

Abstract
The creation of vascularized engineered tissues of clinically relevant size is a major challenge of tissue engineering. While it is known that endothelial and mural vascular cells are integral to the formation of stable blood vessels, the specific cell type and optimal conditions for engineered vascular networks are poorly understood. To this end, we investigated the vasculogenic potential of human mesenchymal stem cell (MSC) populations derived from three different sources: (i) bone marrow aspirates, (ii) perivascular cells from umbilical cord vein, and (iii) perivascular cells from umbilical cord artery. Cell populations were isolated and identified as MSCs according to their phenotypes and differentiation potential. Human umbilical vein endothelial cells (HUVEC) were used as a standard for endothelial cells. A novel co-culture system was developed to study cell-cell interactions in a spatially controlled three-dimensional (3D) fibrin hydrogel model. Using microfluidic patterning, it was possible to localize hydrogel-encapsulated HUVECs and MSCs within separate channels spaced at 500, 1000 or 2000 μm. All three MSC populations had similar expression profiles of mesenchymal cell markers, and similar capacity for osteogenic and adipogenic differentiation. However, bone marrow-derived MSCs (but not umbilical vein or artery derived MSCs) showed strong distance-dependent migration toward HUVECs and supported the formation of stable vascular networks resembling capillary-like vasculature. The presented approach provides a simple and robust model to study cell-cell communication of relevance to engineering vascularized tissues.
Keywords: Human mesenchymal stem cells, human endothelial cells, vascularization, micropatterning, cell co-culture, tissue engineering, hydrogel
1. Introduction
Normal tissue function requires vascular supply. Unravelling mechanisms of blood vessel formation would offer therapeutic solutions to a large variety of disorders (Carmeliet 2003). In particular, immediate vascularization of implanted tissues is critical for their survival and function (Laschke et al., 2006), due to the limitations in oxygen and nutrient supply (Jain et al., 2005). During culture, tissue constructs can be supplied with nutrients by medium perfusion (Radisic et al., 2008). However, vascular ingrowth in vivo is usually too slow to ensure graft survival (Rouwkema et al., 2008).
One approach to provide functional blood supply would be the in vitro creation of vascular infrastructure that could anastomose with the host vasculature (Jain et al., 2005). In general, a better understanding of the interplay between the cells, signalling molecules and the physical microenvironment is necessary to develop effective techniques for vascularization (Johnson et al., 2007). Previous studies have used microtechnologies to probe the angiogenic microenvironment (Sarkar et al., 2006, Sudo et al., 2009), with control of cell positioning (Vickerman et al,. 2008) and focus on the effects of cytokines and smooth muscle cells on endothelial sprouting in these studies (Chung et al., 2009).
Endothelial cells (ECs) self-assemble into vascular tubes when surrounded by extracellular matrix (Davis et al., 2002) and exposed to angiogenic factors (Conway et al., 2001). To complete the process of new blood vessel formation, ECs must functionally interact with mural cells, such as pericytes or vascular smooth muscle cells (SMCs) that in turn provide for vascular maturation and stability (Gerhardt and Betsholtz 2003).
In vitro models of angiogenesis provide an excellent tool for studying cell-cell interactions in the context of paracrine and autocrine signaling, extracellular matrix, and heterotypic cell-cell contacts (Hirschi et al., 1998; Ding et al., 2004; Sorrell et al., 2007, 2009, Vickerman et al., 2008). Growth factors released from ECs induce the recruitment of MSCs along angiogenic sprouts (Lindahl et al., 1997; Hellstrom et al., 1999). Upon contact with ECs, mesenchymal precursors from bone marrow (but not from adipose tissue) differentiate into pericyte cells, by processes involving local activation of transforming growth factor (TGF)-β (Hirschi et al., 1998; Ding et al., 2004) and resulting in EC quiescence (Antonelli-Orlidge et al., 1989; Sato and Rifkin, 1989) and ECM assembly (Pepper, 1997; Davis et al., 2002). These studies have shown that MSCs from certain sources support the formation of stable vascular tubes by providing both angiogenic (Sorrell et al., 2009; Au et al., 2008) and stabilization factors (Armulik et al., 2005).
We investigated functional interactions between human mesenchymal and endothelial cells, using a simple yet highly controllable hydrogel system. Cells were encapsulated in 3-dimensional (3D) hydrogel that was printed using microfluidic patterning, to enable precise control of the distance between the two cell populations (500, 1000 or 2000 μm). This way, we were able to vary the relative contributions of cell contact and diffusing signals. To explore the capacity of different mesenchymal sources to support endothelial vascular networks, we isolated and characterized human MSC populations from three different sources: bone marrow aspirates, perivascular cell populations from umbilical cord vein and artery, and studied their supporting roles to vasculogenesis in the micropatterned hydrogel model.
2. Materials and methods
Cell isolation
HUVEC and MSC cells from umbilical cord vein (UCV)
Fresh umbilical cords (UC) of term delivery were processed according to a protocol approved by the Columbia University Institutional Review Board. Endothelial and MSC-like cells were harvested by a standard protocol for HUVEC isolation (Marin et al., 2001; Romanov et al., 2003). Briefly, the umbilical cord segment was disinfected in 70% ethanol and rinsed with phosphate-buffered saline (PBS; Gibco, USA). The vein lumen was then infused with PBS, 0.1% type VI collagenase solution (Sigma, USA) and the cord segment was clamped at both ends. Enzymatic digestion was performed for 15 min at 37°C. The cell suspension was collected by extensive washing with PBS and pelleted by centrifugation at 250 g for 5 min. Cells were suspended in endothelial cell medium (EGM-2; Cambrex, USA) and plated in 75 cm2 tissue culture flasks. Heterogeneous culture of attached cells was subsequently sorted by florescence activated cell sorting (FACS) and the CD90+ cells were designed as UCV cells.
