

Abstract
The Myc transcription factor regulates fundamental processes in a cell’s life: its growth, division, and survival. Myc is conserved throughout metazoan phyla, and its identification in the fruit fly, Drosophila melanogaster has led to new insights in Myc’s physiological roles. In this review, we describe recent research on the biology of Myc and its family members in Drosophila, paying particular attention to its role in the control of growth during development.
Keywords: Myc, Growth, Cell division, Ribosome biogenesis, Cell competition
1. Introduction
Myc proteins have fascinated biologists for almost 25 years, yet despite the enormous literature that documents their biological functions they still remain mysterious. Myc is the founding member of a family of transcription factors of the basic-helix-loop-helix-leucine zipper (BHLH-LZ) class, charged with directing some of the most basic aspects of a cell’s life: its growth, division, and survival. As such, it is not surprising that this family has ancient roots, with members of the Myc “network” – which includes c-, L-, and N-Myc, Myc’s binding partner Max, and its functional antagonists, Mad/Mxi/Mnt) – encoded in the genomes of most metazoan phyla [1]. Years of research on c-Myc in mammalian systems have led to a dauntingly diverse range of potential genetic and functional targets. Yet it has remained difficult to evaluate Myc’s function in its entirety due to redundancy among family members, tissue specificity, and complex phenotypes.
These problems spurred the search for Myc in genetically tractable animal models, and led to the identification of Myc family members in zebrafish, in the nematode Caenorhabditis elegans, and in Drosophila. The zebrafish Danio rerio genome encodes several transcript variants of Max, as well as homologs of c-Myc, N-Myc, L-Myc, and Mxi/Mnt [2,3]. Interestingly, although C. elegans has functional Max and Mnt orthologs, Myc is conspicuously absent from its genome [4]. Drosophila, on the other hand, has one homolog each of myc, max, and mnt. The relatively simple genome and the highly developed genetics that Drosophila offers have made it the model of choice for study of Myc family members. In fact, the first myc mutant of any organism was identified as a spontaneous mutation in Drosophila in the 1930s by Eleanor Nichols-Skoog and Calvin Bridges, which they called diminutive (dm), for its smaller body size [5]. dm was identified as a mutation in the Drosophila myc (dmyc) gene in 1996, and since then more than a dozen mutations of dmyc have been characterized [6,7]. In the last several years, work on dMyc has clarified some of the protein’s more enigmatic functions and also led to the identification of new functional roles. In this review, we focus primarily on work carried out in Drosophila, and highlight recent findings regarding the roles dMyc plays during development, their biological importance, and parallels with what is known about Myc in vertebrates.
2. dMyc, dMax, and dMnt: the network in Drosophila
The Max protein network – Myc, its antagonists Mad/Mxi/Mnt, and their common binding partner, Max – is conserved in Drosophila, but exists in a simple form, with one member of each class. dMyc was the first of the network to be identified, as a binding partner for human Max in two-hybrid screens [6,7]. The isolation of dMyc allowed the subsequent isolation of dMax, and demonstration that as in vertebrates, dMyc and dMax heterodimerize, and bind as a complex to the canonical E-box sequence CACGTG [6]. The last family member to be isolated was dMnt, the sole member of the Mad/Mxi/Mnt cohort of Myc-antagonizing factors [8].
2.1. dMyc
As its physical interaction with Max suggests, dMyc is structurally related to c-Myc and contains its important functional protein domains, including a well-conserved C-terminal BHLH-LZ domain, which mediates DNA-binding and dimerization with Max. The N-terminus of dMyc is substantially longer than that of c-Myc, and less well conserved (for a more detailed account of sequence conservation in dMyc, dMax, and dMnt see ref. [1]). Within the N-terminus of c-Myc are two short motifs that are highly conserved among vertebrate Myc proteins, known as Myc-box I (MBI) and Myc-box II (MBII). MBII is essential for most biological functions of c-Myc [9]. Both MBI and MBII form part of c-Myc’s transactivation domain, interact with a variety of cellular proteins, and are responsible for control of the cellular concentration of c-Myc [10]. Not surprisingly, these regions are also hotspots for mutations that transform c-Myc into an oncoprotein. Despite tight conservation among vertebrates, in dMyc the amino acid sequence of this region is only loosely homologous to c-Myc [1]. dMyc has a recognizable MBII, but MBI is not well conserved. Even so, dMyc can functionally substitute for c-Myc in transactivation assays in human cell culture [6], can transform rat embryo fibroblasts when expressed along with human RasV12 [7], and can rescue growth defects of mouse embryo fibroblasts derived from c-myc conditional knock-outs [11].
In humans and mice there are three isoforms of c-Myc, translated from distinct start sites, which differ in the length of their N-termini. MycS is the shortest variant and lacks most of the N-terminal transactivation domain, but retains the essential MBII. In Drosophila, some variation in dmyc transcript length has been reported, but whether the dmyc gene has more than one promoter or multiple translation start sites is unknown, and only one protein form of dMyc has been identified [6,12]. However, two c-Myc isoforms – c-Myc2 and MycS – can rescue the lethality of flies carrying a strong dmyc allele, allowing their development into fertile adults [12]. It has been hypothesized that the single dMyc protein performs all of the various biological roles that in human cells are carried out by different c-Myc protein isoforms [12], although some controversy exists about their actual physiological role(s) [13].
2.2. dMnt
dMnt was also isolated in a two-hybrid screen, for dMax interacting proteins [8]. While dMnt lacks the proline- and proline/histidine-rich domains characteristic of mouse Mnt, its size, and overall organization is more similar to Mnt than to the Mad/Mxi paralogs [1,8]. In transactivation assays, dMnt/dMax heterodimers bind to, and repress transcription from canonical E-box sequences, and like human Mad proteins, dMnt represses transcription through its association with the corepres-sor Drosophila Sin3 [8]. Three Mnt isoforms are synthesized in Drosophila: the full-length dMnt, dMntΔZip, which lacks the leucine zipper, and dMntΔSID, lacking the Sin3-interaction domain [8]. Expression of the full-length dMnt during fly development results in a marked cell size reduction and reduces growth of cell clones. Neither dMntΔZip nor dMntΔSID affect cell size when overexpressed, but do reduce clonal growth, suggesting that interaction with dMax and dSin3 are necessary for dMnt to regulate cell size. These variant dMnts occur naturally and are the result of differential splicing, but their physiological role is currently unknown; additional dmnt mutants, which specifically remove individual isoforms will be needed to understand the roles of these dMnt proteins.
