

Abstract
Arsenic is a human bladder carcinogen. Arsenic is methylated to both monomethyl and dimethyl metabolites which have been detected in human urine. The trivalent methylated arsenicals are more toxic than inorganic arsenic. It is unknown if these trivalent methylated metabolites can directly cause malignant transformation in human cells. The goal of this study is determine if monomethylarsonous acid (MMAIII) can induce malignant transformation in a human bladder urothelial cell line. To address this goal, a non-tumorigenic human urothelial cell line (UROtsa) was continuously exposed to 0.05 μM MMAIII for 52 weeks. Hyperproliferation was the first phenotypic change observed in exposed UROtsa (URO-MSC). After 12 weeks of exposure, doubling time had decreased from 42 h in unexposed control cells to 27 h in URO-MSC. Hyperproliferation continued to be a quality possessed by the URO-MSC cells after both 24 and 52 weeks of exposure to MMAIII, which had a 40–50% reduction in doubling time. Throughout the 52-week exposure, URO-MSC cells retained an epithelial morphology with subtle morphological differences from control cells. 24 weeks of MMAIII exposure was required to induce anchorage-independent growth as detected by colony formation in soft agar, a characteristic not found in UROtsa cells. To further substantiate that malignant transformation had occurred, URO-MSC cells were tested after 24 and 52 weeks of exposure to MMAIII for the ability to form tumors in SCID mice. Enhanced tumorigenicity in SCID mouse xenografts was observed after 52 weeks of treatment with MMAIII. These observations are the first demonstration of MMAIII-induced malignant transformation in a human bladder urothelial cell line and provide important evidence that MMAIII may be carcinogenic in human tissues.
Keywords: Arsenic methylation, Monomethylarsonous acid, Bladder cancer, Cell culture, UROtsa
Introduction
Arsenic is a naturally occurring metalloid of the geosphere that is associated with numerous diseases in humans. Human exposure commonly occurs upon consumption of drinking water contaminated with arsenic leachate. Exposure to low concentrations of arsenic in drinking water is associated with an increased risk for the development of cancers of the skin, lung, and bladder (IARC, 1980; NRC, 2000). Arsenic is also known to be an occupational hazard. Employees of the mining industry are exposed to inorganic arsenite after its liberation during metal smelting. Inhalation of arsenic is associated specifically with lung cancer development (Enterline et al., 1995). Upon inhalation or ingestion, inorganic arsenic is enzymatically reduced and methylated to a number of metabolites (Fig. 1). Not only is the mechanism of arsenic-induced carcinogenesis unknown, it is not known which of the arsenic metabolites function as carcinogens (Kitchin, 2001).
Fig. 1.
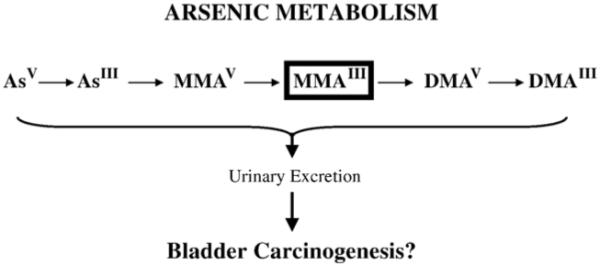
Metabolism of inorganic arsenic in mammals.
Inorganic arsenate (AsV) and arsenite (AsIII) are the forms of arsenic most commonly consumed by humans. Arsenate is readily reduced to arsenite by arsenate reductase in a glutathione-dependent reduction. Arsenite is methylated by arsenic methyltransferase to monomethylarsonic acid (MMAV) (Aposhian, 1997; Zakharyan et al., 1995). MMAV is reduced by MMAV reductase to monomethylarsonous acid (MMAIII) a substrate for subsequent methylation by MMAIII methyltransferase for the production of dimethylarsinic acid (DMAV) (Cullen and Reimer, 1989; Zakharyan and Aposhian, 1999). DMAV is then reduced to dimethylarsinous acid (DMAIII). DMAIII is converted to a final trivalent arsenic compound trimethylarsine oxide (TMAO), which can be reduced to trimethylarsine (TMAIII). However, it has been suggested that this metabolite is produced in significant quantities only in rats (Kitchin, 2001).
With the exception of TMAIII, all arsenic metabolites have been detected in humans exposed to inorganic arsenic (Aposhian et al., 2000a; Aposhian et al., 2000b; Le et al., 2000; Valenzuela et al., 2005). Although many mammals methylate arsenic, humans possess a very different metabolic profile than most mammals in that they produce and excrete a rather large amount of MMA (Vahter, 1994). Thus, monomethylated arsenic species are of particular interest to human health.
In general, there are only a few studies that examined the carcinogenicity of the methylated arsenicals. The previously conducted studies primarily focused on the carcinogenicity of the pentavalent, mono- and di-methylated species probably due to reduced stability and availability of the trivalent, methylated arsenicals. DMAV was demonstrated to be a bladder carcinogen in F334 rats by Wei et al. (1999). F334/DuCrj rats developed cancers of multiple organ systems only after being pretreated with a carcinogen followed by DMAV exposure (Yamamoto et al., 1995). Xie et al. (2004) observed no tumors in v-Ha-rastransgenic (Tg.AC) mice treated with either MMAV or DMAV for 17 weeks. Studies where tumors have been successfully produced after DMAV exposure in rodents have been criticized for the doses required to produce neoplasia (50–200 ppm).