MSC-like cells from umbilical cord artery (UCA)
Cells from the vessel wall of the UCA were isolated by tissue explant method (Ray et al., 2001). Briefly, the segment of the artery was extracted and the surrounding tissue was removed. The perivascular tissue was dissected into small pieces (1–2 mm), that were placed into to a Petri dish, and the culture medium was added. The MSC culture medium consisted of Dulbecco’s Modified Eagle’s Medium–high glucose (DMEM-HG; Gibco), 10% fetal bovine serum (FBS; Gibco), 1% Penicillin-Streptomycin solution (Pen-Strep; Gibco) and 5 ng/mL bFGF (Invitrogen). Cells that spread out of the explants were harvested by trypsin/EDTA (Sigma) and reseeded at the density of 7,000 cells/cm2. EGM-2 (Clonetics) was used for later cultures. Cells were isolated by florescence activated cell sorting (FACS) and the CD90+ cells were designed as UCA cells.
MSC from bone marrow
Cryopreserved mononuclear cells (designated as MSC) were obtained from a commercial source (Cambrex, USA) and plated in 150 cm2 flasks at a density of 30,000 cells/cm2. Non-adherent cells were removed with subsequent media changes and the adherent cells were cultured to 80% confluence. Subconfluent cells were harvested by trypsin/EDTA and reseeded at the density of 5,000 cells/cm2.
Fluorescence-activated cell sorting (FACS) of morphologically heterogeneous UCV-derived cells was performed by MoFlo high-speed cell sorter (Dako, Italy). Confluent cells were harvested by trypsin/EDTA, pelleted and resuspended (105–106 cells in 100 μL of PBS). Cells were stained with anti-human CD90 (Thymocyte differentiation antigen-1 Thy-1) conjugated with R-Phycoerythrin (RPE) (Dako, Italy) for 30 min, or indirectly labelled with mouse anti-human CD90 for 20 min (Millipore, USA), washed with PBS and incubated for 15 min with the secondary antibody goat anti-mouse immunoglobulin G (IgG), fluorescein-isothiocyanate (FITC) conjugate (Millipore, USA). Labelled cells were washed with PBS, passed through 40 μm cell strainer, and suspended in PBS for sorting (106 cells per 100 μL PBS). CD90 positive and negative populations were gated and sorted into collection tubes. Cells were suspended in EGM-2 medium and plated in tissue culture flasks at the density of 8,000 cells/cm2. The two sorted cell populations, CD90+ and CD90−, were expanded in EGM-2 medium and characterized to verify the MSC character of CD90+ cells and the endothelial character of CD90− cells.
Differentiation study
MSCs (passage 3) and the perivascular MSC-like populations from UCV and UCA (passages 6, 7) were treated with induction media as described below to stimulate their osteogenic and adipogenic differentiation. Cells cultured in MSC culture medium were used for control. The medium was changed two times a week and cell differentiation was evaluated over 3 weeks of cultivation.
Adipogenic Differentiation was induced on confluent cells by adipogenic medium containing DMEM-HG, 10% FBS, 1 μM dexamethasone (Sigma, USA), 5 μg/ml recombinant human insulin (Sigma, USA), 0.5 mM 3-isobutyl-1-methyl-xanthine (IBMX; Sigma, USA), 0.2 mM indomethacin (Sigma, USA) and 1% Pen-Strep. Adipogenic cell differentiation was confirmed by Oil Red O staining. After 1, 2 and 3 weeks of differentiation, cells were fixed with 4% formaldehyde for 1 hour, rinsed with distilled water and stained with a working solution of 0.5% Oil Red O (Sigma, USA) for 20 min. Intracellular accumulation of lipid-rich vacuoles was visualized by light microscopy and the images were taken with Qimage colour camera.
Osteogenic Differentiation was induced by culturing confluent cells in osteogenic medium containing DMEM-HG, 10% FBS, 100 nM dexamethasone (Sigma, USA), 10 mM β-glycerophosphate (Sigma, USA), 0.1 mM ascorbic acid-2-phosphate (Sigma, USA) and 1% Pen-Strep. Mineralized matrix was stained by von Kossa after 3 weeks of culture. The cells fixed in 4% formaldehyde were incubated in 5% silver nitrate (Sigma, USA) for 1 hour under intense light, and incubated in 3% sodium thiosulfate (Sigma, USA) for 5 min to develop the signal. Quantitative determination of total calcium was done using the colorimetric Calcium (CPC) LiquiColor assay (Stanbio Laboratory, USA). Calcium was extracted by 5% trichloroacetic acid (200 μL per P48 well) overnight and determined spectrophotometrically at 550 nm following the reaction with o-cresolphthalein complexone. All assays were done in triplicate and expressed as absolute values (μg of Ca2+ per well) according to the standard curve.
Microfabrication of the mold
The design of the desired hydrogel micropattern was created in AutoCAD (Autodesk Inc.) and printed at high-resolution (CAD/Art Services, Inc., USA). The pattern consisted of two parallel channels, 1 cm long and 1000 μm wide, with a spacing of 500, 1000 or 2000 μm. The generated transparency mask was used in photolithography of SU-8 50 photoresist (Microchem, USA). The photoresist was spin-coated onto the glass wafer (Laurell).
The master was developed by immersion of the SU-8 wafer into developer propylene glycol methyl ether acetate (PGMEA; Sigma, USA) under strong agitation for 10 minutes to remove the non-crosslinked photoresist, and used for replica molding of degassed elastomer mix, composed of poly(dimethylsiloxane) (PDMS) and the curing agent in ratio 10:1 (Sylgard 184 Silicone elastomer kit; Dow Corning, USA). The curing of PDMS was conducted in an oven at 60°C for 2 hours. Solid PDMS was peeled off the master, the individual microfluidic molds were cut out with holes punched for fluid inlets. Plasma oxidation was used to render the PDMS mold surface hydrophilic, as required to pull the hydrogel through the channels via capillary action. The molds were plasma cleaned for 20 s, with the pattern side up. The PDMS surface was then blocked using a 5% solution of FBS (Gibco) in distilled water.
