

Abstract
Human arginase I is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of L-arginine to generate L-ornithine and urea. We demonstrate that N-hydroxy-L-arginine (NOHA) binds to this enzyme with Kd = 3.6 μM, and nor-N-hydroxy-L-arginine (nor-NOHA) binds with Kd = 517 nM (surface plasmon resonance) or Kd ≈ 50 nM (isothermal titration calorimetry). Crystals of human arginase I complexed with NOHA and nor-NOHA afford 2.04 Å and 1.55 Å resolution structures, respectively, which are significantly improved in comparison with previously determined structures of the corresponding complexes with rat arginase I. Higher resolution structures clarify the binding interactions of the inhibitors. Finally, the crystal structure of the complex with L-lysine (Kd = 13 μM) is reported at 1.90 Å resolution. This structure confirms the importance of hydrogen bond interactions with inhibitor α-carboxylate and α-amino groups as key specificity determinants of amino acid recognition in the arginase active site.
Human arginase I is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of L-arginine to generate L-ornithine and urea. This reaction is the key step of the urea cycle in the liver that allows for the excretion of nitrogenous waste resulting from protein catabolism; healthy adults excrete approximately 10 kg urea per year [1–4]. In extrahepatic tissues arginase serves to regulate L-arginine concentrations for other metabolic pathways. For example, arginase activity can decrease L-arginine concentrations utilized by nitric oxide synthase to generate NO; arginase inhibitors can increase L-arginine concentrations and thereby enhance NO biosynthesis and NO-dependent physiological processes such as smooth muscle relaxation [5]. Accordingly, arginase is a pharmaceutical target for the treatment of diseases associated with aberrant smooth muscle physiology such as erectile dysfunction [6, 7] and asthma [8, 9].
Among substrate analogue inhibitors of arginase, boronic acid and N-hydroxyguanidinium derivatives exhibit the highest affinity (selected inhibitors are shown in Table 1) [7,10–19]. The crystal structures of rat arginase I complexed with 2(S)-amino-6-boronohexanoic acid (ABH) [6], dehydro-ABH [17], and S-(2-boronoethyl)-L-cysteine (BEC) [7], human arginase I complexed with ABH and BEC [18], and human arginase II complexed with BEC [20] reveal that the planar boronic acid moiety of the inhibitor undergoes nucleophilic attack by the metal-bridging hydroxide ion to yield a tetrahedral boronate anion that mimics the tetrahedral intermediate and its flanking transition states in catalysis. In contrast, the low resolution crystal structures of rat arginase I complexed with N-hydroxy-L-arginine (NOHA) and an analogue bearing a side chain shortened by one methyl group (designated nor-NOHA) [21] indicate that the N-OH moiety of the inhibitor displaces the metal-bridging hydroxide ion. Although interactions with the binuclear manganese cluster are strikingly different for the best N-hydroxyguanidinium and boronic acid inhibitors, each binds with comparable affinity. With regard to product binding, early studies indicated that L-ornithine and L-lysine are competitive inhibitors of rat and bovine arginase I [22–24]. Moreover, hydroxylation of the side chain amino group of L-lysine yields a potent inhibitor of bovine arginase I with Ki = 4 μM [14]. Here, too, the N-OH group likely displaces the metal-bridging hydroxide ion of the native enzyme.
Table 1.
Arginase Inhibitors
| Inhibitor | Structure | Affinity, rat arginase I (nM) (Kd or Ki, pH 8.5) | Affinity, human arginase I (nM) (Kd, pH 8.5) |
|---|---|---|---|
| 2(S)-amino-6-boronohexanoic acid (ABH) | 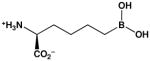 |
100a | 5b |
| (S)-(2-boronoethyl)-L-cysteine (BEC) | 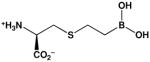 |
2,200c | 270b |
| Nω-hydroxy-L-arginine (NOHA) | 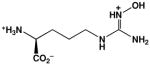 |
10,000d | 3,600e |
| Nω-hydroxy-L-nor-arginine (nor-NOHA) | 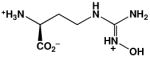 |
500d | 517e; 47, 51f |
| L-lysine | 1,100,000g | 13,100e |
Given the appreciable inhibitory activity observed for amino acid derivatives of the substrate L-arginine and the product L-ornithine, and given the growing pharmaceutical importance of human arginase isozymes, we now report affinity measurements and X-ray crystal structures of the complexes between human arginase I and nor-NOHA, NOHA, and L-lysine. Importantly, crystals of human arginase I afford 1.55 Å and 2.04 Å resolution structures of complexes with nor-NOHA and NOHA, respectively, which are significantly improved in comparison with previously determined structures of rat arginase I complexed with these inhibitors at 2.8 Å and 2.9 Å resolution, respectively [21]. The crystal structure of the complex with inhibitor L-lysine is determined at 1.90 Å resolution. The higher resolution structures of complexes with human arginase I clarify features that contribute to high affinity, particularly with regard to the geometry of metal coordination interactions in the binuclear manganese cluster as well as hydrogen bond interactions with the α-carboxylate and α-amino groups of each inhibitor. Taken together, these results serve to clarify the specificity determinants of amino acid recognition in the active site of human arginase I [25].