Carcinogenicity of the trivalent methylated arsenic metabolites has been recently investigated in rodents. It is plausible that these arsenicals, DMAIII and MMAIII, are potentially carcinogenic since they are more genotoxic that inorganic arsenic (Kitchin, 2001). Recently, Shen and colleagues (2006) observed that female F344 rats produce more MMAIII when treated with DMAV and had a higher degree of pathological changes occurring in their urinary bladder. Delker et al. (2006) treated K6/ODC mice with 10, 50, and 100 ppm MMAIII in drinking water. After 26 weeks of MMAIII treatment, mice developed skin papillomas. Differential gene expression data was collected and found that genes expressed in resultant papillomas were consistent with tumor formation including increased oncogene expression (c-myc, fra-1, and v-maf) and decreased tumor suppressor gene expression (p27) (Delker et al., 2006). In addition, Krishnamohan et al. (2006) conducted a 2-year rodent bioassay and found MMAIII to be carcinogenic in C57BL/6J mice at doses of 500 μg/l (500 ppb). Both treated and untreated groups of mice developed tumors. However, the incidence of tumor formation doubled from 26% to 54%. Autopsy on treated animals revealed that MMAIII-treated mice developed tumors in multiple organ systems including lymph nodes, pancreas, liver, spleen, uterus, lung, kidney, ovary, thymus, and gut (Krishna-mohan et al., 2006). These findings were the first demonstrate that MMAIII is carcinogenic in vivo. However, it is unknown if MMAIII is a carcinogen in human tissue.
Methylated, trivalent arsenicals are more toxic than AsIII to cultured human cells (Petrick et al., 2000; Styblo et al., 2000). However, the mechanism(s) by which methylated, trivalent arsenicals affect cells is not well-defined. Early research investigating the mechanism of MMAIII-induced toxicity demonstrated that this metabolite is a more potent inhibitor of key cellular proteins such as glutathione reductase (Styblo et al., 1997), thioredoxin reductase (Lin et al., 1999), and pyruvate dehydrogenase (Petrick et al., 2001). These findings not only illustrate that MMAIII can affect enzyme activity, but may affect cellular redox state given the identity of some of these enzymes.
A number of studies investigated the genotoxicity of the methylated trivalent arsenicals. Mass et al. (2001) found that MMAIII nicked naked (ΦX174) DNA and found this metabolite to be more potent than other arsenicals, DMAIII being the exception, at generating alkaline-labile sites and/or DNA strand breaks in human peripheral lymphocytes. MMAIII is believed to damage DNA by ROS formation (Ahmad et al., 2002; Nesnow et al., 2002, Wang et al., 2002). Schwerdtle et al. (2003) also observed oxidative DNA damage via MMAIII in PM2 DNA and HeLa cells. In a number of experiments, MMAIII was found to be highly clastogenic, but not, a gene mutagen (Kligerman et al., 2003).
Since many studies found strong evidence that MMAIII is capable of inducing genetic damage and changes in signal transduction consistent with many carcinogens, there is a need to assess the ability of this arsenic metabolite to function as a carcinogen in vivo and in vitro. Only one published study has investigated the effects of chronic MMAIII treatment on human cells. Mure et al. (2003) exposed human osteosarcoma cells to particularly low concentrations (0.00625, 0.0125, 0.025, 0.05 μM) of MMAIII over 6 and 8 weeks and observed no evidence for mutagenesis or transformation. To our knowledge, no other studies have investigated long-term exposures of human cells to MMAIII.
The model chosen to investigate the long-term exposure of a human cell line to MMAIII is human bladder urothelial cells (UROtsa). These cells are derived from urothelium lining the ureter and were immortalized via temperature-sensitive SV40 large T-antigen gene construct. UROtsa cells do not exhibit anchorage-independent growth or tumorigenicity in nude mice (Petzoldt et al., 1995; Sens et al., 2004). However, UROtsa cells can be transformed to anchorage-independent growth and tumorigenicity by AsIII and cadmium. AsIII-induced transformation was conducted at a low concentration (1 μM) for approximately 52 weeks. This was the first study conducted in a human bladder cell line that showed direct malignant transformation of urothelium by AsIII. Because humans excrete appreciable amounts of MMAIII and previous studies have indicated that UROtsa have a small capacity to methylate AsIII (Bredfeldt et al., 2004), it is of interest to determine if this metabolite can directly induce malignant transformation in this human urothelial cell line.
Methods
Reagents
Sodium arsenite, methylthiazoletetrazolium (MTT, 3-(4,5-dimethylthizol-2-yl)-2,5-diphenyltetrazolium bromide), phenazinemethylsulfate, isopropanol, hydrochloric acid, and trypan blue were purchased from Sigma Chemical Company (St. Louis, MO). Dulbecco’s Modified Eagle Medium (DMEM), fetal calf serum (FBS), antibiotic–antimycotic, and 1× trypsin–EDTA (0.25%) were acquired from Gibco Invitrogen Corporation (Carlsbad, CA). Noble agar was purchased from Amersham Biosciences (Piscataway, NJ). Diiodomethylarsine (MMAIII iodide, CH3AsI2) was prepared using the method of Millar et al. (1960). Water used in studies was distilled and de-ionized.
Dosing solutions
Pure MMAIII iodide was stored in ampules at 4 °C. Fresh stock solutions of 25 mM MMAIII were made and diluted to a final concentration of 5 μM for dosing. All dosing solutions were sterile filtered with a 0.2 μm acrodisc and stored in sealed, sterile tubes that were opened only for dosing in a sterile cell culture hood. As previously reported by Gong et al. (2001), MMAIII solutions in distilled, de-ionized water were stable for approximately 4 months at 4 °C with no degradation observed when monitored using HPLC-ICP MS.