Unlike its mouse counterpart, dmnt null mutants are viable, thus it is not essential for animal development. This is noteworthy since the mouse Mnt knock-out is lethal shortly after birth, and the absence of strong phenotypes in Mad1, Mad3, Mxi1 knock-out MEFs had been attributed in part to functional overlap between the several Mad/Mxi proteins and Mnt [14]. However, dmnt mutants do have growth phenotypes that in general are complementary to dmyc mutants (described in detail below). Cells mutant for dmnt are larger than normal and consequently, dmnt mutant adults are heavier than wildtype flies in overall body weight. Unexpectedly, while dmnt adults are healthy, they have a shortened life-span [8]. An intriguing related observation was made in nematodes: the C. elegans Mnt/Mad ortholog mdl1 was identified as a gene that is up-regulated with loss of daf-2, which encodes the Insulin/IGF-1 receptor, and down-regulated with daf-16 RNAi treatment [15]. Daf-2 signaling regulates the activity of Daf-16, a Foxo-family transcription factor, which influences metabolism and lifespan in worms, flies, mice, and humans. The inclusion of a Mad/Mnt homolog in a list of potential Daf-16 targets provides a provocative link between Insulin/IGF signaling and the Myc/Max/Mnt network.
dMnt is expressed in the developing central and peripheral nervous systems coincident with the onset of their differentiation [8]. However, dMnt expression is not strictly limited to differentiating cells; in the salivary glands and fat body, two endoreplicating organs, dMnt is expressed in both replicating and non-replicating cells. Also, dMnt is expressed in the peripodial epithelium encasing imaginal discs at a time when they are proliferating. In some tissues, the expression of dMyc and dMnt proteins is complementary and non-overlapping, but this is not always the case [16].
Indeed, the relationship between dMyc and dMnt is not yet well defined. Whether dMyc and dMnt antagonize one another in the same cell or have opposing roles in separate cells (or both) awaits more detailed study and the generation of dmyc, dmnt double mutant animals. In general, the various dMnt expression patterns, over-size mutant phenotype, and suppression of growth when overexpressed are certainly consistent with a role in counteracting the growth promoting function of dMyc.
2.3. dMax
Little is known about dMax beyond its roles as a binding partner for dMyc and dMnt. Thus far, no dmax mutants have been characterized, but such mutations are expected to eliminate most of the activities of dMyc and dMnt. Along with the dmyc and dmnt null mutants already available, fly mutants of dmax would make possible a thorough genetic epistasis analysis of the Max network. There are some hints, however, that the function of dMax may not be limited to partnering dMyc and dMnt. For example, in experiments designed to map genomic binding by dMyc, dMax, and dMnt, it was found that dMax bound to a large number (365) of genes not bound by dMnt or dMyc [17]. This could be due to binding by dMax homodimers, or, dMax could partner other, unidentified factors. Intriguingly, the unique binding of dMax to these genes did not correlate with the presence of E-boxes, perhaps pointing to the latter possibility [17].
3. The genetics of dmyc mutations: dMyc controls growth
The presence of c-myc, N-myc, and L-myc has complicated genetic assessments of Myc’s function in mammals, thus the expectation was that a genetic model system, such as Drosophila would provide a more straightforward Myc loss of function system. This has been the case, and importantly, has refocused attention on the role of Myc in regulating growth. One of the greatest advantages Drosophila provides to Myc biology is the ease with which growth can be studied in a living animal.
All dmyc mutations show profound growth defects. Null mutant embryos hatch into larvae at the same time as wild-type animals, but fail to grow and die early in the second larval instar [16]. Hypomorphic alleles are lethal at progressively later stages of development, depending upon their severity [16,18]. In animals bearing the weakest alleles, such as the original dm1 mutation and dmycP0, development is delayed and yields small flies – the result of smaller cells – with short, thin bristles [19]. Animals carrying the slightly stronger dmycP1 allele also have a significant reduction in cell number. Interestingly, although the hypomorphic mutant flies are smaller than normal they appear normally proportioned (Fig. 1) [19,20]. Many of these same defects are characteristic of flies with growth deficits, and also appear in flies with mutations in genes encoding ribosomal proteins (the Minute class of mutations) and other components of ribosome biogenesis.
Fig. 1.

dMyc controls animal growth. Flies expressing less dmyc, such as a viable dmyc hypomorph (dmycP0), are smaller in overall body size than wildtype flies (yw), and wing size is reduced approximately 15% (a and b). Conversely, flies with increased dmyc expression, from a Tubulin promoter-dmyc transgene (Tub > dmyc), are approximately 30% larger in overall body size than wildtype flies (b and c).
The existence of a broad dmyc allelic series with a range of phenotypes suggests that some developmental processes in the fly require dmyc more than others. dmyc is expressed in numerous tissues throughout fly development, and its expression occurs in a dynamic pattern (Wu and Johnston, unpublished data) [6,19]. It is expressed in both endoreplicating cells, which oscillate between G1 and S phase but do not divide, and in mitotic tissues [16,19]. Use of the FLP/FRT technique of mitotic recombination to generate somatic dmyc mutant cells in an otherwise normal animal demonstrated that in all tissues examined, clones of dmyc mutant cells result in pronounced defects in cellular growth. This defect is manifest by a small cell size and by impaired progress through the cell cycle. In endoreplicating larval cells that can normally achieve a DNA content upwards of 2000C, dmyc null cells fail to increase in size as development progresses, a defect that is directly related to the number of endocycles a cell undergoes [16]. Since the dramatic cellular growth of these cells is required to sustain the overall growth of a fly larva, the impaired growth of dmyc mutant endoreplicating tissue probably accounts for the growth arrest and subsequent death of these null mutant larvae [16]. dmyc is also required for growth of polyploid cells within the female germline, and in the diploid imaginal cells that give rise to the adult body structures of the fly [18,19]. Proliferating dmyc mutant imaginal cells are markedly reduced in size and spend a disproportionate amount of the cell cycle in the G1 phase; these cells are also smaller in S, G2, and M phases [19]. Conversely, overexpression of dMyc in imaginal cells increases cell size by accelerating cellular growth, and when expressed throughout the animal the size of the fly is increased by nearly 30% (Fig. 1) [19,21]. Collectively, these mutant phenotypes provide solid evidence that dMyc is required in vivo for cellular growth, and that it acts in a dose-sensitive manner.