Micropatterning of cell-loaded hydrogels
The elastic PDMS mold was used to pattern the 3D cell-loaded hydrogel channels onto tissue culture plastics. Fibrin hydrogel was generated by polymerization of fibrinogen (Fraction I from pig plasma; Sigma, USA) by thrombin (from bovine plasma; Sigma, USA). The PDMS molds were reversibly sealed on 100 × 20 mm culture plates (BD Falcon, USA), with the patterns facing down. Endothelial cells and MSC populations (passage 3 for bone marrow-derived MSCs, passage 6 for UCVs, and passage 7 for UCAs) were encapsulated into separate channels, as follows. An endothelial cell pellet was suspended in a thrombin solution (8 U/mL) at high cell density, approximately 2 × 106 cells per 100 μL and mixed with a 1% fibrinogen solution in ratio 1:5. The suspension was immediately injected into one of the microchannels, and the suspension of one of the MSC populations was injected into the opposite channel using the same procedure. Approximately 20 μL of cell suspension was used per channel. The fibrinogen was allowed to polymerize for 15 minutes. The PDMS mold was carefully taken off the culture dish leaving the patterned hydrogel with encapsulated cells attached to the plastic surface. The hydrogel channels were then encapsulated in another layer of fibrin to provide uniform hydrogel matrix.
Cell migration assay
Mesenchymal cell populations from the three sources were assessed for their capacity to migrate through fibrin toward ECs and stabilize the vascular network. Cell sprouting was monitored within the first three days after encapsulation to assess chemotactic effect between the cells. Bright-field images of the channels were acquired, and a single cell sprout length was measured as the perpendicular distance of mesenchymal cell migration from their channel edge toward the opposite channel containing HUVECs, at a 200X magnification, using Metamorph Software caliper. For each cell type, the average and total lengths of all the sprouts in each image (showing 330 μm of the channel length) were determined. Three images were evaluated for each experimental condition, at 24 hr intervals. Single channels of mesenchymal cell populations in fibrin matrix were used as controls.
Live/dead assay for visualizing vascular network formation (LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells; Invitrogen) was used for simultaneous determination of live and dead cells. The co-culture systems were stained two weeks after encapsulation according to the manufacturer’s protocol. Live cells converted the virtually nonfluorescent calcein AM to the intensely green fluorescent calcein, while ethidium homodimer (EthD-1) bound to nucleic acids of cells with damaged membranes, thereby producing a bright red fluorescence in dead cells. The Live Dead staining was effective in visualizing vascular network formations due to its ability to highlight living cells
Immunofluorescence staining of the starting cell populations
Cells were characterized after 3 to 6 passages by immunofluorescence staining. Cells on cover glasses in 12- well culture plates were fixed with 4% formaldehyde for 15 minutes and washed with PBS. For intracellular antigen staining cell permeabilization with 0.1% Triton X-100 (Sigma) in PBS for 10 minutes was performed. After 1 hour-incubation in blocking serum (10% goat serum; Gibco, USA) cells were labeled with a primary antibody diluted 1:100 in PBS for 1 hour at room temperature. The following mouse anti-human antibodies were used: anti-CD90 (Millipore, USA), anti-CD105 (Abcam, USA), anti-CD31 (Sigma, USA) and anti-Smooth Muscle Actin α-isoform (α-SMA; Invitrogen, USA), anti-von Willebrand Factor (vWF, Invitrogen, USA). After several washings with PBS the cells were incubated with the secondary antibody anti-mouse IgG-FITC at 1:100 dilution in PBS for 30 minutes at room temperature. Finally, cells were washed with PBS, cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma) and cover slips were mounted using Fluoromount-G (Southern Biotech, USA).
Immunofluorescence staining of hydrogel-encapsulated cells
Micropatterned co-cultures of cells were immunostained for α-SMA and von Willebrand factor, with incubation times adjusted for the presence of the surrounding fibrin matrix. The samples were fixed with 4% formaldehyde for 30 minutes, rinsed and the cells were permeabilized with 0.1% Triton X-100 in PBS for 20 minutes. After incubation in the blocking serum (10% goat serum) for 2 hours, the mixture of primary antibodies: mouse anti-human α-SMA and rabbit anti-human von Willebrand factor (Sigma, USA) diluted 1:200 in PBS was added. Incubation was done overnight at room temperature under slow agitation. The samples were washed three times with PBS for 1 hour, stained with secondary antibodies: goat anti-mouse IgG-FITC and goat anti-rabbit IgG-RPE (Sigma, USA) at 200X dilution in PBS for 3 hours at room temperature, and washed three times with PBS (for 1 hour each time). Cell nuclei were counterstained with DAPI and the samples were observed under the fluorescent microscope immersed in PBS without any mounting.
Statistical analysis
The measured values were expressed as Average ± Standard Deviation. Statistical evaluation by unpaired t-test was used to assign statistically significant differences between the groups at defined levels of significance (p < 0.05 or p < 0.01).
3. Results
Cell phenotypes
Cell populations were characterized for the expression of the MSC markers (CD90, CD105), an early SMC marker (α-SMA) and an endothelial cell marker (CD31). The CD90+ fractions of mesenchymal cells from unmbilical vein, umbilical artery and bone marrow, designated as UCV, UCA and MSCs, respectively, had similar expression profiles of mesenchymal cell markers (Figure 1). All cells were uniformly positive for CD90 and CD105, partially positive for α-SMA, and negative for CD31, suggesting that the UCV and UCA perivascular cell populations are of a mesenchymal phenotype. As expected, HUVECs were strongly positive for CD105 and CD31, but negative for CD90 and α-SMA (Figure 2), consistent with the endothelial phenotype.