Cell culture
UROtsa cells, an immortal, non-tumorigenic human bladder cell line, were obtained from the laboratory of Drs. Donald and Mary Ann Sens (University of North Dakota). URO-ASSC cells were generously provided by Drs. Donald and Mary Ann Sens (University of North Dakota). URO-ASSC cells, AsIII-transformed human urothelial cells, were developed from UROtsa cells chronically treated with 1 μM sodium arsenite (Sens et al., 2004). Cells were maintained on 75 cm2 tissue culture flasks. Culturing conditions were adapted from those described by Rossi et al. (2001). Cells were cultured in a growth medium of DMEM containing 5% v/v FBS and 1% antibiotic–antimycotic. Growth medium was changed every 2 days. Cultured cells were incubated in an atmosphere that was 5% CO2:95% air at 37 °C. Confluent cells were removed from plates with trypsin–EDTA (0.25%) and subcultured at a ratio of 1:3. MMAIII-treated cells were continuously cultured in a medium enriched with 0.05 μM MMAIII.
Cell viability
Alterations in mitochondrial activity were used as an indicator of cell viability. The methylthiazoletetrazolium (MTT, 3-(4,5-dimethylthizol-2-yl)-2,5-diphenyltetrazolium bromide) assay measures mitochondrial activity (Loveland et al., 1992). Cells were plated in 6-well plates and treated with 0.5 μM to 10 μM MMAIII for 24, 48, and 72 h.
Cell growth kinetics
Growth curves for UROtsa and URO-MSC after 12, 24, and 52 weeks of exposure to 0.05 μM MMAIII were obtained via trypan blue exclusion assay. Cells were plated in 6-well plates at a density of 2 × 105 cells per well. Cells were removed from the plates via trypsin and counted. Growth curves were generated based on increases in cell population per 24 h periods for a total time of 96 h. These growth curves were then used to calculate doubling time.
Cell morphology
Cells were cultured on Delta T dishes (Bioptech, Butler, PA) at 500,000 cells per plate and allowed to reach 90% confluency before visualization. Morphology was evaluated weekly via light microscopy. Photographs were taken to assess changes in morphology of all cell lines. Plated cells were prepared for photography by removal of culture media and rinse with phosphate-buffered saline (PBS). Fresh culture medium containing no MMAIII was added to cells. Images of cells treated for 12, 24, and 52 weeks were obtained and compared with untreated parental cell line. To obtain photographs via confocal microscopy, the cells plated on Biotech culture dishes were attached to a temperature controller to maintain the cultures at 37 °C and mounted on the microscope stage. A Zeiss LSM 510 confocal microscope (Carl Zeiss Microimaging Inc., Thornwood, NY) with a 40× “dipping” lens was used to obtain images of the parental and treated cells using differential interference contrast (DIC).
Colony formation in soft agar
Anchorage-independent growth was detected in cells treated with MMAIII for 24 and 52 weeks by colony formation in soft agar. For colony formation in soft agar, cells were removed from culture flask with trypsin and suspended in culture medium supplemented with 0.3% agar. The agar enriched with cells was overlaid onto 0.6% agar medium in a 24-well plate with a density of 1 × 104 cells per well. Plated cells were monitored for growth at 1, 7, and 14 days. After 14 days of incubation, colonies were manually counted with an Olympus CK2 microscope (Olympus America, Inc. Melville, NY). Data represent colonies formed in single plane of agar. Photographs were obtained with an Olympus IX70 microscope coupled to an Olympus camera (Olympus America, Inc. Melville, NY). Analysis of photographs was conducted with Magnafire software (Optronics, Goleta, CA).
SCID mouse colony
A SCID mouse colony was developed at the University of Arizona using original SCID (C.B-17/IcrACCscid) obtained from Taconic (Germantown, New York). The mice were housed in microisolator cages (Allentown Caging Equipment Company, Allentown, New Jersey) and maintained under specific pathogen-free conditions. The mice ate NIH-31 irradiated pellets (Tekland Premier, Madison, Wisconsin) and drank autoclaved water. Sentinel mice were screened monthly for mycoplasma, mouse hepatitis virus, pinworms, and Sendai virus via ELISA. Male mice 6–8 weeks of age were bled (200 μl) by retro-orbital puncture in order to screen for the presence of mouse immunoglobulin (Ig) using ELISA. Only mice with Ig levels ≤20 μg/ml were used for the xenograft experiments.
Tumorigenicity in SCID mouse xenografts
To assess malignant transformation, UROtsa, URO-ASC, URO-MSC24, and URO-MSC52 cell injections (10 × 106) were given subcutaneous in the lower right flank of the mouse in a total volume of 100 μl of sterile saline using a 27-gauge needle (Becton Dickinson, Franklin Lakes, NJ). As tumors developed, subcutaneous tumors were measured twice weekly for tumor volume estimation (mm3) in accordance with the formula (a2 × b / 2) where a is the smallest diameter and b is the largest diameter. Tumors were harvested from each animal after approximately 10 weeks, and 1/2 of the tumor was placed in 10% neutral buffered formalin (NBF) for 24 h then transferred to 70% EtOH. The other half was snap frozen in OCT. The tissues were taken to Tissue Acquisition and Cellular/Molecular Analysis Shared Service (TACMASS) at the Arizona Cancer Center for analysis. To evaluate histology, tumor samples were paraffin-embedded, sectioned, and stained with hematoxylin and eosin (H&E) and analyzed via light microscopy by Dr. Raymond B. Nagle (University of Arizona, Department of Pathology). All procedures were performed in accordance with approved protocols of the University of Arizona Institutional Animal Care and Use Committee.
Evaluation of tumor proliferation via Ki-67 immunohistochemistry
A representative paraffin block was selected from UROtsa and URO-MSC52 xenografts. Ki-67 was visualized using the Discovery XT platform (Ventana Medical Systems, Inc., Tucson, AZ). All reagents for detection, including Ki-67 primary antibody (3 mg/ml), and on-line antigen retrieval with cell conditioner 1, were obtained from Ventana Medical Systems. Counterstaining of slides was conducted on-line using H&E solutions. Lastly, sections were dehydrated through graded alcohols, cleared by xylene, and coverslipped with Pro-Texx mounting medium. Images were collected using an Olympus BX50 microscope (Olympus America, Inc. Melville, NY) mounted with a SPOT RT Slider (Model 2.3.0) camera (Diagnostic Instruments, Sterling Heights, MI). Collected images were standardized for light intensity. No further automated analysis of data was performed.