3.1. Flies and mice: the same but not the same
Both flies and mice carrying myc mutations are small in size, but the basis for this effect appears to be different. Like dmyc mutant flies, c-myc null mice generated with conventional germline “knock-out” techniques die at early stages as small, delayed embryos with a variety of developmental defects [22]. Conditional flox-induced knock-outs of c-myc within individual tissues avoids the early lethality and has allowed an analysis of the growth phenotypes with a series of increasingly severe c-myc alleles [11,23]. Mice carrying hypomorphic alleles of c-myc are smaller in overall body size, and like the fly mutants, stronger c-myc alleles result in progressively smaller sizes [11].
A striking difference between flies and mice is that even in the strongest hypomorphic c-myc mouse, cell size in several tissues is normal [11]. The small body size of the mutant mice thus appears to be due solely to a dearth of cells. Drosophila hypomorphic dmyc mutants also have fewer cells, but the cells are significantly smaller than normal [19]. This disparity suggests that there may be inherent differences in the way mice and flies regulate cell (and body) size [11]. However, other observations argue against this generalization. Tissue specific excision of a c-myc flox allele to completely inactivate c-myc leads to greatly diminished cell size in epidermal keratinocytes and liver hepatocytes [24,25]. The difference in effects on cell size of c-myc hypomorphic and null mutations could thus be due to allele strength. Alternatively, the requirement for c-myc during mammalian cellular growth may differ with cell type. This is a real possibility since specific inactivation of c-myc in a mosaic B-lymphocyte population prevents activation-induced cell growth and proliferation upon mitogen stimulation (although many of the early steps of activation are still induced) [23]; in contrast, a very similar protocol has little effect on cell growth of T-lymphocytes after T-cell receptor activation [11]. Regardless, since growth is a measure of mass accumulation whether scaled by cell number or cell size, the central observation – that both c-myc mutant mice and dmyc mutant flies have less mass than controls – indicates that Myc is required for animal growth.
4. dMyc and the cell cycle
Given its pivotal role in human cancer and in developmental control of growth, understanding how Myc regulates the cell cycle has historically been of great interest. Like vertebrate Myc, dMyc is required for efficient transit through G1 into S phase, as dmyc mutations stall cells in G1, and overexpression of dMyc accelerates G1 [16,18,19]. However, dMyc is not essential for cell cycle progression. This is clearly demonstrated in the female fly germline, where mitotically dividing cytoblast cell clones carrying a strong allele of dmyc still undergo all the normal divisions, producing the expected 16 cells of a normal germline cyst [18]. On the other hand, as mentioned above, endoreplicating cells mutant for dmyc undergo significantly fewer rounds of S phase [16,18]. The molecular nature of the defect in endoreplication is unclear, as dmyc cells still periodically express the G1 Cdk2 regulators Cyclin E and Dacapo, the fly p27cip/kip homolog, and are able to complete the entire endocycle [18]. One possibility is that dMyc regulates the frequency of S phase entry in these cells. Consistent with this idea, overexpression of dMyc increases the rate of endoreplication in cells of the Drosophila fat body, and this effect is accompanied by (and dependent upon) oscillating activity of Cyclin E/Cdk2 [16]. Moreover, dMyc expression in endocycling cells can partially reverse a growth arrest imposed by expression of the phosphoinositol-3-kinase (PI3K) adaptor p60, rescuing both endoreplication and cellular growth.
4.1. How does dMyc regulate G1?
Wing imaginal cells undergo canonical mitotic cell cycles in which the rate limiting regulators of G1/S and G2/M are Cyclin E and the Cdc25 phosphatase, String, respectively [26]. Overexpression of dMyc in these cells increases Cyclin E levels and accelerates the G1/S transition [19,27]. Cyclin E mRNA levels are moderately enhanced in response to dMyc expression, but Cyclin E protein is increased disproportionately, suggesting that most of the increase is due to post-transcriptional regulation [27,28]. This post-transcriptional regulation could be mediated by signaling from the small GTPase, Ras, as it is in mammals, since in ras mutant cells dMyc expression no longer induces high Cyclin E levels [27]. dMyc also regulates the activity of the E2F/Rb axis. Overexpression of dMyc increases the expression of dE2F targets, such as proliferating cell nuclear antigen (PCNA), ribonucleotide reductase (RNR), and Cyclin E; it also increases expression (both mRNA and protein) of the Rb regulator, Cyclin D, and its obligate cyclin dependent kinase partner, Cdk4 [28]. Cyclin D/Cdk4 is not essential for cell cycle regulation in Drosophila, and does not have a strong regulatory role in G1/S transition [29,30]. However, the complex is required for growth, as animals completely mutant for either Cyclin D or Cdk4 are 25% smaller than wildtype, and their overexpression drives “balanced” growth, where cellular growth rates and cell division rates are in synch (in contrast to dMyc, see below) [21,30]. It appears that G1 cyclins and stimulation of E2F activity are both important effectors of dMyc’s ability to promote G1 progression. Nonetheless, dMyc expression cannot bypass a cell cycle arrest in either diploid or polyploid cells. Expression of the vertebrate cyclin dependent kinase inhibitor, p21, which blocks Cyclin E/Cdk2 activity, arrests Drosophila endocycling cells, and dMyc expression is not sufficient to overcome that arrest [16]. Likewise, ectopic expression of dMyc is not able to drive arrested diploid cells of the developing wing or eye back into the cell cycle [19].
4.2. G2 regulation is independent of dMyc
When expressed in cell clones in the developing wing, dMyc causes an increase in cell size. This size increase occurs because dMyc accelerates the cellular growth rate (the rate at which a cell accumulates mass), but is not sufficient to speed up their division rate [19]. The failure of dMyc to promote progression through the entire cell cycle was initially perplexing since c-Myc is a potent driver of cell proliferation in cell culture and when de-regulated in cancer. However, the inability of overexpressed dMyc to drive a faster cell cycle is explained at least in part by developmental constraints imposed in vivo. In most (if not all) mitotic Drosophila cells the Cdc25 homolog, String (Stg), is not regulated by dMyc, but is under developmentally regulated transcriptional control [19,31]. Thus, even though ectopic dMyc expression drives cells through G1 and into S phase quickly, sufficient levels of Stg for passage through G2 require the appropriate developmental cues. Co-expression of dMyc with Stg does result in a faster cell division rate, matching it to the increased cellular growth rate, and yielding more cells of normal size [19].