Figure 1. Phenotypic characterization of starting cell populations.
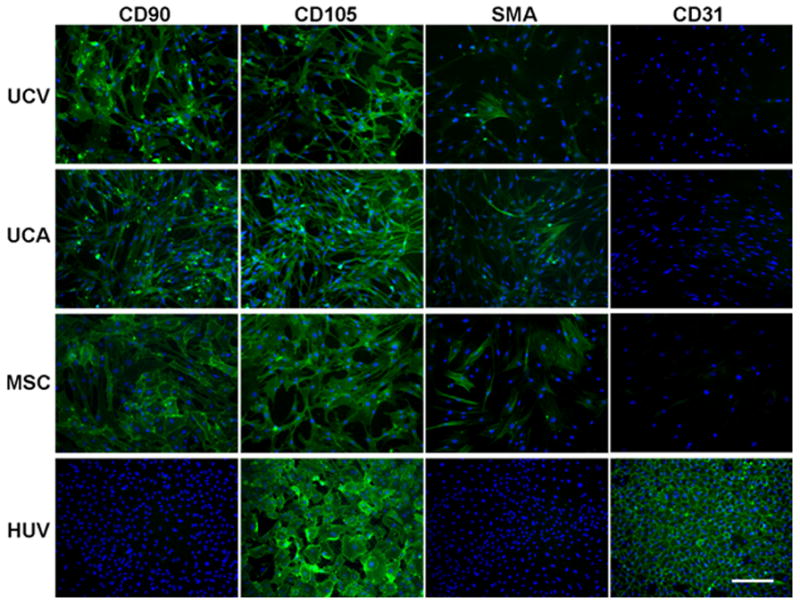
Immunofluorescence staining of umbilical cord vein (UCV) and artery (UCA) MSC-like cells, BM-derived MSCs (MSC) and HUVECs (HUV) for mesenchymal stem cell markers CD90 and CD105, early smooth muscle cell marker α-SMA (SMA) and endothelial cell marker CD31. Magnification 100×, calibration bar 200 μm.
Figure 2. Differentiation of mesenchymal stem cells into adipogenic and osteogenic lineages.

A Oil red O staining, visualizing lipid vacuoles in the cytoplasm of adipogenic treated cells, and von Kossa staining (scale bar: 1 mm) of osteogenic treated cultures of umbilical cord vein cells (UCV), artery cells (UCA) and bone marrow derived mesenchymal stem cells (MSC) showing Ca2+ deposits after 3 weeks of treatment (scale bar: 100 μm). The respective untreated controls are shown in the insets. B Bar charts showing Ca2+ contents in the cultures treated with EGM-2 (EGM), MSC culture medium (DMEM) and osteogenic medium (OST) after 3 weeks of treatment. ** represent a statistically significant difference between the osteogenic and the control cultures (p<0.01).
Osteogenic and adipogenic capacity of mesenchymal cell populations
To further assess the mesenchymal character of the UCV and UCA cells, their potential for differentiation into adipogenic and osteogenic lineages was compared to that of bone marrow derived MSCs. In all adipogenic cultures, the cells changed their morphology from fibroblast-like to large, rounded cells containing intracellular lipid vacuoles (Figure 2A). In contrast, non-treated cells remained fibroblast-like and no lipid accumulation was observed. These results demonstrated that similarly to bone marrow derived MSCs, the UCV and UCA cells exhibit adipogenic differentiation potential.
In osteogenic cultures, the accumulation of mineralized matrix was demonstrated by von Kossa staining after three weeks of treatment (Figure 2A), whereas no deposition was observed in the respective controls. Calcium deposits were more broadly distributed in cultures of MSCs in comparison to the UCV and UCA cells, where the depositions were more concentrated and coincided with the regions of multilayered cell condensations. The elevated calcium content in osteogenic cultures was further confirmed by the calcium colorimetric assay that showed a gradual increase of calcium content over the course of three weeks, in contrast to negligible levels of calcium in cultures of MSCs in EGM-2 medium (Figure 2B). After 3 weeks of osteogenic treatment, calcium content was slightly but not significantly higher in cultures of MSCs as compared to UCV and UCA cells. However, all three cell populations showed markedly and significantly higher calcium content than the control cultures.
Taken together, the data in Figure 2 show that the mesenchymal stem cell populations, although obtained with different number of passages (P3 for bone marrow MSCs, P6 for UCVs, P7 for UCAs), responded rather similarly to adipogenic and osteogenic induction, as verified by Oil red O staining (for adipogenic), and the von Kossa staining and measurements of the Ca2+ content (for osteogenic differentiation). In contrast, control cells from all groups lacked to accumulate lipid vacuoles or form Ca2+ deposits.
Co-cultures of mesenchymal and endothelial cells in micropatterned hydrogel
Microfluidic patterning techniques were employed to arrange cell-loaded hydrogel into two parallel channels spaced apart at 500 μm, 1000 μm or 2000 μm) (Figure 3A). Fibrin was used as a hydrogel, because of its appropriate biochemical and mechanical properties as a 3D matrix for angiogenensis in vitro (Lafleur et al., 2002), as well as due to its unique characteristic of rapid assembly, which was desirable for the generation of co-cultures. Fluorescent labelling of HUVECs and mesenchymal cells with red and green dyes, respectively, confirmed that it was possible to encapsulate the different cell types into separate fibrin channels (Figure 3B).
Figure 3. Micropatterned hydrogel system for 3-dimensional co-culture of mesenchymal and endothelial cells.