Statistical methods
Data analysis was carried out using GraphPad Prism 4.0 software (GraphPad Software, Inc., San Diego, CA). Graphs were generated in Microsoft Office Excel (Microsoft Corp., Redmond, WA). Data from the MTT assay, trypan blue assay, and soft agar assay are expressed as the average of three experiments. Data from tumorigenicity studies represent average of four experiments. These data are represented as the mean ± SEM. Specific statistical tests used to determine significance are described in figure legends.
Results
Monomethylarsonous acid toxicity in UROtsa cells
Before attempting MMAIII-induced transformation of UROtsa cells, toxicity was assessed to identify a non-toxic, low concentration of MMAIII for chronic exposure. UROtsa cell viability was determined with the MTT assay (Loveland et al., 1992). Cells plated in 6-well plates were exposed to MMAIII in concentrations of 0.5 to 10 μM for 24, 48, and 72 h. MMAIII (0.5–2 μM) exposure had no significant effect on UROtsa cell viability. In fact, lower doses of MMAIII are often stimulatory to mitochondrial activity (Fig. 2). Concentrations of MMAIII exceeding 5 μM were always cytotoxic to UROtsa cells. The IC50 value for MMAIII was determined to be approximately 5 μM in 24 h, making MMAIII 20 times more toxic than AsIII to UROtsa cells (Bredfeldt et al., 2004).
Fig. 2.
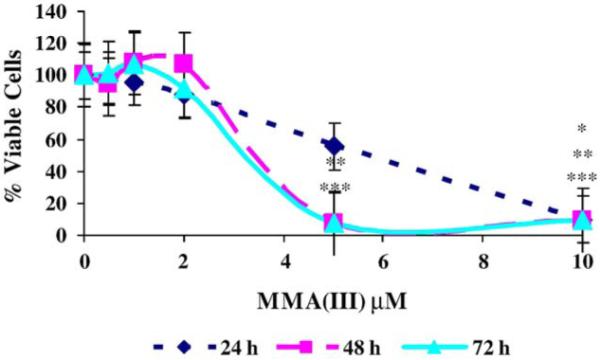
Cytotoxicity of MMAIII to UROtsa cells. Cells were treated with 0.5–10 μM MMAIII for 24, 48, and 72 h. Values represent means ± SEM. Assay results are portrayed as percentage value of untreated samples (n = 3). Significant changes in viability were identified with ANOVA followed by Bonferroni’s multiple comparisons test. P < 0.05 was considered significant and marked by asterisk(s) *(24 h), **(48 h), and ***(72 h).
Microscopic evaluation of treated and untreated cells revealed a similar finding that only cells exposed to MMAIII doses >2 μM had significant changes in viability and morphology. Morphology of untreated UROtsa cells is similar to that reported by Rossi et al. (2001). While morphology remained unchanged at lower doses (<2 μM) of MMAIII, large zones of apoptotic/necrotic cells develop throughout the monolayer with increasing concentrations (>2 μM) after 24, 48, and 72 h exposures. A general thinning of cell density was accompanied by the extensive cell death as concentration was increased. Cells did not survive high doses of MMAIII. These cytotoxicity studies not only identified an approximate IC50 for MMAIII, but illustrate that UROtsa cells have very little tolerance for MMAIII. There is an apparent threshold for cytotoxicity in UROtsa exposed to MMAIII in that doses exceeding 2 μM are cytotoxic.
Monomethylarsonous acid causes hyperproliferation in UROtsa cells
Transformation was conducted with cells exposed to 0.05 μM MMAIII. The concentration of 0.05 μM was chosen for several reasons. Previous work demonstrated that doses as low as 1 μM AsIII were sufficient to malignantly transform UROtsa cells (Sens et al., 2004). Our data indicate that MMAIII is roughly 20–25 times more toxic than AsIII. Therefore, a dose that is significantly lower than 1 μM MMAIII would be necessary to avoid clonal selections based on survival of overt cytotoxicity. The concentration of 0.05 μM is 20 times lower concentration than the 1 μM concentration previously used for malignant transformation by Sens et al. (2004). The dose of 0.05 μM MMAIII did not cause cytotoxicity or induce immediate morphological changes in treated cells. Cells were grown to 90–100% confluence before being subcultured at a ratio of 1:3. During the first 8 weeks of exposure, MMAIII-treated cells were phenotypically similar to untreated cells. Following 12 weeks of exposure, MMAIII-treated cells gained a noticeably reduced doubling time. Due to the increased growth rate, URO-MSC cells were then split at a ratio of 1:5 after 24 weeks and 1:8 after 52 weeks. A higher splitting ratio was not selected because we chose to avoid extensive differences in culturing conditions between the groups of cells.
After URO-MSC cells became hyperproliferative, doubling time was used as a method for investigating changes in cell growth kinetics. Arsenite-transformed UROtsa, URO-ASSC, were used as a positive control group since Sens et al. (2004) showed that this cell line has a reduced doubling time. Untreated control cells have a doubling time of approximately 42 h. Treated cells had a doubling time of approximately 27 h after 12 weeks of exposure to 0.05 μM MMAIII, which is a 40% reduction in doubling time (Table 1). The URO-MSC cells had a growth rate comparable to the URO-ASSC cells (Sens et al., 2004). Since these cells were showing possible changes consistent with transformation, growth in soft agar was performed to determine cellular transformation. However, cells treated for 12 weeks did not grow in soft agar. Therefore, MMAIII treatment was continued and doubling time and growth on soft agar were reconsidered 24 weeks after exposure. Cells treated for 24 weeks with 0.05 μM MMAIII had growth kinetics similar to the 12-week exposure group. Doubling time for 24-week treated URO-MSC cells was approximately 25 h. Similarly, cells treated for 52 weeks with 0.05 μM had a doubling time of 21 h (Table 1).