Materials and methods
Crystalography
Crystals of the human arginase I-L-lysine complex were prepared by soaking crystals of the native enzyme in buffer solutions containing L-lysine. Crystals of human arginase I were prepared by the hanging drop vapor diffusion method. Typically, drops containing 3 μL protein solution [3.5 mg/mL human arginase I, 50 mM bicine (pH 8.5), 2 mM thymine, 100μM MnCl2] and 3 μL precipitant solution [0.1 M bis-Tris (pH 6.5), 28% PEG monomethyl ether 2000] were equilibrated over a 1 mL reservoir of precipitant solution at 21° C. Crystals generally appeared within 3–4 days, as previously described [26]. Crystals were harvested and soaked in a precipitant solution augmented with 20 mM L-lysine for two days, and then cryoprotected in a precipitant solution containing 32% Jeffamine prior to flash cooling in liquid nitrogen.
Crystals of the human arginase I-NOHA and -nor-NOHA complexes were prepared by cocrystallization. Hanging drops containing 3 μL protein solution [3.5 mg/mL protein, 50 mM bicine (pH 8.5), 2 mM NOHA or nor-NOHA, 100μM MnCl2] and 3 μL precipitant solution [0.1 M bis-Tris (pH 6.5), 28% PEG monomethyl ether 2000] were equilibrated over a 1 mL reservoir of precipitant solution at 21° C. Crystals appeared overnight and grew to typical maximal dimensions of 1.0 mm × 0.3 mm × 0.3 mm. Crystals were further soaked for 24 hours in a precipitant solution augmented with 5 mM NOHA or nor-NOHA and cryoprotected in a precipitant solution containing 32% Jeffamine prior to flash cooling in liquid nitrogen.
X-ray diffraction data from all crystals were collected at the Advanced Photon Source (APS) (Argonne National Laboratory, Argonne, IL) at the NE-CAT beamline 24-ID-C. Diffraction intensities measured from all crystals exhibited symmetry consistent with apparent space group P6 (unit cell parameters a = b = 91.0 Å, c = 69.8 Å) (Table 2). As with crystals of other human arginase I-inhibitor complexes [18, 26], deviations from ideal Wilson statistics were observed with 〈I2〉/〈I〉2 = 1.5, indicating perfect hemihedral twinning. The structure of each enzyme-inhibitor complex was solved by molecular replacement using the program Phaser [27] with chain A of the human arginase I-ABH complex (PDB accession code 2ZAV, less water molecules) [18] used as a search probe against twinned data. In order to calculate electron density maps, structure factors amplitudes (|Fobs|) derived from twinned data (Iobs) were deconvoluted into structure factor amplitudes corresponding to twin domains A and B (|Fobs/A| and |Fobs/B|, respectively) using the structure-based algorithm of Redinbo and Yeates [28] implemented in CNS [29].
Table 2.
Data Collection and Refinement Statistics
| Human arginase I complex | nor-NOHA | NOHA | L-Lysine |
|---|---|---|---|
| Data Collection | |||
| Resolution, Å | 50.0–1.55 | 50.0–2.04 | 50.0–1.90 |
| Total/Unique reflections measureda | 135232/90987 | 81841/40118 | 117869/49010 |
| Rmergea,b | 0.032 (0.302) | 0.089 (0.186) | 0.081 (0.433) |
| I/σ(I)a | 14.4 (2.0) | 28.9 (8.7) | 17.4 (2.5) |
| Completeness (%)a | 98.2 (96.2) | 98.6 (91.8) | 97.1 (98.9) |
| Refinement | |||
| Reflections used in refinement/test set | 85281/4185 | 39744/1587 | 46106/1897 |
| Rtwina,c | 0.144 | 0.124 | 0.155 |
| Rtwin/freea,c | 0.178 | 0.174 | 0.204 |
| Protein atomsd | 4782 | 4782 | 4782 |
| Water moleculesd | 302 | 269 | 209 |
| Inibitor atomsd | 24 | 26 | 20 |
| Manganese ionsd | 4 | 4 | 4 |
| R.m.s. deviations | |||
| Bond lengths, Å | 0.006 | 0.006 | 0.006 |
| Bond angles,° | 1.34 | 1.29 | 1.33 |
| Average B-factors, Å2 | |||
| Main chain | 18 | 26 | 23 |
| Side chain | 20 | 28 | 26 |
| Manganese ions | 13 | 17 | 18 |
| Inhibitors | 16 | 41 | 27 |
| Solvent | 23 | 30 | 25 |
Number in parentheses refer to the outer 0.1 Å shell of data.