Still, there is much that is not understood about how dMyc controls the cell cycle and how its growth promoting activities are linked to cell cycle regulation. For example, whether regulation of Cyclin D and Cdk4 levels by dMyc are important for dMyc’s control of growth and the cell cycle is not known. Why, if dMyc overexpression increases the expression of Cyclin D and Cdk4 (which in Drosophila promote progression through both G1 and G2 [30]), does not the cell cycle progress faster as a whole? Indeed, in some cases, as in during cell competition (see below), expression of dMyc can increase proliferation rates (Senoo-Matsuda and Johnston, unpublished data). These and other discrepancies imply that dMyc has additional, undefined roles in cell cycle regulation. The fact that dMyc expression can overcome an endoreplicative block due to growth inhibition but not one due to cell cycle arrest suggests that much of dMyc’s influence on the cell cycle occurs indirectly, through its role in regulating growth.
5. How does dMyc make cells grow?
The genetic studies in both the fly and the mouse have stressed the importance of Myc’s role in regulating growth, yet how Myc does this is still largely unknown. Both genetic experiments in vivo and target identification approaches in vitro have been taken in Drosophila to get at this problem. By far, the strongest candidate mechanism for growth regulation by dMyc appears to be through its transcriptional control of key regulators of ribosome biogenesis.
5.1. Identifying dMyc target genes
Several groups have set out to clarify dMyc’s mechanism of growth regulation by cataloging dMyc target genes through expression profiling, and both loss- and gain-of-function microarray experiments have been done using cell culture, isolated tissues, or whole animals. A novel approach involving DNA methylation was taken to identify Drosophila genes that are direct targets of dMyc, a tactic that was possible because the Drosophila genome is otherwise unmethylated. The DamID technique employs DNA adenine methyltransferase (Dam), fused to a protein of interest, to methylate, and thereby physically mark the protein’s binding sites in cultures of Drosophila Kc cells, which are derived from wildtype embryos [32]. This technique was used to uncover genomic binding sites of dMyc, dMax, and dMnt [17]. The DamID experiments indicate that when overexpressed, dMyc, dMax, and dMnt bind extensively throughout the genome–to about 15% of loci on arrays that included half of all Drosophila coding sequences. This binding occurred with specificity, as genomic regions bound by these proteins were significantly enriched in two motifs, the canonical E-box, and the DNA Replication Element. However, of all genes activated by dMyc, only about 36% were bound by either dMyc or dMnt (either alone or in combination with Max), suggesting that a large number of genes are activated indirectly upon dMyc overexpression [17]. The DamID experiments also indicate that genomic binding by dMyc is very sensitive to dMax levels. Surprisingly, at either low or high dMax levels, a large proportion of dMyc genomic sites (approximately 2/3) were not bound by dMax. It is not yet clear what accounts for dMyc’s Max-independent presence, particularly in light of DNA-binding experiments in vitro, where dMyc did not bind to E-box sequences without dMax [6]; however, it is still possible that dMyc can also be recruited to DNA by another, unidentified factor.
In cultured Drosophila Schneider’s 2 (S2, a cell line derived from embryos) cells, conditional RNA-interference (RNAi) has been used to reduce dMyc activity [33]. Remarkably, especially given the large number of sites bound by dMyc in the genome, under these conditions just 30 genes continuously require dMyc for stable expression. Most of these dMyc-dependent targets encode factors involved in RNA binding, rRNA processing, nucleolar function, and ribosome biogenesis, and include, for example, the nucleolar proteins Fibrillarin and CG1542, which are involved in processing of the 35S primary transcript and of 27S pre-rRNA, respectively [33]. A majority of the targets that are down-regulated by dmyc RNAi are induced by overexpression of dMyc in S2 cells, or in experiments using RNA from whole larvae in which dMyc is overexpressed [17,34]. Again, genes involved in ribosome biogenesis dominate the targets increased by dMyc: one-fifth of transcripts up-regulated by dMyc expression encode factors used in ribosome biogenesis [17,33,34]. These targets include RNA Polymerase I- and II-transcribed genes, and many appear to be directly responsive to dMyc activity. For example, RpI135 mRNA, encoding a Pol I subunit, is rapidly increased within 4 h of dMyc expression, as is dTIF-IA, a growth-regulated Pol I-associated factor. This rapid response occurs even before an increase in the synthesis of pre-processed rRNA is detected [34].
5.2. dMyc activity makes more ribosomes
The results of expression profiling experiments are backed-up by genetic manipulations in the developing fly. The increase in expression of pre-rRNA by dMyc overexpression is accompanied by a dramatic increase in nucleolar size, a good indicator of increased ribosome activity [34]. This effect is not observed when other growth regulating factors, such as the PI3K, Dp110, or Cyclin D/Cdk4 are overexpressed (de la Cova & Johnston, unpublished data) [34]. Expression of Fibrillarin is also increased in these larger nucleoli, and the cytoplasm of larval salivary gland cells expressing dMyc are packed with ribosomes and polysomes, with a dense network of rough endoplasmic reticulum (Fig. 2) [34]. By contrast, in dmyc mutant larvae, pre-rRNA levels are low compared to controls despite even levels of rDNA, and nucleolar size is reduced [34]. As a whole, the data indicate that modulation of ribosome biogenesis is an important effector of dMyc during normal growth, with dMyc-dependent transcriptional regulation of ribosome biogenesis resulting in greater translational activity within the cell. Similar observations have been made in vertebrate cells, pointing to the control of ribosome biogenesis as a fundamental and conserved part of Myc biology [35,36].
Fig. 2.