Table 1.
Growth kinetics of UROtsa and URO-MSC cells after treatment with MMAIII
| Cell line | Doubling time (h) |
|---|---|
| UROtsa | 42 ± 1.9 |
| URO-MSC, 12 weeks | 27 ± 0.3* |
| URO-MSC, 24 weeks | 25 ± 0.2* |
| URO-MSC, 52 weeks | 21 ± 0.2* |
UROtsa were continuously exposed to 0.05 μM MMAIII for 12, 24, and 52 weeks. Trypan blue exclusion assay was used to enumerate cells at 24, 48, 72, and 96 h after plating. Then, growth curves were generated and used to estimate doubling times of treated and untreated cells. Data represent results from one experiment (n = 3). ANOVA followed by Bonferroni’s multiple comparisons test was used to identify significant changes in doubling time.
Significant deviations from control cells (P < 0.05) were marked with an asterisk.
Morphological changes in UROtsa cells chronically exposed to monomethylarsonous acid
Cells were examined at each passage for changes in morphology. Parental cell morphology has been extensively characterized and is comparable to that reported by Rossi et al. (2001) (Fig. 3A). Cell morphology was not modified by MMAIII treatment during the first 8 weeks of exposure. Minor morphological changes were observed 12 weeks after treatment with 0.05 μM MMAIII. Examination via light microscopy revealed that treated cultures maintained an epithelial morphology. However, URO-MSC morphology was unlike that of the untreated parent cell line or URO-ASSC. These cells are similar in size to the parental cell line, but with a less defined cell membrane, making individual cells difficult to distinguish by cell membrane (Figs. 3B and C). This morphology remained the same after 24 weeks of incubation with MMAIII. Another notable feature of MMAIII-treated cells was the increased appearance of multinucleated cells that contain abundant cytoplasm and two to four nuclei among cells of the monolayer. Such cells are considered an indicator of cellular transformation (Walen, 2004).
Fig. 3.

Evaluation via confocal microscopy of UROtsa cells before, during, and after transformation with MMAIII. Morphology of untreated UROtsa cells (A). Morphology of URO-MSC cells treated with 0.05 μM MMAIII for 12 (B), 24 (C), and 52 (D) weeks. Magnification used for photographs is ×200.
52 weeks of treatment resulted in a population of cells that appeared as squamous epithelium, being flat and wide. URO-MSC52 cells also had poorly defined cell membranes. In addition, multinucleate cells were present in URO-MSC52 cell cultures (Fig. 3D).
URO-MSC growth in soft agar is similar to URO-ASSC
The ability to form colonies in soft agar is a characteristic of many cancer cell lines. In this study, the soft agar assay was used to determine if UROtsa cells were transformed by MMAIII. URO-MSC cells were tested after 24 and 52 weeks of exposure to MMAIII and were able to form colonies in soft agar. Colony formation in soft agar was comparable to URO-ASSC cells. Colonies were counted via microscopy and demonstrated that number of colonies formed per 20 microscope fields on a single plane of agar (Fig. 4). Shorter exposure times, including a 12-week treatment 0.05 μM MMAIII, were not sufficient to induce anchorage-independent growth in UROtsa cells (data not shown).
Fig. 4.

Anchorage-independent growth of URO-MSC cells. Light microscopy of colony growth 14 days after plating for negative control, UROtsa, positive control, URO-ASSC, URO-MSC24, and URO-MSC52 cells (A). Colonies were photographed at ×200. (B). A single plane of agar was selected, and colonies were manually counted in twenty randomly selected microscope fields. Bars represent mean ± SEM (n = 3). However, error bars are too small to visualize. Data were further analyzed with Student’s t test to identify significant changes in the ability to form colonies in soft agar. Statistically significant values (P < 0.05) are marked with an asterisk (*).
Chronic MMAIII treatment enhanced tumorigenicity in UROtsa cells
To determine if malignant transformation had occurred in UROtsa cells chronically exposed to 0.05 μM MMAIII, URO-MSC cells were injected into the flank of SCID mice. Subcutaneous tumor formation in xenograft is evidence of malignant transformation. UROtsa and URO-ASSC cells were used as negative and positive control cell lines, respectively. Tumors were formed by all injected cell lines. UROtsa cells have been previously described as non-tumorigenic (Petzoldt et al., 1995; Sens et al., 2004). However, these studies did not inject high passage cells into rodents to determine if there was some small degree of underlying tumorigenicty in this SV40 large T-antigen transformed cell line. Tumors formed in UROtsa cells after 24 weeks of treatment with MMAIII were not statistically significant from negative control (Fig. 5).
Fig. 5.