Rmerge = Σ|I−〈I〉|/ΣI, where I is the observed intensity and 〈I〉 is the average intensity calculated for replicate data.
Rtwin = Σ|[|Fcalc/A|2+|Fcalc/B|2]1/2−|Fobs||/Σ|Fobs| for reflections contained in the working set. |Fcalc/A| and |Fcalc/B| are the structure factor amplitudes calculated for the separate twin domains A and B, respectively. Rtwin underestimates the residual error in the model over the two twin-related reflections by a factor of approximately 0.7. The same expression describes Rtwin/free, which was calculated for test set reflections excluded from refinement.
Per asymmetric unit.
Crystallographic refinement of each enzyme-inhibitor complex against twinned data was performed using CNS [29] as previously described [18, 26]. In the later stages of each refinement after the majority of water molecules were located, gradient omit maps clearly showed each substrate and product analogue bound to the active site of each monomer in the asymmetric unit. The atoms of nor-NOHA, NOHA and L-lysine were refined with full occupancy and atomic B factors consistent with the average B-factor calculated for the entire protein. Disordered segments at the N- and C-termini were excluded from the final models (M1-S5 and N319-K322). The quality of each refined model was assessed using PROCHECK [30]. Data collection and refinement statistics are recorded in Table 2.
Surface plasmon resonance
The binding of L-lysine, NOHA, and nor-NOHA to human arginase I was studied by surface plasmon resonance using a BIAcore 3000 instrument with identical procedures to those previously reported [31]. All measurements were performed at pH = 8.5 and the analyte concentrations typically ranged 0–200 μM. Binding data for NOHA and nor-NOHA were fit to the equation describing a 1:1 Langmuir interaction model [32, 33], where R is the concentration of the molecular complex formed, CA is the concentration of ligand A, and Rmax is the total concentration of immobilized human arginase I: dR/dt = kaCA(Rmax − R) − kdR This fit yielded the association rate (ka), the dissociation rate (kd), and the dissociation constant (Kd = kd/ka). Since the binding kinetics for L-lysine were too rapid to confidently resolve kon and koff by standard fitting of association and dissociation phases, affinities were determined using the values of Req from the association phase and fitting the Req values using the program GraphPad Prism, version 4.0 (Prism Software, Irvine, CA). The quality of curve fitting was assessed by residual plots and χ2 values for the 1:1 Langmuir interaction model, and by assessment of R2 for the steady state analysis.
Isothermal titration calorimetry
Calorimetry experiments were performed on an MCS isothermal titration calorimeter (Microcal Software, Northampton, MA) using previously reported procedures [7,18]. Briefly, human arginase I was dialyzed into 50 mM bicine (pH 8.5), 100 μM MnCl2 and diluted to a concentration of 0.036 mM. A solution of 1.5 mM nor-NOHA was prepared in the same buffer. The inhibitor solution was titrated into the cell containing the protein in a series of 5 μL injections. Data were analyzed using Origin software and were best fit with the equation describing two sets of independent sites, i.e., a model in which binding to the first monomer of the trimer (association constant K1) has slightly lower affinity than binding to the second and third monomers of the trimer (association constant K23):
(q is the heat evolved during the course of the reaction, [E]t is the total enzyme concentration, V is the cell volume, n1 is the number of inhibitor equivalents required to saturate the first binding site, n23 is the number of equivalents to saturate the second and third sites, ΔH1 and ΔH23 are the binding enthalpy per mole of ligand to the first monomer and to the second and third monomers, respectively, and [L] is inhibitor concentration). Note that dissociation constants Kd1 = 1/K1 and Kd23 = 1/K23.
Results
The binding affinities of NOHA, nor-NOHA, and L-lysine to human arginase I as measured by surface plasmon resonance are recorded in Table 1, and sensorgrams are shown in Figure 1. The highest affinity analogue is nor-NOHA, with Kd = 517 nM as determined by surface plasmon resonance. Interestingly, isothermal titration calorimetry indicates that the binding of nor-NOHA is consistent with binding to one arginase monomer with Kd1 = 47 nM and the remaining two arginase monomers with Kd23 = 51 nM (Figure 1). Possibly, weak cooperativity of inhibitor binding may contribute to the ~10-fold difference with the Kd value determined by surface plasmon resonance.
Figure 1.