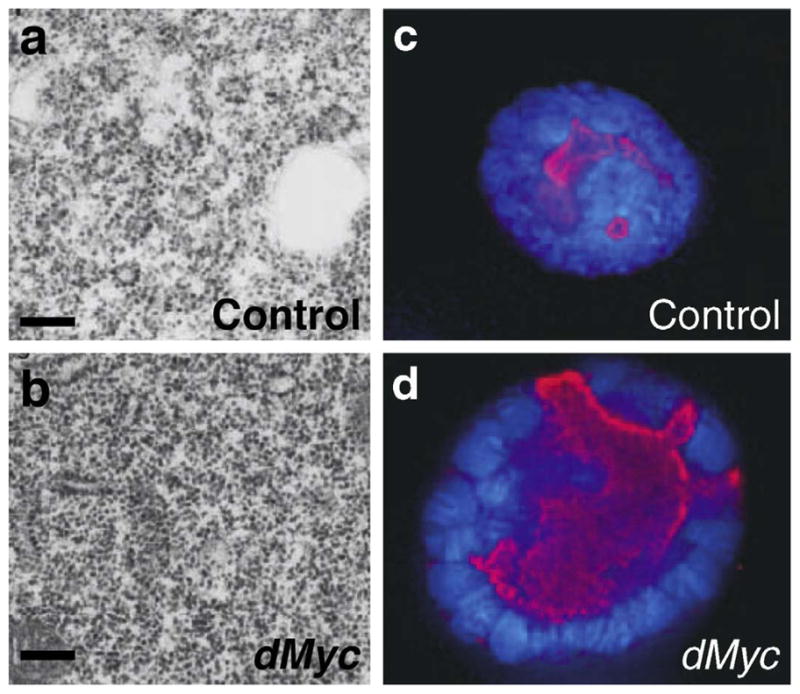
dMyc regulates nucleolar size and ribosome biogenesis in vivo. Cells of the fly salivary gland overexpressing dMyc have increased ribosome content (dark dots in a and b), and larger nucleolar size (c and d) as visualized by Fibrillarin (red), and nuclear size (c and d) as seen by DAPI (blue). (Adapted from Grewal, et al. 2005 and used with permission of the authors. See Grewal et al. [34] for more details.)
5.3. Transcriptional repression by dMyc
Like c-Myc, dMyc can repress transcription [33,37]. In vertebrates, c-Myc-induced transcriptional repression is not associated with the presence of E-boxes at target genes, and may occur when c-Myc binds and inhibits other transcriptional activators, such as Miz-1 [38]. In Drosophila, conditional removal of dmyc is not sufficient to activate repressed targets [33]. It is possible that repression of target genes does not continuously require the presence of dMyc, or that other factors act redundantly with dMyc in repression. One dMyc-repressed target, which has been confirmed with experiments in vivo is the dmyc locus itself [37]. To identify cellular factors required for repression of the dmyc locus, a large-scale genetic screen was conducted using a transposon insertion that, by virtue of its insertion site in the dmyc locus, acts as a visible eye-color reporter of dmyc transcription. Mutations in Polycomb (Pc), as well as Posterior sex combs, another Pc group gene, derepress dmyc transcription. Furthermore, Pc is required for dMyc repressive ability because when dMyc is overexpressed in the absence of Pc, 73% of dMyc-repressed targets are derepressed [37]. The Pc group complex can mediate long-term “memory” of transcriptional repression and its role in dMyc repression is consistent with the possibility that dMyc is not continuously required for repression of its targets. Why dMyc inhibits expression of itself is unclear, but it is a strategy that could be used to limit growth in some circumstances, or in some cell types.
6. dMyc in development: control of tissue growth and links with pattern formation
One of the biggest mysteries of animal development is how growth is coordinated with pattern formation. By controlling cellular growth and cell proliferation, dMyc also has a major impact on the regulation of animal size. In contrast to the growth-regulating network of Insulin/PI3K and dTOR, dmyc expression is regulated by at least two of the major developmental signaling pathways that regulate pattern in Drosophila, Wingless (Wg)/Wnt, and Decapentaplegic (Dpp)/BMP/TGF-β. While Insulin/PI3K/dTOR signaling controls growth in response to nutrients, the responsiveness of dMyc to Wg and Dpp, which are also required for growth of many fly organs, suggests a model wherein developmental signals contribute to tissue growth, and ultimately body size and proportion, by regulating dMyc activity. We explore this idea here by describing two specific developmental processes that involve dMyc: a patterned cell cycle arrest, and cell competition.
6.1. Repression of dMyc enforces a developmental growth arrest
Developing wing cells all exit the cell cycle at the end of development, but specific cells at the dorsal-ventral (DV) boundary, a developmental compartment boundary in the wing, arrest more than a day earlier than the rest as part of a neural differentiation program and have been called the zone of non-proliferating cells (ZNC) [39]. A significant fraction of the ZNC arrests in G1, and requires the Drosophila Rb family member, Rbf, as cells lacking rbf, or overexpressing Drosophila E2F1 (dE2F1), fail to arrest [28,39]. Similarly, overexpression of Cyclin E, which is an inhibitor of Rbf activity, is sufficient to prevent the normal G1 arrest [39]. Since dMyc regulates Cyclin E and Cyclin D expression, and the dE2F1 targets PCNA and RNR, dMyc activity must be inhibited in the ZNC to allow unhindered Rbf activity to enforce the cell cycle arrest.
Earlier in development, during the growth phase, dmyc is expressed throughout the growing wing, but is later lost in the ZNC through the activity of Wg, which is expressed in cells immediately flanking the DV boundary of the wing [19,39]. Inhibition of dMyc by Wg is necessary to allow Rbf to lock in the cell cycle arrest, and loss of dMyc also ensures that cellular growth is reduced in the arrested cells [19,28]. Wg may inhibit dmyc expression by inducing Halfpint (Hfp), a homolog of human FBP interacting repressor (FIR), a pre-mRNA splicing factor also called PUF60 [40]. In human cells, FIR inhibits c-myc transcription and interacts with FBP, a protein that binds an upstream element at the c-myc locus [41]. In the fly, hfp mutant cells have elevated dmyc transcript levels and the ZNC fails to form [40]. Wg signaling thus initiates a program utilizing Hfp and Rbf that restricts dmyc expression and ensures that dE2F1 activity remains off. Although, it is currently an open question whether Hfp regulates dmyc expression by the same mechanism as human FIR, the ZNC provides an excellent system to examine the relationship between developmental signals, such as Wg and control of dMyc expression.
6.2. A competitive edge: dMyc defines the winners
Recently, work in Drosophila has revealed a new aspect of dMyc function: high levels of dMyc provide cells with a competitive edge that allows them to kill nearby cells that have less dMyc. In the fly, cell competition is a process that is operationally defined by the progressive elimination of normally viable (but less “competitive”) cell types. When cell clones overexpressing dMyc are generated in the developing wing, such clones grow faster than the surrounding wildtype cells, which in turn actually grow less than expected and die more frequently [21,42]. The ability of dMyc to induce cell competition is a remarkable property that is not shared by all growth regulators [21].