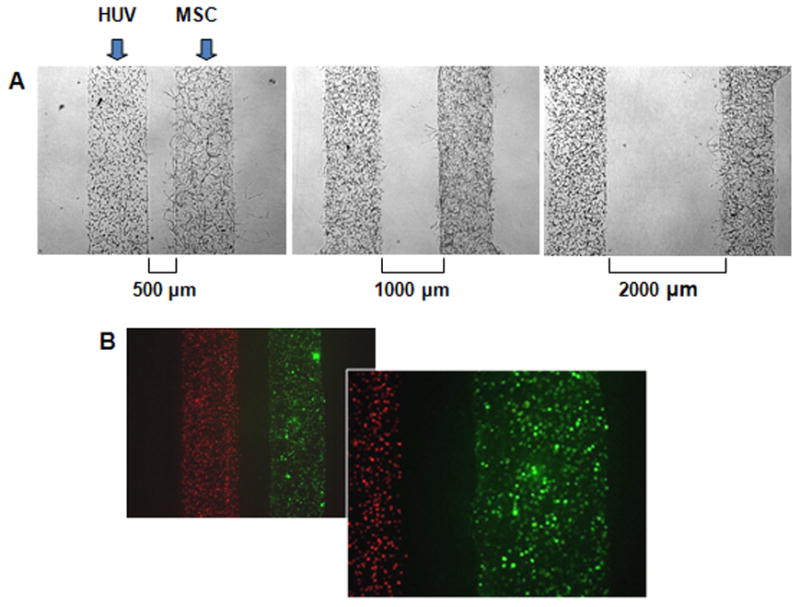
A Micropatterned fibrin channels loaded with HUVECs (left channel) and MSCs (right channel) with the distances between channels of 500, 1000 and 2000 μm. B Encapsulated HUVECs labelled with red intracellular dye (left channel) and MSCs labelled with green dye (right channel).
Directional migration of mesenchymal cells
Bone marrow derived MSCs demonstrated strong distance-dependent directional migration toward HUVECs, with highest extent of sprouting when these cells were placed at the closest (500 μm) initial distance from HUVECs (Figure 4). The extent of sprouting decreased as the channel-to-channel distance increased, and became indistinguishable from the control at the distance of 2000 μm. The total length of sprouts forming at the 500 μm channel-to-channel distance was already significantly different from the control after one day of culture (4,460 vs. 2,140 μm, respectively, p<0.05). By day 3 of culture, the total length of sprouts reached 86,800 μm for the 500 μm channel distance, and 30,300 μm for the control MSCs cultured without HUVECs (p < 0.01). For the 1,000 μm channel distance, the total length of sprouts was 52,000 μm, as compared to 30,300 μm for the control (p<0.01). These data suggest that the total length of sprouts depended on both the MSC-HUVEC distances and time in culture. Notably, the average length of sprouts was not significantly dependent on channel-to-channel distance. Taken together, the distance-dependent migration studies suggest that the chemotactic effects increase the number of cells undergoing sprouting, rather than causing the existing sprouts to increase in length.
Figure 4. Effects of cell source, channel spacing and culture time on directed migration of mesenchymal cells.
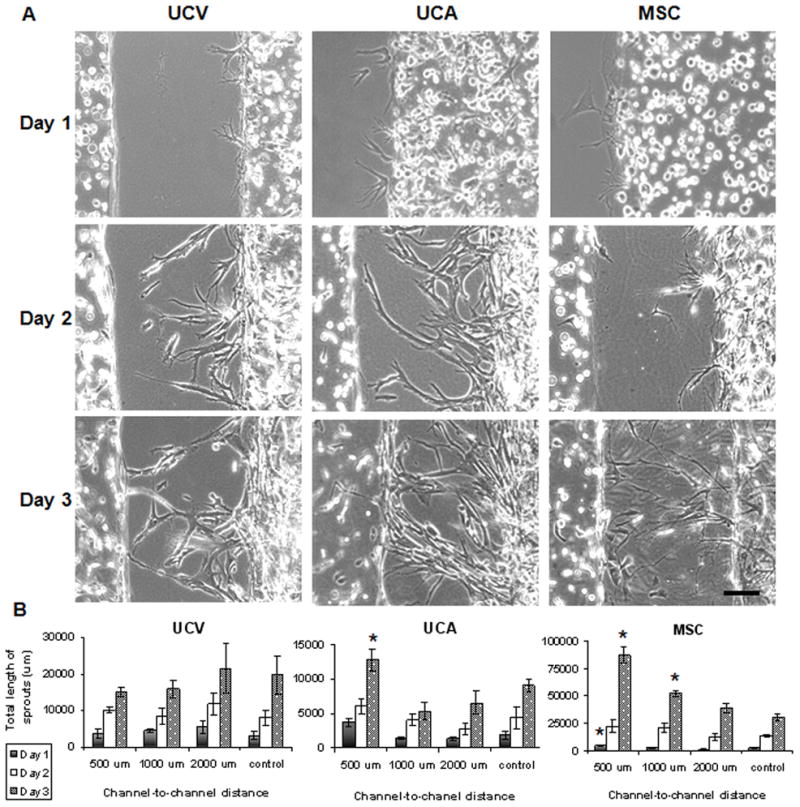
A Micropatterned fibrin channels loaded with HUVECs (left channel) and MSCs (right channel) 500 μm apart, demonstrating sprouting of the UC vein cells (UCV), artery cells (UCA) and BM-derived mesenchymal stem cells (MSC) for the first three days after cell encapsulation in hydrogel. Light microscopy, scale bar 100 μm. B The total lengths of sprouts for mesenchymal cells from different sources: UC vein cells (UCV), artery cells (UCA) and BM-derived mesenchymal stem cells (MSC). * statistically significant differences between groups (p < 0.05).
In contrast, the UCV cells showed no distance-dependent sprouting (Figure 4). The total lengths of sprouts were in the range of 3,100 to 5,450 μm on day one, and increased to 15,200–21,500 μm by day three. Likewise, the relative distance from HUVECs had only a modest effect on directed migration of UCA cells (Figure 4). The distance-dependence noticed on day one (3,770 versus 1,870 μm, p<0.05) diminished with time in culture (12, 800 versus 9,050 μm, p>0.05). Taken together, these results demonstrate significant differences in the migration behaviour of MSC populations in co-culture with the HUVECs. While MSCs exhibited strong chemotactic migration, UCA cells showed only moderate responsiveness, and UCV cells were unresponsive to the presence of HUVECs.