(a) Sensorgram showing the interaction of nor-NOHA with human arginase I, yielding Kd = 517 nM. (b) Sensorgram showing the interaction of NOHA with human arginase I, yielding Kd = 3.6 μM. (c) Plot of equilibrium concentrations (Req) determined by surface plasmon resonance for the complexation of human arginase I with L-lysine yields Kd = 13.1 μM; binding kinetics are too rapid to facilitate direct determination of association and dissociation rate constants (inset). (d) Isothermal titration calorimetry of nor-NOHA binding to human arginase I. Shown are the raw data obtained by titration of 0.036 mM arginase with 30 × 5 μL injections of 1.5 mM nor-NOHA (top). The area of each peak is integrated and plotted against [nor-NOHA]/[arginase] (bottom); nonlinear least-squares fitting of the data to a two-site model (solid line) yields Kd values of 51 nM and 47 nM.
Comparisons of the affinity data in Table 1 suggest that NOHA and possibly nor-NOHA bind slightly more tightly to human arginase I than to rat arginase I. This is reminiscent of affinity trends previously measured using isothermal titration calorimetry for the binding of ABH and BEC to the human and rat enzymes (Table 1) [18]. However, while high resolution structures of ABH complexes with rat arginase I and human arginase I suggest that shorter and hence stronger hydrogen bonds contribute to the ~20-fold higher affinity of the human arginase I-ABH complex [18], a similar comparison cannot be made reliably for the binding of NOHA or nor-NOHA due to the low resolution of crystal structure determinations of their complexes with rat arginase I [21]. Notably, L-lysine binds to human arginase I with Kd = 13.1 μM based on our surface plasmon resonance measurements, suggesting a much higher affinity than that implied by the Ki, value of 2.5 mM reported by Ikemoto and colleagues [34]. We cannot explain this apparent discrepancy.
The 1.55 Å resolution crystal structure of the human arginase I complex with nor-NOHA (Figure 2a) reveals that inhibitor binding does not cause any significant conformational changes in the active site, and the r.m.s. deviation is 0.20 Å for 312 Cα atoms in comparison with the unliganded enzyme. Superposition with the 2.80 Å resolution crystal structure of the rat arginase I-nor-NOHA complex [21] shows reasonable overlap (Figure 2b), but the higher resolution structure of the human arginase I-nor-NOHA complex clearly allows for a more accurate description of inhibitor binding interactions. For example, the previously-determined low resolution structure did not reveal the complete array of hydrogen bond interactions expected for the α-carboxylate and α-amino groups of nor-NOHA [21], but the higher resolution structure clearly reveals that these substituents are anchored in the active site of human arginase I by three direct and four water-mediated hydrogen bonds with protein residues (Figure 2a). Additionally, while the higher resolution structure confirms that the Nζ-OH group of nor-NOHA displaces the metal-bridging hydroxide ion and nearly symmetrically bridges the binuclear manganese cluster, metal ion coordination geometry is much improved with Mn2+A---O and Mn2+B---O coordination distances of 2.1 Å and 2.2 Å, respectively. Additionally clarified is the hydrogen bond between the hydroxyguanidinium Nζ–H group and D128. Finally, the hydroxyguanidinium Nδ-H group does not make any hydrogen bond interactions.
Figure 2.

(a) Stereoview of a simulated annealing gradient omit map contoured at 3.3σ, in which nor-NOHA bound in the active site of human arginase I (monomer A) was omitted from the structure factor calculation. Manganese coordination and hydrogen bond interactions are indicated by red and green dashed lines, respectively. Atom color codes: carbon (yellow), oxygen (red), nitrogen (blue), manganese (violet). (b) Superposition of the human arginase I-nor-NOHA complex (color coded as in (a)) and the rat arginase I-nor-NOHA complex (blue) [21].
Interestingly, the hydroxyguanidinium group is sandwiched between the imidazole side chains of H141 and H126 and appears to make π–π stacking interactions (Figure 2a). Given its pKa value of 8.1, the hydroxyguanidinium group may be in the protonated, positively-charged state. However, given that the hydroxyguanidinium group interacts with the side chain of H141, which may have an elevated pKa due to its hydrogen bond with E277, the protonation of both the hydroxyguaninium group of nor-NOHA and the imidazole side chain of H141 would result in a repulsive interaction. If the hydroxyguanidinium group of nor-NOHA is not protonated, its packing interactions with H141 and H126 would be reminiscent of favorable packing interactions in stacked amide-π complexes [35]. Alternatively, H141 may be in the neutral imidazole state and the inhibitor hydroxyguanidinium group may be protonated. This possibility is outlined in further detail in the Discussion section.