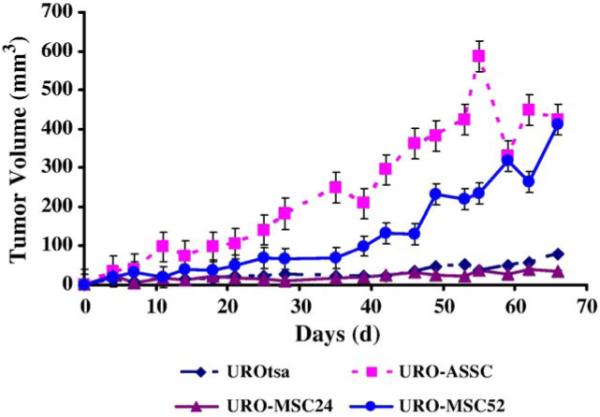
Tumorigenicity of URO-MSC cells in SCID mice. Tumor volumes were measured two times per week. Data are tumor volume (mm3) per time (days). Bars represent mean ± SEM (n = 4). However, error bars in UROtsa and URO-MSC24 tumors are too small to visualize. Significant changes in tumor volume were identified with ANOVA followed by Bonferroni’s multiple comparisons test. P < 0.05 was considered significant. URO-ASC tumors are statistically different in tumor volume from UROtsa 30 days after injection. URO-MSC52 tumors are significantly different from UROtsa 40 days after injection.
Harvested tumors were evaluated for histology by a pathologist with no prior knowledge regarding the treatment groups. All tumors formed by UROtsa, URO-ASSC, URO-MSC24, and URO-MSC52 were moderately differentiated squamous cell carcinoma (data not shown). The histology of squamous cell carcinoma produced by URO-ASSC cells was extensively characterized by Sens et al. (2004). The tumors in a number of hallmark features found in squamous cell carcinoma including deposits of keratin were called “keratin pearls”.
Highly proliferative tumors form from UROtsa cells chronically treated with MMAIII
To further illustrate that MMAIII treatment enhanced the tumorigenic qualities of UROtsa, tumor proliferation was investigated. When tumors were stained for Ki-67 as a biomarker of proliferation, URO-MSC52 tumors strongly stained for Ki-67 demonstrating that they were significantly more proliferative than those originating from UROtsa, which weakly stained for Ki-67. In addition, strong Ki-67 staining suggests that chronic MMAIII treatment induced permanent changes that causing UROtsa cells to form large tumors (Fig. 6).
Fig. 6.
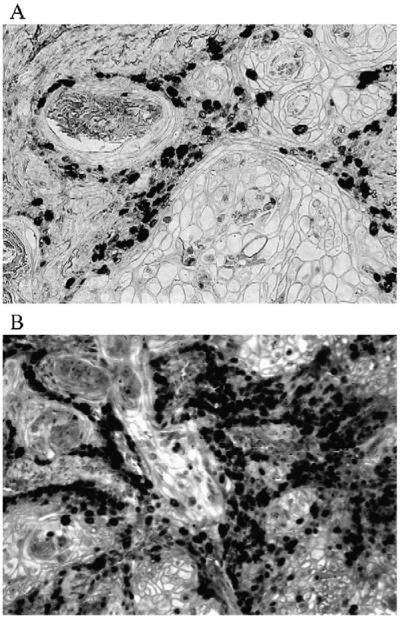
Ki-67 staining of tumor tissue from UROtsa (A) and URO-MSC52 (B) cells. Representative photograph of Ki-67-stained tissue sections demonstrates the increased proliferation found in URO-MSC52 tumors. Magnification used for photographs is ×400.
Discussion
Epidemiological studies demonstrated that human exposure to inorganic arsenic causes an increased risk for the development of a number of internal cancers, including bladder cancer. In addition, a number of in vitro and in vivo studies found that inorganic arsenite is able to transform human cell lines at low, biologically relevant concentrations (Chien et al., 2004; Lee et al., 1985; Mure et al., 2003; Qu et al., 2002; Sens et al., 2004; Takahashi et al., 2002). However, an issue important to the field of arsenic toxicity is clarification of the potential carcinogenicity of arsenic metabolites. The study presented herein is, to our knowledge, the first demonstration of monomethylarsonous-acid-induced transformation of a human cell line.
Studies investigating the carcinogenicity of methylated arsenicals have been few in number. Commonly, this research was conducted in rodents where tumors have been difficult to induce (Basu et al., 2001; Kitchin, 2001; Rossman et al., 2001). The focus of many of these studies has been on the carcinogenicity of the pentavalent methylated arsenicals MMAV and DMAV. F344/DuCrj rats pretreated with carcinogens (diethyl nitrosamine, N-methyl-N-nitrosourea, 1, 2-dimethyl hydrazine, N-butyl-N-(4-hydroxybutyl)nitrosamine, or N-bis(2-hydroxypropyl)nitrosamine) developed cancers of the kidney, liver, thyroid gland, and urinary bladder after exposure to DMAV (Yamamoto et al., 1995). Wei et al. (1999) demonstrated that F334 rats treated solely with DMAV developed tumors of the urinary bladder. Investigations of p53 knockout and wild-type C57BL/6J mice treated for 18 months with DMAV found this metabolite to be a tumor-causing agent. The tumors generated in the knockout mice were mostly osteosarcomas, soft tissue sarcomas, and lymphomas. Lower incidences of other tumors such as hepatocellular carcinomas, skin squamous cell carcinomas, and lung adenomas were also observed in the p53 knockout mice. Fewer tumors were produced in the wild-type mice presumably due to a reduced susceptibility for neoplasia formation. Tumors formed in wild-type mice were mainly malignant lymphomas with a very low occurrence of the previously mentioned other types of tumors. Interestingly, the urinary tract in all treated and untreated mice revealed no changes in DMA-treated groups (Salim et al., 2003). Another study investigating the effects of a chronic exposure found that V-Ha-ras transgenic (Tg.AC) mice did not develop tumors after a 17-week treatment with either MMAV or DMAV (Xie et al., 2004).
Evidence for the in vivo carcinogenicity of the trivalent methylated arsenic metabolite MMAIII has begun to accumulate. Since MMAIII is more genotoxic than inorganic arsenic, it is likely that it may act as a carcinogen in vivo (Kitchin, 2001). Preliminary evidence in vivo for the carcinogenicity of MMAIII came from Shen and colleagues (2006) who found that female F344 rats treated with DMAV had a higher degree of pathological changes occurring in their urinary bladder. The authors suggested that the changes in the urinary bladder were due to the higher levels of MMAIII in female F344 rat urine following DMAV treatment.