Competition can be induced whenever neighboring cells differ in levels of dMyc. Wildtype cells, containing endogenous dmyc, are only killed when they reside near cells overexpressing dMyc [21,42]. Likewise, although dmyc hypomorphic cells are viable when surrounded by each other, when they exist in somatic clones surrounded by wildtype cells, they are eliminated from the wing [19]. Similar competitive outcomes result from other manipulations that allow some cells to have higher dMyc levels than their neighbors. For example, loss of archipelago (ago), which encodes a Drosophila F box protein homologous to human Fbw7, results in elevated dMyc protein levels and a competitive advantage that allows ago mutant cells to overtake whole body structures while wildtype cells are eliminated [20]. Also, overexpression of the dMyc antagonist dMnt slows growth of cell clones, and these cells are eventually eliminated [8]. Surprisingly, however, cell competition does not always occur when faster and slower growing cells are neighbors, as overexpression of Cyclin D/Cdk4 or Dp110 does not lead to competition, and cells lacking these regulators in mosaics grow very slowly but are not killed by their wildtype neighbors [21,29,43,44]. Enabling cells with a competitive edge therefore appears be a specific effect of dMyc activity.
How are less competitive cells eliminated from the growing fly? Cells with mutations in receptors for patterning factors, such as Dpp, Wg, or EGF, or those deficient in ribosome biogenesis, such as Minutes, a large class of mutations in genes encoding ribosomal proteins, are also subject to competitive elimination [45–48]. Cells lost in competition die by apoptosis, but how the apoptotic program is initiated is not clear [21,42,49]. Two models have been proposed to explain how cell competition occurs. In one model, less competitive cells are deprived of growth factors due to the capture of such factors by their more competitive neighbors [50]. The elimination of cells unable to receive Dpp, Wg, or EGF is certainly consistent with this idea, as is a report that some Minute cells are deficient in responding to Dpp [49]. However, the predictions that this model makes do not always hold. For example, dMyc expression neither enhances a cell’s response to signals such as Dpp or Wg, nor alters the response of neighboring cells, as would be predicted from a ligand-capture model (de la Cova, Vargas, and Johnston, unpublished data) [21]. A second model posits that competition is due to secretion of a factor that initiates an apoptotic program in neighboring cells. This model comes from the observation that although physical contact is not necessary for wildtype cells to be killed, they must be in close proximity to dMyc-expressing cells [21]. Thus, dMyc may allow cells to sense each other’s presence, and induce competition via a short-range signal (Fig. 3) [21]. Interestingly, many of the Minute genes, as part of the large number of genes involved in ribosomal biogenesis, are dMyc transcriptional targets. Also, as mentioned above, dMyc expression is influenced by Wg and Dpp [19,21,51]. These observations might imply that all cell competition – competition involving dMyc, a reduction in ribosome biogenesis, or lack of patterning factors – operates by the same mechanism. However, this has not been demonstrated, and the molecular mechanism of cell competition remains to be established.
Fig. 3.
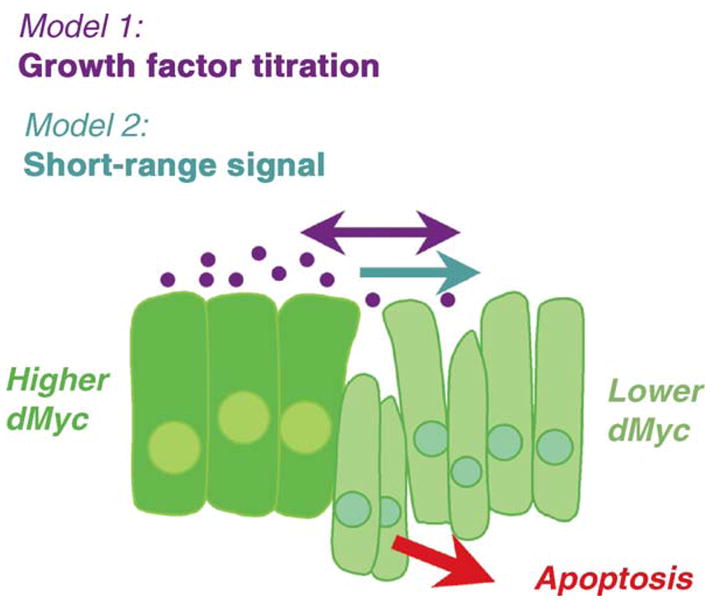
dMyc provides growing cells with a competitive advantage. Two models that can explain the competitive advantage of dMyc-expressing cells are: (1) that cells expressing more dMyc deprive their neighbors of growth and/or survival factors and (2) that dMyc expressing cells induce a short-range signal that kills nearby cells that express less dMyc.
Cell competition is not an anomaly of flies, as it also occurs in mice. Cells with a mutation in the ribosomal protein L24 are competitively eliminated when wildtype cells are introduced to a mutant mouse blastocyst, a situation reminiscent of the Minute mutations of Drosophila [52]. Also, somatic inactivation of mouse c-myc in B- or T-cell mosaics results in the progressive loss of c-myc null lymphocytes [11,23]. Given the deregulation of c-Myc that occurs in many tumors, an exciting possibility that remains to be tested is that a tumor cell population in which c-Myc is activated may out-compete nearby wildtype cells. A Myc-induced competitive advantage, and the ability to kill neighboring cells, could facilitate growth of an incipient tumor. However, there are instances in both mice and flies where differences in Myc levels do not lead to competition. For example, c-myc null hepatocytes persist at stable frequencies in mosaic livers [53]. Furthermore, in the developing fly wing, dmyc expression is down-regulated in hinge cells as development proceeds, yet these cells are not competed away by nearby cells with higher dMyc levels (Wu & Johnston, unpublished data). Furthermore, post-mitotic cells in the fly seem to be protected from competition. The properties that allow some cells to ignore Myc level differences and others be gravely affected by them is unknown, but should reveal information about how cells perceive themselves as constituents of a growing tissue.