The 2.04 Å resolution crystal structure of the human arginase I complex with NOHA (Figure 3a) similarly reveals that inhibitor binding does not cause any significant conformational changes in the active site, and the r.m.s. deviation is 0.30 Å for 313 Cα atoms in comparison with the unliganded enzyme. Superposition with the 2.90 Å resolution crystal structure of the rat arginase I-NOHA complex [21] shows reasonable overlap (Figure 3b), but here too the higher resolution structure of the human arginase I-NOHA complex clearly allows for a more accurate description of inhibitor binding interactions. Specifically, the higher resolution structure clearly reveals that the α-carboxylate and α-amino substituents make three direct and four water-mediated hydrogen bonds with protein residues (Figure 3a), exactly as observed for the binding of nor-NOHA (Figure 2a) and other inhibitors such as ABH [18]. Additionally, the geometries of metal coordination interactions in the binuclear manganese cluster are significantly improved: the Nη-OH group of NOHA displaces the metal-bridging hydroxide ion and nearly symmetrically bridges the binuclear manganese cluster with Mn2+A---O and Mn2+B---O coordination distances of 2.0 Å and 2.2 Å, respectively. A new interaction is also revealed in the higher resolution structure: the hydroxyguanidinium Nη-H group donates a hydrogen bond to D128, as also observed in the complex with nor-NOHA. Additionally new in the higher resolution structure is the hydrogen bond between the hydroxyguanidinium η-NH2 group of NOHA and T246. The hydroxyguanidium Nε-H group does not make any hydrogen bond interactions. As observed for nor-NOHA, the hydroxyguanidinium group is sandwiched between the imidazole side chains of H141 and H126 and appears to make π–π stacking interactions (Figure 3a).
Figure 3.

(a) Stereoview of a simulated annealing gradient omit map contoured at 2.6σ, in which NOHA bound in the active site of human arginase I (monomer A) was omitted from the structure factor calculation. Manganese coordination and hydrogen bond interactions are indicated by red and green dashed lines, respectively; atoms are color coded as in Figure 2. (b) Superposition of the human arginase I-NOHA complex (color coded as in (a)) and the rat arginase I-NOHA complex (blue) [21].
The 1.90 Å resolution crystal structure of the human arginase I complex with L-lysine (Figure 4a) reveals that the binding of this product analogue does not cause any significant conformational changes in the active site; the r.m.s. deviation is 0.24 Å for 313 Cα atoms in comparison with the unliganded enzyme. Superposition with the 2.50 Å resolution crystal structure of arginase from B. caldovelox complexed with L-lysine [36] shows excellent overlap (Figure 4b), and inhibitor binding interactions in the active site of the human enzyme are identical to those observed in the bacterial enzyme active site. The L-lysine side chain binds with an extended conformation, and the Nζ atom is presumably protonated to make a hydrogen bonded salt link interaction with the metal-bridging hydroxide ion. As observed for the binding of nor-NOHA and NOHA, the α-carboxylate and α-amino groups of L-lysine are anchored by three direct and four water-mediated hydrogen bonds.
Figure 4.

(a) Stereoview of a simulated annealing gradient omit map contoured at 2.8σ, in which L-lysine bound in the active site of human arginase I (monomer A) was omitted from the structure factor calculation. Manganese coordination and hydrogen bond interactions are indicated by red and green dashed lines, respectively; atoms are color coded as in Figure 2. (b) Superposition of the human arginase I-L-lysine complex (color coded as in (a)) and the B. caldovelox arginase-L-lysine complex (blue) [36].
Discussion
Since the crystal structures of human arginase I complexed with nor-NOHA and NOHA are determined at much higher resolutions than the corresponding complexes with rat arginase I [21], several features of enzyme-inhibitor binding interactions that were vague or ambiguous in the previously-determined structures are now clarified as summarized schematically in Figure 5 for nor-NOHA. Specifically, it is clear that the hydroxyl group of each inhibitor displaces the metal-bridging hydroxide ion of the native enzyme (Figures 2 and 3), and metal ion coordination geometry is significantly improved with Mn2+---O coordination distances of 2.0–2.2 Å. Also clarified in the higher resolution structure determinations are hydrogen bond interactions between each N-hydroxyguanidinium group and the side chains of D128 and T246: D128 accepts a hydrogen bond from the hydroxyguanidinium η-NH group, and T246 accepts a hydrogen bond from the hydroxyguanidinium η-NH2 group. These interactions were not readily apparent in the previous low resolution crystal structures [21].
Figure 5.

Summary of intermolecular interactions in the human arginase I-nor-NOHA complex. Metal coordination and hydrogen bond interactions are indicated by red and green dashed lines, respectively. We speculate that the zwitterionic form of the N-hydroxyguanidine moiety may bind as illustrated.
In the human arginase I complexes with nor-NOHA and NOHA, the average N-O---Mn2+ coordination angle is 121°. It is not clear whether metal coordination facilitates ionization of the N-OH moiety to generate a metal-bridging oxyanion. Although we are not aware of a pKa determination for the ionization of N-hydroxyguanidine to form the N-hydroxyguanidine oxyanion, the pKa of a structurally similar oxime moiety (C=N-OH) is ~11, so it is possible that the pKa of the hydroxyguanidinium hydroxyl group is sufficiently lowered by metal coordination to facilitate ionization. Moreover, since the pKa of the positively-charged N-hydroxyguanidinium moiety is 8.1 [37], it is possible that the hydroxyguanidinium group binds as a zwitterion. Such a binding mode would enhance charge-charge interactions with the binuclear manganese cluster as well as D128 as illustrated in Figure 5.
