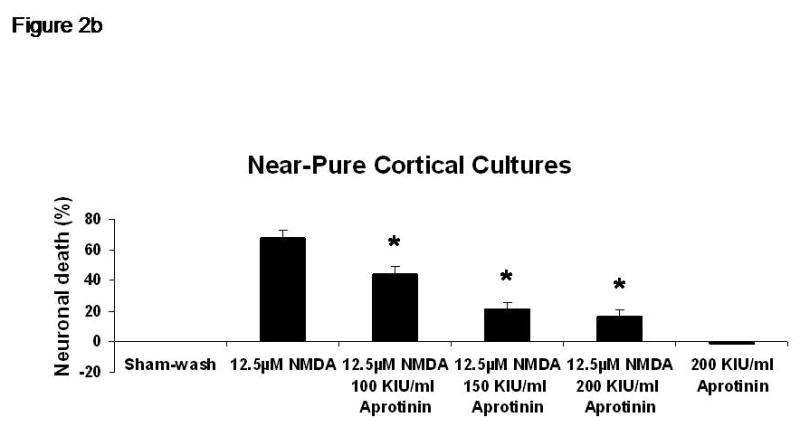
% neuronal cell death induced by exposure to 12.5 μM NMDA revealed about 70% neuronal cell death by LDH assay measuring LDH levels in bathing medium in both cultures. Ibuprofen at a concentration of 25, 50 and 100 μg/ml reduced % neuronal cell death significantly from 74.5±3.8% to 56.1±4.0%, 38.7±2.8% and 12.3±1.4% in mixed cultures, respectively, revealing a strong dose-response (P < .001). However low concentration Ibuprofen failed to protect neurons in near-pure cultures.
A: mixed cultures, B: near pure cultures. * indicates significantly different from NMDA alone by ANOVA with Tukey correction (mean ± SE).
Figure 3.
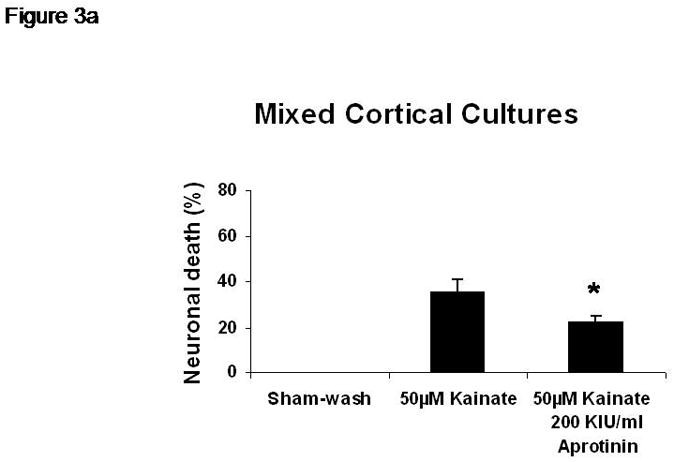
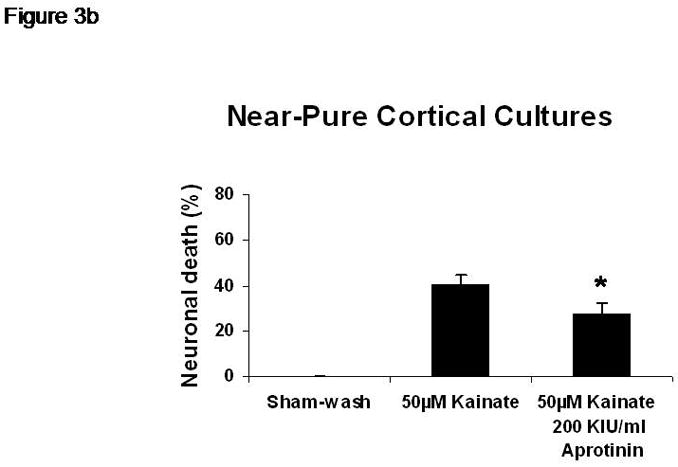
% neuronal cell death induced by exposure to 50 μM kainate revealed about 30% neuronal cell death. Ibuprofen reduced % neuronal cell death induced by exposure to 50 μM kainate from 36.1±3.5% to 20.0±3.1% in mixed cultures but failed in near-pure cultures.
A: mixed cultures, B: near pure cultures. * indicates significantly different from Kainate alone by ANOVA with Tukey correction (mean ± SE).
Discussion
This study has demonstrated that Ibuprofen protects neurons against glutamatergic excitotoxic neuronal cell death induced by NMDA or kainate in mixed cortical cultures. However Ibuprofen fails to reduce neuronal cell death in near-pure cultures. This result suggests that protection of neurons by Ibuprofen requires interaction with glial cells and is not a result of a direct effect on neurons. Further evidence for the role of glial cells is the observation that % neuronal cell death was the same whether induced by NMDA or kainate in both cultures. It is possible that Ibuprofen stimulates glial cells to release a neuroprotective mediator such as a cytokine or it may inhibit the deleterious effects of mediators released by glial cells.
It is thought that inflammation contributes to post-ischemic brain injury. Although cytokines are produced in the CNS by other cells including astrocytes, neurons and endothelial cells, microglia rapidly respond to injury by secreting inflammatory cytokines upon injury. Pro-inflammatory cytokines such as IL-1β and TNF-α are involved in ischemia/hypoxia and trauma-induced brain injury [13, 14]. Ibuprofen is a non-steroidal anti-inflammatory drug (NSAID) and is widely used to reduce pain, fever and inflammation. The drug inhibits cyclooxygenases and activates peroxisome proliferators-activated receptors (PPARs); both of these actions result in reduced inflammation [1–3]. Park and co-workers described Ibuprofen protecting neurons against ischemia via up-regulating interleukin-1 receptor antagonist expression [5]. However, inhibition of cyclooxygenase (COX) has been controversial in adult cardiac surgery regarding cardiac protection. McGuinness and co-workers reported that glutamine pretreatment conferred infarct protection through up-regulation of COX-2 [15]. Wong and colleague reported that a selective COX-2 inhibitor did not affect the myocardium in patients undergoing coronary artery bypass grafting [16].