Stronger evidence for the carcinogenicity of MMAIII came from recent studies investigating the effects of chronic MMAIII treatment in mice. Delker et al. (2006) treated K6/ODC mice with 10, 50, and 100 ppm MMAIII in drinking water for 26 weeks. Following MMAIII treatment, mice developed skin papillomas. Genes expressed in MMAIII-induced papillomas were consistent with tumor formation including decreased tumor suppressor gene expression (p27) and increased oncogene expression (c-myc, fra-1, and v-maf) (Delker et al., 2006). In addition, Krishnamohan et al. (2006) conducted a 2-year rodent bioassay and in C57BL/6J mice at MMAIII doses of 500 μg/l (500 ppb). The incidence of tumor formation doubled from 26% to 54%. MMAIII-treated mice specifically developed tumors of the pancreas, lymph nodes, liver, kidney, lung, intestine, spleen, uterus, ovary, and thymus (Krishnamohan et al., 2006). These findings were the first demonstrate carcinogenicity of MMAIII in vivo. Despite this strong evidence of carcinogenicity of MMAIII, it is unknown if MMAIII can induce malignant transformation in human cells.
Scientific interest in the potential effects of the trivalent, methylated arsenicals has grown significantly. Research focusing on MMAIII and DMAIII has consistently found these metabolites to be more cytotoxic and genotoxic (Ahmad et al., 2002; Mass et al., 2001; Petrick et al., 2000; Styblo et al., 2000). However, there are few investigations examining the carcinogenicity of trivalent, methylated arsenicals in rodents or human cell lines. Mure et al. (2003) found that exposure of human osteosarcoma cells to MMAIII for duration of 6 and 8 weeks did not produce cellular transformation, which was detected as anchorage-independent growth in the soft agar assay. In contrast, our study demonstrates MMAIII-induced cellular transformation to anchorage-independent growth and enhanced tumorigenicity over a 52-week exposure period.
Since the goal of this study was to conduct a clonal selection based on exposure to a non-cytotoxic concentration of MMAIII, it was necessary to investigate the toxicity of MMAIII in our model to identify a concentration that did not cause overt cytotoxicity. Preliminary findings in this study regarding the cytotoxicity of MMAIII have been similar to observations from other researchers. The IC50 value for MMAIII was determined to be approximately 5 μM for 24 h exposure. MMAIII is roughly 20–25 times more toxic to UROtsa cells than AsIII, which has an IC50 value of approximately 100 μM (Bredfeldt et al., 2004). Other groups have observed similar findings (Cohen et al., 2002; Petrick et al., 2000; Styblo et al., 2000). The concentration of 0.05 μM MMAIII did not affect UROtsa cell viability or plating efficiency, making it appropriate for long-term exposure.
In addition to dose selection, it was important to demonstrate that UROtsa were not metabolizing significant amounts of MMAIII to other toxic metabolites such as DMAIII. UROtsa are able to metabolize AsIII to MMAIII, MMAV, and DMAV (Bredfeldt et al., 2004). However, the most abundant metabolite produced in those studies was MMAIII. When treated for 24 h with MMAIII, UROtsa cells did not metabolize MMAIII to more toxic products, as detected by HPLC/ICP-MS (data not shown). Therefore, MMAIII was the chemical responsible for the observed effects as >90% of the dose remained in the form of MMAIII or MMAV, which is less toxic. No DMAIII was detected in cells treated with 0.05 μM MMAIII.
The first observed phenotypic changes, morphological differences and hyperproliferation, occurred in URO-MSC cells after 12 weeks of exposure to MMAIII. Overall, morphology of URO-MSC cells remained like that of an epithelial cell. The observed increase in multinucleate cells (MNCs) has been defined as a feature of cellular transformation due to alterations in proteins that regulate cell mitosis including formation of spindle apparatus. The exact mechanism of how MNCs form however is not completely understood. Nonetheless, increases in MNC were in treated cells only. The few MNCs that appear infrequently in the parental cell line are known as giant cells. Giant cells are different from MNCs in that they generally have greater than four nuclei and are the result of SV40 transformation (Walen, 2004). The other notable morphological change was a slight flattening of cells with a less visible appearance of cell membrane. At this point in these studies, the reasons for these changes are unknown but suggest possible changes in cell-membrane-associated proteins such as adhesion molecules. URO-MSC hyperproliferation after 12, 24, and 52 weeks of exposure was notable due to a 40–50% reduction in doubling time, suggesting that MMAIII drives cell growth via stimulation of mitogenic signal transduction pathways. The mitogenic effects of MMAIII treatment are consistent with the finding that MMAIII is a potent inducer of c-jun phosphorylation and activator protein-1 (AP-1) binding to DNA in UROtsa cells. Further analysis found that MMAIII treatment caused extracellular-signal-regulated kinase (ERK) activation, which was responsible for the increased AP-1 activity (Drobona et al., 2003). The manner in which MMAIII is causing activation of mitogenic signal transduction is not specifically known. However, the mechanism of ROS-induced damage and alterations in signal transduction in AsIII, MMAIII, and DMAIII-treated cells has gained considerable validity through an increasing number of studies.