Why does cell competition occur? One hypothesis is that cell competition might contribute to an overall size-control mechanism. A striking observation is that although, for example, dMyc expressing clones in the developing wing proliferate more than wildtype cells, they do not change the final size of the adult wing. Overgrowth is prevented because the competitive elimination of wildtype cells compensates for the increased growth of dMyc-expressing cells. If cells are protected from death, or if dMyc is expressed ubiquitously, thereby preventing competitive interactions, size control is overridden, and a larger wing size results (Fig. 1) [21]. Such a size-controlling role of cell competition is intriguing as, paradoxically, it suggests that the competitive killing of wildtype cells allows dMyc-expressing cells to overtake a structure, and yet also keeps overgrowth of that structure in check. A second attractive theory about cell competition is that it represents a quality control mechanism that removes defective or mispatterned cells [54]. Testing of these ideas has only just begun. Genetic or other conditions that specifically prevent competitive cell death would be remarkable and useful tools for uncovering the normal role of cell competition. dMyc-induced cell competition in Drosophila provides a unique model that could lead to the identification of genes that are involved in the earliest steps of cancer progression.
7. Conclusions and perspectives
The simple Myc network, broad allelic series of dmyc mutations, and the ease with which growth and cell proliferation can be studied in a living animal has made Drosophila an ideal model organism for investigating the biological roles of Myc. Not surprisingly, then, dMyc has been the focus of intense scrutiny in the last several years, yielding several insights as well as some surprises. In this review, we have discussed what has been learned from studies in Drosophila and how these findings relate to Myc function in other organisms. Altogether, the data tell us that in all organisms that carry myc homologs, the Myc family of interacting proteins is essential for appropriate cellular and animal growth—a huge responsibility for a relatively small protein network. Myc’s role in regulating growth is probably an ancient one, as most of the network is conserved throughout taxa.
Still, several puzzles remain, and as always when research is informative, many additional questions have been raised. For instance, in Drosophila, high expression levels of Myc confer an advantage on cells that allows them to compete against and kill cells with less Myc. Why is this aspect of Myc function not shared by growth regulators like PI3K or Cyclin D? What is the molecular mechanism underlying Myc-induced competition, and does it occur in vertebrates? Another question raises an evolutionary issue—why does the nematode C. elegans lack myc, but retain max and mnt homologs? The answer to this is unknown, but it is possible that Myc’s ability to integrate complex growth regulatory signals is not necessary in C. elegans; in contrast to flies, mice, and humans, where growth is “regulative” and allows developmental plasticity, growth in C. elegans occurs by proliferation of a fixed developmental lineage of cells [55]. A major gap also remains in our knowledge of how dMyc, dMnt, and dMax function together (or apart) to control growth, and how each contributes to cell cycle regulation. Similarly, we know little of how dMyc expression is controlled during development. In Drosophila, developmental signals, such as the Wnt family member Wg modulates dMyc expression in growing tissues [19], and it has been hypothesized that Myc expression, regulated by developmental signals, may provide a link between patterning information and tissue growth [56]. Further study of how the Myc/Max/Mnt network is regulated and functions in vivo is clearly important, and promises to provide insight into many aspects of developmental control of growth and the cell cycle.
References
- 1.Gallant P. Current topics in microbiology and immunology. Springer-Verlag; 2006. Myc/Max/Mad in invertebrates: the evolution of the Max network; pp. 237–54. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber-Agus N, Horner J, Torres R, Chiu FC, DePinho RA. Zebra fish myc family and max genes: differential expression and oncogenic activity throughout vertebrate evolution. Mol Cell Biol. 1993;13(5):2765–75. doi: 10.1128/mcb.13.5.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber-Agus N, Chin L, Chen K, et al. Evolutionary relationships and functional conservation among vertebrate Max-associated proteins: the zebra fish homolog of Mxi1. Oncogene. 1994;9(11):3167–77. [PubMed] [Google Scholar]
- 4.Yuan J, Tirabassi RS, Bush AB, Cole MD. The C. elegans MDL-1 and MXL-1 proteins can functionally substitute for vertebrate MAD and MAX. Oncogene. 1998;17:1109–18. doi: 10.1038/sj.onc.1202036. [DOI] [PubMed] [Google Scholar]
- 5.Lindsley DLaZ GG. The genome of Drosopahila melanogaster. Academic Press; 1992. [Google Scholar]
- 6.Gallant P, Shiio Y, Cheng PF, Parkhurst SM, Eisenman RN. Myc and Max homologs in Drosophila. Science. 1996;274(5292):1523–7. doi: 10.1126/science.274.5292.1523. [DOI] [PubMed] [Google Scholar]
- 7.Schreiber-Agus N, Stein D, Chen K, Goltz JS, Stevens L, DePinho RA. Drosophila myc is oncogenic in mammalian cells and plays a role in the diminutive phenotype. Proc Natl Acad Sci. 1997;94:1235–40. doi: 10.1073/pnas.94.4.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loo LWM, Secombe J, Little JT, et al. The transcriptional repressor dMnt is a regulator of growth in Drosophila melanogaster. Mol Cell Biol. 2005;25(16):7078–91. doi: 10.1128/MCB.25.16.7078-7091.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Ann Rev Cell Dev Biol. 2000;16:653–99. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 10.Facchini LM, Penn LZ. The molecular role of Myc in growth and transformation: recent discoveries lead to new insights. FASEB J. 1998;12(9):633–51. [PubMed] [Google Scholar]
- 11.Trumpp A, Refaeli Y, Oskarsson T, et al. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 2001;414(6865):768–73. doi: 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- 12.Benassayag C, Monterro L, Colombie N, Gallant P, Cribbs D, Morello D. Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol Cell Biol. 2005;25(22):9897–909. doi: 10.1128/MCB.25.22.9897-9909.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirst SK, Grandori C. Differential activity of conditional MYC and its variant MYC-S in human mortal fibroblasts. Oncogene. 2000;19(45):5189–97. doi: 10.1038/sj.onc.1203904. [DOI] [PubMed] [Google Scholar]
- 14.Queva C, Hurlin PJ, Foley KP, Eisenman RN. Sequential expression of the MAD family of transcriptional repressiors during differentiation and development. Oncogene. 1998;16(8):967–77. doi: 10.1038/sj.onc.1201611. [DOI] [PubMed] [Google Scholar]
- 15.Murphy CT, McCarroll SA, Bargmann CI, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424(6946):277–83. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 16.Pierce S, Yost C, Britton JS, et al. dMyc is required for larval growth and endoreplication in Drosophila. Development. 2004;131(10):2317–27. doi: 10.1242/dev.01108. [DOI] [PubMed] [Google Scholar]