Vascular network formation
Bone marrow derived MSCs supported the formation of long, highly branched and interconnected tubular structures that resembled capillary networks. The initial distance between the MSCs and HUVECs did not affect vascular network formation. Once the MSCs came into direct contact with endothelial tubular structures (within 3–8 days), they were able to stabilize them irrespectively of the starting distance. The stabilized capillary-like networks were maintained in culture for more than two weeks, while tube-like structures formed by HUVECs alone gradually regressed. These vascular networks were also considerably different from the MSC sprouts formed without the contact with HUVECs. Even though the MSC sprouts were elongated and branched, the perimeter was distinctly smaller in comparison with the stabilized vascular tubes (Figure 5B).
Figure 5. Stabilization of the vascular network by mesenchymal stem cells.

Live/dead fluorescent staining of the HUVEC channel (right panels) and the MSC channel (left panels) after two weeks of culture. The images demonstrate cell organization in co-cultures of HUVECs and mesenchymal cells from various sources (UCV, UCA, bone marrow MSC). Stabilized vascular network is seen in the co-culture of HUVECs with MSCs, but not with UCV and UCA cells. Fluorescent microscopy, scale bar 500 μm.
In contrast to bone marrow derived MSCs, the UCV and UCA cells did not demonstrate stabilizing effects on endothelial tubular structures. Interestingly, when these cells populations reached HUVECs, the pre-formed tubular structures reorganized into round-shaped cell clusters. However, the UCV cells seemed to support the survival of HUVECs, even if they did not stabilize endothelial tubules, whereas UCA cells did not exert any apparent supporting effect on endothelial cells.
Differentiation of hydrogel-encapsulated mesenchymal cells into SMC-pericyte lineages
The expression of the early marker of SMC differentiation, α-SMA, showed remarkable differences in terms of the localization and relative quantity of α-SMA positive cells over the course of cultivation (Figure 6). On day 3 after encapsulation, only scarce α-SMA positive mesenchymal cells were observed. After 14 days of culture, strong α-SMA staining was observed at the sites of contact between HUVECs and migrated bone marrow MSCs, while only scarce α-SMA+ cells were present elsewhere. The staining revealed well-organized distribution of α-SMA+ cells that were co-aligned with endothelial tubular structures (Figure 6). Both controls of HUVEC and MSC cells alone showed no SMA staining by day 14 (Figure 6B). The expression of α-SMA and the co-alignment with endothelial tubes indicated pericyte-like manner of bone marrow MSCs (but not the UCA and UCV cells) on stabilization of the vascular networks, with cell-cell contact playing a major role in the process.
Figure 6. The progression of cell sprouting in the hydrogel co-culture system.
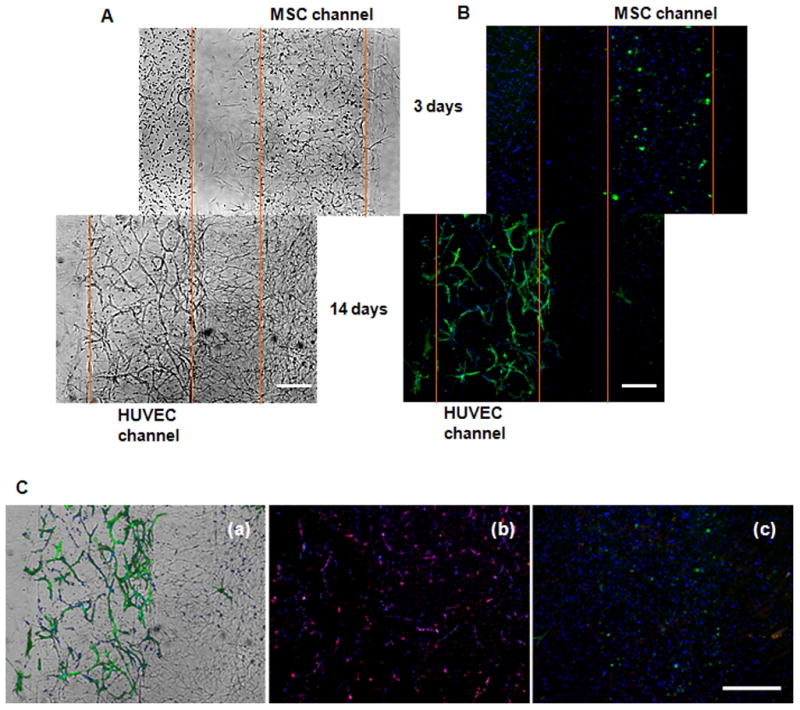
A Co-culture of HUVECs with bone marrow MSCs encapsulated within the separate channels at a 500 μm distance. Bright-field images show cell sprouting at days 3 and 14, as MSCs migrated towards HUVECs. Light microscopy, scale bar 250 μm. B The corresponding immunofluorescence stains. Green staining demonstrates α-SMA+ cells; nuclei are stained blue with DAPI. C Overlay of the bright-field and immunofluorescence images at day 14 (a), illustrating co-alignment of α-SMA+ cells with the endothelial tubular structures. No α-SMA+ cells were present in pure cultures of HUVECs (b) and MSCs (c). Red staining shows von Willebrand factor positive HUVECs (b). Scale bar: 500 μm.