It is notable that neither the hydroxyguanidinium Nδ-H group of nor-NOHA nor the Nε-H group of NOHA make hydrogen bond interactions in the enzyme active site, suggesting that this group is dispensable in the design of tight binding inhibitors. The substitution of these N-H groups with CH2 groups yields N-hydroxy-nor-indospicine and N-hydroxyindospicine, respectively, which exhibit nearly equal inhibitory potencies to those of nor-NOHA and NOHA, respectively [38].
The molecular recognition of the amino acid moiety of inhibitors is a key determinant of enzyme-inhibitor affinity [25], and high resolution X-ray crystal structures of rat arginase I complexed with ABH, and human arginase I complexed with ABH and BEC, reveal that the α-amino and α-carboxylate groups of each inhibitor make 3 direct and 4 water-mediated hydrogen bond interactions with the protein [6,18]. The previously-determined structures of rat arginase I complexed with nor-NOHA and NOHA did not reveal a comparable array of hydrogen bond interactions due to the low resolution of the structure determinations [21]. Here, the higher resolution views of the enzyme-inhibitor complexes reveal that nor-NOHA and NOHA make an identical array of hydrogen bond interactions with their α-amino and α-carboxylate groups; furthermore, these hydrogen bond interactions are identical to those observed in the rat arginase I complex with ABH [6], the human arginase I complexes with ABH and BEC [18], and the human arginase I complex with L-lysine (Figure 4). These hydrogen bond interactions are summarized schematically in Figure 5 for the human arginase I-nor-NOHA complex and serve to rationalize affinity trends measured for derivatives of nor-NOHA and NOHA. In particular, deletion of the α-carboxylate group diminishes the inhibitory potency of nor-NOHA 375-fold, and deletion of the α-amino group diminishes affinity more than 6000-fold [17]. These data indicate that the interactions of the α-amino group are more critical for affinity, perhaps due to the salt link interaction with D183.
With high resolution structures of the human arginase I complexes with nor-NOHA and NOHA now in hand, we can formulate a hypothesis as to why nor-NOHA binds more tightly than NOHA. Overall, it appears that the longer side chain of NOHA compared with nor-NOHA results in the hydroxyguanidinium group being “pushed” more deeply into the active site (Figure 6). Consequently, the metal coordination geometry is less optimal in the complex with NOHA than in the complex with nor-NOHA, in that the metal-bridging hydroxyl group of NOHA is 1.3 Å away from the position of the metal-bridging hydroxide ion of the native enzyme; in comparison, the metal-bridging hydroxyl group of nor-NOHA is only 0.7 Å away (Figure 6). Given that the metal-bridging hydroxyl groups of the best arginase inhibitors ABH and nor-NOHA occupy a position closer to that of the metal-bridging hydroxide ion of the native enzyme, this coordination site appears to be a “hot spot” for tight binding inhibitors.
Figure 6.

Superposition of human arginase I structures: native (red; the metal-bridging hydroxide ion is a smaller red sphere), nor-NOHA complex (blue), and NOHA complex (green). Active site Mn2+ ions are large spheres color coded according to their respective structures. Note that the hydroxyl oxygen of nor-NOHA lies closer to the position of the metal-bridging hydroxide ion of the native enzyme.
In closing, it is interesting to note that NOHA is an intermediate in the reaction catalyzed by nitric oxide synthase [39–42], and the X-ray crystal structure of inducible nitric oxide synthase complexed with NOHA has been reported [43]. Given that the concentration of NOHA in human plasma is 9.1 μM [44], and given that Kd = 3.6 μM for the human arginase I-NOHA complex (Table 1), it is possible that circulating concentrations of NOHA are sufficient to achieve appreciable inhibition of arginase activity in vivo. It is further notable that exogenously administered NOHA significantly reduces arginase activity in cultured endothelial cells [45] and lipopolysaccharide-stimulated macrophages [46]. Thus, the high resolution crystal structures of human arginase I complexed with NOHA and nor-NOHA not only provide a platform for clarifying structure-affinity relationships for tight binding arginase inhibitors, but these structures also illuminate a chemical and physiological relationship between two key enzymes of L-arginine metabolism in living systems.
Acknowledgments
This work was supported by NIH grant GM49758.