Local cerebral glucose utilization decreases after traumatic brain injury as live cells decrease in number. Pappius and Wolfe demonstrated that pre-treatment with Ibuprofen prevented the depression of local cerebral glucose utilization after traumatic brain injury [6]. They suggested that Ibuprofen protected neurons and improved cerebral metabolism after brain injury and that these effects might be associated with suppression of prostaglandin synthesis.
Usually cerebral blood flow decreases after cerebral ischemia because of vasoconctriction resulting from hypoperfusion during ischemia and subsequent inflammation. Grice and colleague reported that Ibuprofen improved post-ischemic hypoperfusion after normothermic global cerebral ischemia using dogs, although cerebral blood flow did not return to normal. Also they demonstrated that Ibuprofen suppressed the production of thromboxane B2 and prostaglandin I2 after global cerebral ischemia [7]. Ibuprofen has been reported to reduce infarct size after temporary cerebral ischemia such as may occur after deep hypothermic circulatory arrest [4]. They suggested that the beneficial effect of cyclooxygenase inhibition occurs during reperfusion.
Conclusion
Ibuprofen provides neuroprotection through glial cells against glutamatergic excitotoxicity as demonstrated by reduced neuronal cell death in mixed cell cultures. Further studies regarding mechanisms at molecular level and with whole animal models are needed to assess the potential of ibuprofen to reduce neurological injury in patients undergoing circulatory arrest and cardiopulmonary bypass.
Acknowledgments
This study was supported by NIH R01HL060922
Footnotes
This paper was presented at the 16th Annual Meeting of the Asian Society for Cardiovascular & Thoracic Surgery in Singapore, March 15, 2008.
References
- 1.Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54(3):601–24. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272(6):3406–10. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 3.Kojo H, Fukagawa M, Tajima K, Suzuki A, Fujimura T, Aramori I, Hayashi K, Nishimura S. Evaluation of human peroxisome proliferator-activated receptor (PPAR) subtype selectivity of a variety of anti-inflammatory drugs based on a novel assay for PPAR delta (beta) J Pharmacol Sci. 2003;93(3):347–55. doi: 10.1254/jphs.93.347. [DOI] [PubMed] [Google Scholar]
- 4.Cole DJ, Patel PM, Reynolds L, Drummond JC, Marcantonio S. Temporary focal cerebral ischemia in spontaneously hypertensive rats: the effect of ibuprofen on infarct volume. J Pharmacol Exp Ther. 1993;266(3):1713–7. [PubMed] [Google Scholar]
- 5.Park EM, Cho BP, Volpe BT, Cruz MO, Joh TH, Cho S. Ibuprofen protects ischemia-induced neuronal injury via up-regulating interleukin-1 receptor antagonist expression. Neuroscience. 2005;132(3):625–31. doi: 10.1016/j.neuroscience.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 6.Pappius HM, Wolfe LS. Effects of indomethacin and ibuprofen on cerebral metabolism and blood flow in traumatized brain. J Cereb Blood Flow Metab. 1983;3(4):448–59. doi: 10.1038/jcbfm.1983.71. [DOI] [PubMed] [Google Scholar]
- 7.Grice SC, Chappell ET, Prough DS, Whitley JM, Su M, Watkins WD. Ibuprofen improves cerebral blood flow after global cerebral ischemia in dogs. Stroke. 1987;18(4):787–91. doi: 10.1161/01.str.18.4.787. [DOI] [PubMed] [Google Scholar]
- 8.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23(9):1261–76. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 9.Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine cortical cell culture. In: Tyson CA, Fraizer JM, editors. In Vitro Biological Methods. San Diego, CA: Academic Press; 1993. pp. 46–60. [Google Scholar]
- 10.Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med. 1995 Mar 30;332(13):848–54. doi: 10.1056/NEJM199503303321303. [DOI] [PubMed] [Google Scholar]
- 11.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20(1):83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 12.Motulsky H. Intuitive biostatistics. New York: Oxford University Press; 1995. pp. 255–67. [Google Scholar]
- 13.Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 1989;491(2):394–7. doi: 10.1016/0006-8993(89)90078-4. [DOI] [PubMed] [Google Scholar]
- 14.Saito K, Suyama K, Nishida K, Sei Y, Basile AS. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett. 1996;206(2–3):149–52. doi: 10.1016/s0304-3940(96)12460-5. [DOI] [PubMed] [Google Scholar]
- 15.McGuinness J, Neilan TG, Cummins R, Sharkasi A, Bouchier-Hayes D, Redmond JM. Intravenous Glutamine Enhances COX-2 Activity Giving Cardioprotection. J Surg Res. 2008 Apr 28; doi: 10.1016/j.jss.2008.03.045. In press. [DOI] [PubMed] [Google Scholar]
- 16.Wong PS, Asmat A, Chan YH, Lee CN. A randomized, double-blind, placebo-controlled trial of a COX-2 inhibitor (Rofecoxib) in patients undergoing coronary artery bypass surgery. Interact Cardiovasc Thorac Surg. 2006;5(2):101–4. doi: 10.1510/icvts.2005.118455. [DOI] [PubMed] [Google Scholar]