- 17.Orian A, van Steensel B, Delrow J, et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101–14. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maines JZ, Stevens LM, Tong X, Stein D. Drosophila dMyc is required for ovary cell growth and endoreplication. Development. 2004;131(4):775–86. doi: 10.1242/dev.00932. [DOI] [PubMed] [Google Scholar]
- 19.Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–90. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moberg KH, Mukherjee A, Veraksa A, Artavanis-Tsakonas S, Hariharan IK. The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr Biol. 2004;14:965–74. doi: 10.1016/j.cub.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 21.de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–16. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- 22.Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7(4):671–82. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- 23.de Alboran IM, O’Hagan RC, Gartner F, et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001;14(1):45–55. doi: 10.1016/s1074-7613(01)00088-7. [DOI] [PubMed] [Google Scholar]
- 24.Zanet J, Pibre S, Jacquet C, Ramirez A, de Alboran IM, Gandarillas A. Endogenous Myc controls mammalian epidermal cell size, hyper-proliferation, endoreplication and stem cell amplification. J Cell Sci. 2005;118(8):1693–703. doi: 10.1242/jcs.02298. [DOI] [PubMed] [Google Scholar]
- 25.Baena E, Gandarillas A, Vallespinos M, et al. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci. 2005;102(20):7286–91. doi: 10.1073/pnas.0409260102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neufeld TP, de la Cruz AFA, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93(7):1183–93. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- 27.Prober DA, Edgar BA. Ras1 promotes cellular growth in the Drosophila wing. Cell. 2000;100(4):435–46. doi: 10.1016/s0092-8674(00)80679-0. [DOI] [PubMed] [Google Scholar]
- 28.Duman-Scheel M, Johnston LA, Du W. Repression of dMyc expression by Wingless promotes Rbf-induced G1 arrest in the presumptive Drosophila wing margin. PNAS. 2004;101(11):3857–62. doi: 10.1073/pnas.0400526101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer CA, Jacobs HW, Datar SA, Du W, Edgar BA, Lehner CF. Drosophila Cdk4 is required for normal growth and is dispensible for cell cycle progression. EMBO J. 2000;19(17):4533–42. doi: 10.1093/emboj/19.17.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Datar SA, Jacobs HW, de la Cruz AFA, Lehner CF, Edgar BA. The Drosophila Cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 2000;19(17):4543–54. doi: 10.1093/emboj/19.17.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehman DA, Patterson B, Johnston LA, et al. Cis-regulatory elements of the mitotic regulator, string/Cdc25. Development. 1999;126(9):1793–803. doi: 10.1242/dev.126.9.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using dam methyltransferase. Nat Biotechnol. 2000;18(4):424–8. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- 33.Hulf T, Bellosta P, Furrer M, et al. Whole-genome analysis reveals a strong positional bias of conserved dMyc-dependent E-boxes. Mol Cell Biol. 2005;25(9):3401–10. doi: 10.1128/MCB.25.9.3401-3410.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7(3):295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 35.Grandori C, Gomez-Roman N, Felton-Edkins ZA, et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7(3):311–8. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 36.Arabi A, Wu S, Ridderstrale K, et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7(3):303–10. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 37.Goodliffe JM, Wieschaus E, Cole MD. Polycomb mediates Myc autorepression and its transcriptional control of many loci in Drosophila. Genes Dev. 2005;19(24):2941–6. doi: 10.1101/gad.1352305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev. 2005;6(8):635–45. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 39.Johnston LA, Edgar BA. Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature. 1998;394(6688):82–4. doi: 10.1038/27925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinn LM, Dickins RA, Coombe M, Hime GR, Bowtell DDL, Richardson H. Drosophila Hfp negatively regulates dmyc and stg to inhibit cell proliferation. Development. 2004;131(6):1411–23. doi: 10.1242/dev.01019. [DOI] [PubMed] [Google Scholar]
- 41.Liu J, He L, Collins I, et al. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell. 2000;5(2):331–41. doi: 10.1016/s1097-2765(00)80428-1. [DOI] [PubMed] [Google Scholar]
- 42.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117(1):117–29. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 43.Emmerich J, Meyer CA, de la Cruz AFA, Edgar BA, Lehner CF. Cyclin D does not provide essential Cdk4-independent functions in Drosophila. Genetics. 2004;168(2):867–75. doi: 10.1534/genetics.104.027417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bohni R, Riesgo-Escover J, Oldham S, et al. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell. 1999;97(7):865–75. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- 45.Burke R, Basler K. Dpp receptors are autonomously required for cell proliferation in the entire developing Drosophila wing. Development. 1996;122(7):2261–9. doi: 10.1242/dev.122.7.2261. [DOI] [PubMed] [Google Scholar]
- 46.Johnston LA, Sanders AL. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat Cell Biol. 2003;5(9):827–33. doi: 10.1038/ncb1041. [DOI] [PubMed] [Google Scholar]
- 47.Baker NE, Yu S-Y. The EGF receptor defines domains of cell cycle porgression and survival to regulate cell number in the developing Drosophila eye. Cell. 2001;104(5):699–708. doi: 10.1016/s0092-8674(01)00266-5. [DOI] [PubMed] [Google Scholar]
- 48.Morata G, Ripoll P. Minutes: mutants of Drosophila autonomously affecting cell division rate. Dev Biol. 1975;42(2):211–21. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- 49.Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416(6882):755–9. doi: 10.1038/416755a. [DOI] [PubMed] [Google Scholar]
- 50.Milan M. Survival of the fittest: cell competition in the Drosophila wing. EMBO Rep. 2002;3(8):724–5. doi: 10.1093/embo-reports/kvf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prober DA, Edgar BA. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002;16(17):2286–99. doi: 10.1101/gad.991102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131(16):3907–20. doi: 10.1242/dev.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baena E, Gandarillas A, Vallespinos M, et al. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci USA. 2005;102(20):7286–91. doi: 10.1073/pnas.0409260102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallant P. Myc, cell competition, and compensatory proliferation. Cancer Res. 2005;65(15):6485–7. doi: 10.1158/0008-5472.CAN-05-1101. [DOI] [PubMed] [Google Scholar]
- 55.Kipreos ETC. elegans cell cycles: invariance and stem cell divisions. Nat Rev Mol Cell Biol. 2005;6(10):766–76. doi: 10.1038/nrm1738. [DOI] [PubMed] [Google Scholar]
- 56.Johnston LA, Gallant P. Control of growth and organ size in Drosophila. Bioessays. 2002;24(1):54–64. doi: 10.1002/bies.10021. [DOI] [PubMed] [Google Scholar]