We thank the laboratory of Prof. Ronen Marmorstein at the Wistar Institute for assistance with the isothermal titration calorimeter, and we thank Dr. Steven Seeholzer, Dr. Hua Ding, and the Protein Core Facility at Children’s Hospital of Philadelphia for assistance with the surface plasmon resonance experiments. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines of the Advanced Photon Source, supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
Atomic coordinates of human arginase I complexed with Nω-hydroxy-nor-L-arginine (nor-NOHA), Nω-hydroxy-L-arginine (NOHA), and L-lysine have been deposited in the Research Collaboratory for Structural Bioinformatics (http://www.rcsb.org/pdb) with the following accession codes: 3KV2, 3LP7, 3LP4.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Christianson DW. Acc Chem Res. 2005;101:191–201. doi: 10.1021/ar040183k. [DOI] [PubMed] [Google Scholar]
- 2.Dowling DP, Di Costanzo L, Gennadios HA, Christianson DW. Cell Mol Life Sci. 2008;65:2039–2055. doi: 10.1007/s00018-008-7554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris SM., Jr Br J Pharmacol. 2009;157:922–930. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krebs HA, Henseleit K. Z Physiol Chem. 1932;210:33–66. [Google Scholar]
- 5.Baggio R, Emig FA, Christianson DW, Ash DE, Chakder S, Rattan S. J Pharmacol Exp Ther. 1999;290:1409–1416. [PubMed] [Google Scholar]
- 6.Cox JD, Kim NN, Traish AM, Christianson DW. Nat Struct Biol. 1999;6:1043–1047. doi: 10.1038/14929. [DOI] [PubMed] [Google Scholar]
- 7.Kim NN, Cox JD, Baggio RF, Emig FA, Mistry SK, Harper SL, Speicher DW, Morris SM, Jr, Ash DE, Traish A, Christianson DW. Biochemistry. 2001;40:2678–2688. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]
- 8.Maarsingh H, Leusink J, Zaagsma J, Meurs H. Eur J Pharmacol. 2006;546:171–176. doi: 10.1016/j.ejphar.2006.07.041. [DOI] [PubMed] [Google Scholar]
- 9.Maarsingh H, Zuidhof AB, Bos IS, van Duin M, Boucher JL, Zaagsma J, Meurs H. Am J Respir Crit Care Med. 2008;178:565–573. doi: 10.1164/rccm.200710-1588OC. [DOI] [PubMed] [Google Scholar]
- 10.Baggio R, Elbaum D, Kanyo ZF, Carroll PJ, Cavalli RC, Ash DE, Christianson DW. J Am Chem Soc. 1997;119:8107–8108. [Google Scholar]
- 11.Zakharian TY, Di Costanzo L, Christianson DW. J Am Chem Soc. 2008;130:17254–17255. doi: 10.1021/ja807702q. [DOI] [PubMed] [Google Scholar]
- 12.Custot J, Moali C, Brollo M, Boucher JL, Delaforge M, Mansuy D, Tenu JP, Zimmermann JL. J Am Chem Soc. 1997;119:4086–4087. [Google Scholar]
- 13.Daghigh F, Fukuto JM, Ash DE. Biochem Biophys Res Commun. 1994;202:174–180. doi: 10.1006/bbrc.1994.1909. [DOI] [PubMed] [Google Scholar]
- 14.Custot J, Boucher JL, Vadon S, Guedes C, Dijols S, Delaforge M, Mansuy D. J Biol Inorg Chem. 1996;1:73–82. [Google Scholar]
- 15.Shin H, Cama E, Christianson DW. J Am Chem Soc. 2004;126:10278–10284. doi: 10.1021/ja047788w. [DOI] [PubMed] [Google Scholar]
- 16.Collet S, Carreaux F, Boucher J-L, Pethe S, Lepoivre M, Danion-Bougot R, Danion D. J Chem Soc, Perkin Trans. 2000;I:177–182. [Google Scholar]
- 17.Cama E, Pethe S, Boucher JL, Han S, Emig FA, Ash DE, Viola RE, Mansuy D, Christianson DW. Biochemistry. 2004;43:8987–8999. doi: 10.1021/bi0491705. [DOI] [PubMed] [Google Scholar]
- 18.Di Costanzo L, Sabio G, Mora A, Rodriguez PC, Ochoa AC, Centeno F, Christianson DW. Proc Natl Acad Sci USA. 2005;102:13058–13063. doi: 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colleluori DM, Ash DE. Biochemistry. 2001;40:9356–9362. doi: 10.1021/bi010783g. [DOI] [PubMed] [Google Scholar]
- 20.Cama E, Colleluori DM, Emig FA, Shin H, Kim SW, Kim NN, Traish AM, Ash DE, Christianson DW. Biochemistry. 2003;42:8445–8451. doi: 10.1021/bi034340j. [DOI] [PubMed] [Google Scholar]