4. Discussion
Understanding the process of new blood vessel formation has become the principal, yet challenging, objective of recent research activities (Rouwkema, et al., 2008). We present a novel 3D micropatterned hydrogel model designed to study interactions between endothelial and mesenchymal cell populations, both important players in neovascularization (Au et al., 2008). Hydrogel micropatterning allows the spatial positioning of these two cell populations at controllable distances (500, 1000 or 2000 μm). The proposed hydrogel model was developed to explore the nature of HUVEC-MSC communication and to compare the vasculogenic potential of different mesenchymal cell populations (from bone marrow, umbilical vein and umbilical artery). These cell populations were characterized for their phenotype, differentiation potential and capacity to directionally migrate and support the vascular network formation by HUVEC cells.
Previous studies have suggested that growth factors released from endothelial cells induce migration of undifferentiated mesenchymal cells toward endothelial cells. We hypothesized that EC-secreted factors establish a distance-dependent chemotactic gradient in fibrin matrix that stimulates directional migration of mesenchymal cells. The model enables study of paracrine interactions, by measuring the extent of directional migration of mesenchymal cells as a function of their distance from HUVECs and time in co-culture.
All three mesenchymal cell populations (MSC, UCV and UCA, Figure 1 & 2) demonstrated capacity for sprouting in fibrin hydrogel (Figure 5). Quantitative data revealed significant differences in their distance-dependent responses to HUVECs (Figures 4&5). MSCs from bone marrow demonstrated strong distance-dependent response, as the migration rate was highest at the closest (500 μm) initial distance from HUVECs, and decreased with the increasing distance (Figure 4). This behaviour is consistent with the model where endothelial cell-secreted chemokines regulate the migration process, in a concentration-dependent manner. In contrast to MSCs, UCV and UCA cells migrated relatively independently of the initial distance from HUVECs (no statistical difference was observed in either case). These results indicated that only bone marrow derived MSCs demonstrate strong chemotactic responses. Further studies should confirm the specific mechanism of mesenchymal cell migration to explain the different chemotactic responses of different mesenchymal cell populations to the proximate endothelial cells.
HUVECs are known to readily organize themselves into tubular structures (Lefleur et al., 2002), with the formation of intracellular vacuoles being an important step in the capillary lumen creation (Bayless et al., 2000; Davis et al., 2002; Kamei et al., 2006). Our culture system provided a suitable microenvironment for EC assembly into tubular network in terms of biochemical cues and biomechanical properties (Figures 3–6). As in other studies (Sorrell et al., 2007; 2009), the tubular network gradually regressed if not stabilized by the surrounding perivascular cells. In contrast, bone marrow MSCs greatly supported the formation of stable vascular networks, with long and highly branched tubular structures resembling a capillary network, for more than two weeks of culture (Figure 5).
The initial distance between the two cell types did not considerably affect the vascular network formation, even though MSCs reached HUVECs at different stages of their tubular assembly. In contrast to bone marrow MSCs, the UCV and UCA cells did not exert stabilizing effect on EC tubular structures, and instead caused the reorganization of HUVECs into cell clusters (Figure 5). Still, UCV cells better supported the survival of endothelial cells, even if they did not stabilize endothelial tubules, whereas UCA cells did not exert any supporting effect on endothelial cells.
Because UCV and UCA cell populations are derived from a perivascular tissue of large vessels, their adopted specialization could make them less efficient in supporting the microvasculature formation. Nonetheless, these two perivascular cell populations, especially UCA cells, exhibited pluripotency and outstanding 3D growth, with aligned and densely packed cells when cultured in a 3D fibrin hydrogel. This suggests their potential for some other tissue-engineering applications, where organized and highly condensed growth of cells is required, such as engineering of blood vessel grafts, which has also been an attractive area of tissue engineering research (Gong and Niklason, 2006).
Since the regressed tubular structures in HUVEC cultures did not express α-SMA, it is reasonable to assume that the surrounding MSCs were the source of α-SMA+ cells. Because bone marrow MSCs did not express α-SMA in early co-cultures, neither in the later cultures at the sites with no direct contact with ECs, their differentiation toward vascular SMC/pericyte lineages was presumably induced by direct contact with endothelial tubes. Our study supports the notion that mesenchymal cells function as vascular pericytes (Au et al., 2008; Sorrell et al., 2009). According to previous studies, local activation of TGF-β could be involved, as the same mechanism directs the differentiation of embryonic mesenchymal precursors 10T1/2 toward SMC lineage (Hirschi et al., 1998; Ding et al., 2004).
In summary, the hydrogel co-culture system developed in this study could serve as a novel platform for studying vasculogenesis under spatially controlled conditions. The ability to precisely define the arrangement of different cell types in a 3D hydrogel system provides a simple and robust model for studying cell-cell interactions as a function of distance between the two cell types, without using elaborate and expensive equipment. When applied to interactions between the populations of mesenchymal and vascular cells, this model enabled screening of the vasculogenic and supportive roles of different cell populations. This approach has potential for a variety of applications, such as drug models, and vascular biological studies that can lead towards conditions for rapidly enhancing vascularization of implanted engineered tissues.
Acknowledgments
The authors would like to acknowledge the funding received from NIH (R01 HL076485 and P41-EB002520 to GV-N), the University of Padua (stipend to ST) and Columbia University (stipend to GE).
Funding sources: NIH (R01 HL076485 and P41-EB002520 to GV-N), the University of Padua (stipend to ST) and Columbia University (stipend to GE).