- 21.Cox JD, Cama E, Colleluori DM, Pethe S, Boucher JL, Mansuy D, Ash DE, Christianson DW. Biochemistry. 2001;40:2689–2701. doi: 10.1021/bi002318+. [DOI] [PubMed] [Google Scholar]
- 22.Hunter A, Downs CE. J Biol Chem. 1945;157:427–446. [Google Scholar]
- 23.Cittadini D, Pietropaolo C, de Cristofaro D, D’Ayjello-Caracciolo M. Nature. 1964;203:643–644. doi: 10.1038/203643a0. [DOI] [PubMed] [Google Scholar]
- 24.Fuentes JM, Campo ML, Soler G. Arch Physiol Biochem. 1994;102:255–258. doi: 10.3109/13813459409003940. [DOI] [PubMed] [Google Scholar]
- 25.Shishova EY, Di Costanzo L, Emig FA, Ash DE, Christianson DW. Biochemistry. 2009;48:121–131. doi: 10.1021/bi801911v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Costanzo L, Pique ME, Christianson DW. J Am Chem Soc. 2007;129:6388–6389. doi: 10.1021/ja071567j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCoy AJ, Grosse-Kunstleve RW, Stroni LC, Read RJ. Acta Crystallogr, Sect D. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 28.Redinbo MR, Yeates TO. Acta Crystallogr, Sect D. 1993;49:375–380. doi: 10.1107/S090744499300294X. [DOI] [PubMed] [Google Scholar]
- 29.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr, Sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J App Cryst. 1993;26:283–291. [Google Scholar]
- 31.Cama E, Shin H, Christianson DW. J Am Chem Soc. 2003;125:13052–13057. doi: 10.1021/ja036365b. [DOI] [PubMed] [Google Scholar]
- 32.Roden LD, Myszka DG. Biochem Biophys Res Comm. 1996;225:1073–1077. doi: 10.1006/bbrc.1996.1297. [DOI] [PubMed] [Google Scholar]
- 33.Karlsson R, Fält A. Immunol Methods. 1997;2000:121–133. doi: 10.1016/s0022-1759(96)00195-0. [DOI] [PubMed] [Google Scholar]
- 34.Ikemoto M, Tabata M, Miyake T, Kono T, Mori M, Totani M, Murachi T. Biochem J. 1990;270:697–703. doi: 10.1042/bj2700697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imai YN, Inoue Y, Nakanishi I, Kitaura K. J Comput Chem. 2009;30:2267–2276. doi: 10.1002/jcc.21212. [DOI] [PubMed] [Google Scholar]
- 36.Bewley MC, Jeffrey PD, Patchett ML, Kanyo ZF, Baker EN. Structure. 1999;7:435–448. doi: 10.1016/s0969-2126(99)80056-2. [DOI] [PubMed] [Google Scholar]
- 37.Fukuto JM. Methods Enzymol. 1996;268:365–375. doi: 10.1016/s0076-6879(96)68039-2. [DOI] [PubMed] [Google Scholar]
- 38.Moali C, Brollo M, Custot J, Sari MA, Boucher JL, Stuehr DJ, Mansuy D. Biochemistry. 2000;39:8208–8218. doi: 10.1021/bi992992v. [DOI] [PubMed] [Google Scholar]
- 39.Pufahl RA, Nanjappan PG, Woodard RW, Marietta MA. Biochemistry. 1992;31:6822–6828. doi: 10.1021/bi00144a024. [DOI] [PubMed] [Google Scholar]
- 40.Stuehr DJ, Kwon NS, Nathan CF, Griffith OW, Feldman PL, Wiseman J. J Biol Chem. 1991;266:6259–6263. [PubMed] [Google Scholar]
- 41.Masters BSS, McMillan K, Sheta EA, Nishimura JS, Roman LJ, Martasek P. FASEB J. 1996;10:552–558. doi: 10.1096/fasebj.10.5.8621055. [DOI] [PubMed] [Google Scholar]
- 42.Pfeiffer S, Mayer B, Hemmens B. Angew Chem Int Ed. 1999;38:1714–1731. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1714::AID-ANIE1714>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Crane BR, Arvai AS, Ghosh S, Getzoff ED, Stuehr DJ, Tainer JA. Biochemistry. 2000;39:4608–4621. doi: 10.1021/bi992409a. [DOI] [PubMed] [Google Scholar]
- 44.Meyer J, Richter N, Hecker M. Anal Biochem. 1997;247:11–16. doi: 10.1006/abio.1997.2008. [DOI] [PubMed] [Google Scholar]
- 45.Buga GM, Singh R, Pervin S, Rogers NE, Schmitz DA, Jenkinson CP, Cederbaum SD, Ignarro LJ. Am J Physiol Heart Circ Physiol. 1996;271:H1988–H1998. doi: 10.1152/ajpheart.1996.271.5.H1988. [DOI] [PubMed] [Google Scholar]
- 46.Hecker M, Nematollahi H, Hey C, Busse R, Racké K. FEBS Lett. 1995;359:251–254. doi: 10.1016/0014-5793(95)00039-c. [DOI] [PubMed] [Google Scholar]