References
- 1.Antonelli-Orlidge A, Saunders KB, Smith SR, et al. An activated form of transforming growth factor β is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci USA. 1989;86:4544–4548. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 3.Au P, Tam J, Fukumura D, et al. Bone marrow-derived mesenchymal stem cells facilitate engineering of long-lasting functional vasculature. Blood. 2008;111:4551–4558. doi: 10.1182/blood-2007-10-118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayless KJ, Salazar R, Davis GE. RGD-dependent vacuolation and lumen formation observed during endothelial cell morphogenesis in three-dimensional fibrin matrices involves the alpha(v)beta(3) and alpha(5)beta(1) integrins. Am J Pathol. 2000;156:1673–1683. doi: 10.1016/s0002-9440(10)65038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 6.Chung S, Sudo R, Mack PJ, Wan CR, Vickerman V, Kamm RD. Cell migration into scaffolds under co-culture conditions in a microfluidic platform. Lab Chip. 2009 Jan 21;9(2):269–75. doi: 10.1039/b807585a. Epub 2008 Oct 31. [DOI] [PubMed] [Google Scholar]
- 7.Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–521. doi: 10.1016/s0008-6363(00)00281-9. [DOI] [PubMed] [Google Scholar]
- 8.Davis GE, Bayless KJ, Mavila A. Molecular basis of endothelial cell morphogenesis in three-dimensional extracellular matrices. Anat Rec. 2002;268:252–275. doi: 10.1002/ar.10159. [DOI] [PubMed] [Google Scholar]
- 9.Ding R, Darland DC, Parmacek MS, et al. Endothelial-mesenchymal interactions in vitro reveal molecular mechanisms of smooth muscle/pericyte differentiation. Stem Cells Dev. 2004;13:509–520. doi: 10.1089/scd.2004.13.509. [DOI] [PubMed] [Google Scholar]
- 10.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]
- 11.Gong Z, Niklason LE. Blood vessels engineered from human cells. Trends Cardiovasc Med. 2006;16:153–156. doi: 10.1016/j.tcm.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Hellström M, Kalen M, Lindahl P, et al. Role of PDGF-B and PDGFR-β in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 13.Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141:805–814. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain RK, Au P, Tam J, et al. Engineering vascularized tissue. Nat Biotechnol. 2005;23:821–823. doi: 10.1038/nbt0705-821. [DOI] [PubMed] [Google Scholar]
- 15.Johnson PC, Mikos AG, Fisher JP, et al. Strategic directions in tissue engineering. Tissue Eng. 2007;13:2827–2837. doi: 10.1089/ten.2007.0335. [DOI] [PubMed] [Google Scholar]
- 16.Kamei M, Saunders WB, Bayless KJ, et al. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. 2006;442:453–456. doi: 10.1038/nature04923. [DOI] [PubMed] [Google Scholar]
- 17.Lafleur MA, Handsley MM, Knäuper V, et al. Endothelial tubulogenesis within fibrin gels specifically requires the activity of membrane-type-matrix metalloproteinases (MT-MMPs) J Cell Sci. 2002;115:3427–3438. doi: 10.1242/jcs.115.17.3427. [DOI] [PubMed] [Google Scholar]
- 18.Laschke MW, Harder Y, Amon M, et al. Angiogenesis in tissue engineering: breathing life into constructed tissue substitutes. Tissue Eng. 2006;12:2093–2104. doi: 10.1089/ten.2006.12.2093. [DOI] [PubMed] [Google Scholar]
- 19.Lindahl P, Johansson BR, Levéen P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 20.Marin V, Kaplanski G, Grès S, et al. Endothelial cell culture: protocol to obtain and cultivate human umbilical endothelial cells. J Immunol Methods. 2001;254:183–190. doi: 10.1016/s0022-1759(01)00408-2. [DOI] [PubMed] [Google Scholar]
- 21.Radisic M, Marsano A, Maidhof R, Wang Y, Vunjak-Novakovic G. Perfusion bioreactors for controlling cellular environments. Nature Protocols. 2008;3:719–738. doi: 10.1038/nprot.2008.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 23.Ray JL, Leach R, Herbert JM, et al. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:185–188. doi: 10.1023/a:1016357510143. [DOI] [PubMed] [Google Scholar]
- 24.Romanov YA, Svintsitskaya VA, Smirnov VN. Searching for alternative sources of postnatal human mesenchymal stem cells: candidate MSC-like cells from umbilical cord. Stem Cells. 2003;21:105–110. doi: 10.1634/stemcells.21-1-105. [DOI] [PubMed] [Google Scholar]
- 25.Rouwkema J, Rivron NC, van Blitterswijk CA. Vascularization in tissue engineering. Trends Biotechnol. 2008;26:434–441. doi: 10.1016/j.tibtech.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Sarkar S, Dadhania M, Rourke P, Desai TA, Wong JY. Vascular tissue engineering: microtextured scaffold templates to control organization of vascular smooth muscle cells and extracellular matrix. Acta Biomater. 2005 Jan;1(1):93–100. doi: 10.1016/j.actbio.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Sato Y, Rifkin DB. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J Cell Biol. 1989;109:309–315. doi: 10.1083/jcb.109.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorrell JM, Baber MA, Caplan AI. A self-assembled fibroblast-endothelial cell co-culture system that supports in vitro vasculogenesis by both human umbilical vein endothelial cells and human dermal microvascular endothelial cells. Cells Tissues Organs. 2007;186:157–168. doi: 10.1159/000106670. [DOI] [PubMed] [Google Scholar]
- 29.Sorrell MJ, Baber MA, Caplan AI. Influence of Adult Mesenchymal Stem Cells on In Vitro Vascular Formation. Tissue Eng Part A. 2009 doi: 10.1089/ten.tea.2008.0254. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sudo R, Chung S, Zervantonakis IK, Vickerman V, Toshimitsu Y, Griffith LG, Kamm RD. Transport-mediated angiogenesis in 3D epithelial coculture. FASEB J. 2009 Jul;23(7):2155–64. doi: 10.1096/fj.08-122820. Epub 2009 Feb 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vickerman V, Blundo J, Chung S, Kamm R. Design, fabrication and implementation of a novel multi-parameter control microfluidic platform for three-dimensional cell culture and real-time imaging. Lab Chip. 2008 Sep;8(9):1468–77. doi: 10.1039/b802395f. Epub 2008 Jul 18. [DOI] [PMC free article] [PubMed] [Google Scholar]