The majority of the evidence supporting MMAIII and DMAIII-induced reactive oxygen species (ROS) comes from studies investigating the DNA damaging potential of these two metabolites. MMAIII and DMAIII are the only arsenic species able to nick ΦX174 DNA. The DNA-nicking action of MMAIII and DMAIII is instigated by ROS generation (Ahmad et al., 2002; Nesnow et al., 2002). Schwerdtle et al. (2003) observed DNA-damaging activity of MMAIII and DMAIII in PM2 DNA and HeLa cells at nanomolar concentrations, suggesting that the dose chosen in this study is potentially genotoxic. MMAIII has been studied as a gene mutagen and appears to have poor mutagenic activity (Kligerman et al., 2003; Mure et al., 2003). However, MMAIII and DMAIII were found to be the most potent clastogenic agents causing chromosomal aberrations in human peripheral blood lymphocytes (PBL) and mutation at the Tk+/− locus in mouse lymphoma cells (Kligerman et al., 2003). From these studies, it can be concluded that MMAIII and DMAIII induce DNA strand breaks leading to chromosomal aberrations. The mechanism of DNA damage is believed to be due to generation of ROS in treated cells.
Few studies have investigated the impact of long-term exposure of animals or human cell lines to MMAIII and DMAIII. Based on studies investigating the DNA damaging potential of these metabolites, it is probable that they are able to induce malignant transformation. However, Mure et al. (2003) found no increases in transformation of HOS cells after 6 and 8 weeks of exposures. In this study, UROtsa cells exposed to 0.05 μM MMAIII for 24 and 52 weeks were able to grow in soft agar, which was the same end point sought to positively identify cellular transformation by Mure and colleagues. The longer exposure period may be the reason why we observed colony formation in soft agar. The degree of growth exhibited by URO-MSC cells was comparable to that of URO-ASSC cells. URO-ASSC and URO-MSC cells originate from the same parental cell line UROtsa. URO-ASSC cells were malignantly transformed with 1 μM AsIII during 52 weeks of exposure (Sens et al., 2004).
Similar to the AsIII transformation described by Sens et al. (2004), MMAIII induced tumorigenicity in UROtsa cells 52 weeks of exposure. Although UROtsa cells have been previously described as completely non-tumorigenic, high passage UROtsa cells did form small tumors in SCID mice despite the fact that they did not form colonies in soft agar. Injections of immortalized human cells can occasionally result in the formation of small tumors, which may be an artifact of the rodent model system itself. However, based on this data, MMAIII did significantly enhance the tumorigenicty of UROtsa cells.
The tumors produced by MMAIII-treated cells were squamous cell carcinoma. This is a novel observation since the majority (~90%) of bladder cancer patients have transitional cell carcinoma. It remains unknown if this squamous differentiation is an artifact of the UROtsa cell model. However, UROtsa cells treated with cadmium formed transitional cell carcinoma when injected into nude mice, suggesting that transitional differentiation is a possible histology (Sens et al., 2004). Further evidence that supports the squamous differentiation is arsenic-induced tumors of the skin and lung (Guo et al., 2004; Rossman et al., 2002). In addition, chronic bladder inflammation such as cystitis caused by chronic Schistosomiasis infection is strongly associated with the development of squamous cell carcinoma of the bladder (Johansson and Cohen, 1997). Although it is unknown if arsenic causes chronic inflammation in the bladder, AsIII and MMAIII stimulate proinflammatory, mitogenic signal transduction pathways in human keratinocytes and in UROtsa cells (Vega et al., 2001; Drobona et al., 2003). Therefore, a similar mechanism may play a role in the development of squamous cell carcinoma of the bladder (Vega et al., 2001).
To further support that MMAIII treatment had affected the tumorigenicty of UROtsa cell, Ki-67 staining was performed to demonstrate that MMAIII caused UROtsa cells to form large, highly proliferative tumors. Ki-67 staining in URO-MSC52 cells was significantly higher than that of UROtsa. Since Ki-67 stains only cells that are actively growing, it is apparent that MMAIII treatment not only induced transformation but resulted in the production of substantially proliferative tumors.
Taken together, our data demonstrated that MMAIII induced tumorigenicity in UROtsa cells, which was measured as hyperproliferation, growth in soft agar, and significant tumorigenicity in SCID mouse xenografts. To our knowledge, this is the first study to demonstrate that MMAIII induces malignant transformation to anchorage-independent growth and tumorigenicity in SCID mice in a human cell line. The results of this study are particularly compelling for several reasons. Since MMAIII has been detected in human urine, urothelial cells of humans consuming arsenic are exposed to MMAIII (Aposhian et al., 2000a; Le et al., 2000). Likewise, the concentration of MMAIII used to transform UROtsa cells in this study is similar to concentrations found in arsenic-exposed humans. MMAIII has been found in concentrations of 4.8–6.9 μg/l (0.06–0.09 μM) (Aposhian et al., 2000a) and 3–30 μg/l (0.04–0.4 μM) (Mandal et al., 2001) in human urine. The concentration chosen for these investigations was 0.05 μM MMAIII, which falls within the ranges that have been detected in urine. These observations suggest that MMAIII can induce malignant transformation at biologically relevant concentrations.
In conclusion, this study demonstrated that chronic exposure to low concentrations of MMAIII can induce hyperproliferation, anchorage-independent growth, and tumorigenicity in a cell line originating from tissue naturally exposed to this arsenic metabolite. Although the mechanism of malignant transformation is not known, it is probable that ROS are causal to DNA damage that results in the build up of genetic aberrations ultimately leading to cellular transformation. In addition, MMAIII stimulates mitogenic signal transduction pathways, particularly ERK, which promote tumorigenesis (Drobona et al., 2003). The resultant cells represent a model that should be further studied to identify changes that have occurred in human cells chronically exposed to MMAIII. Such investigations will provide insight into the molecular targets of MMAIII in human urothelial cells as well as identify potential biomarkers of exposure that could ultimately be used in human risk assessment studies. Based on the observations from this study, which strongly imply that MMAIII can itself function as a carcinogen, future studies should be focused on this end point. Thus, there can be a better understanding of the complex nature of inorganic-arsenic-induced carcinogenesis. Clearly, it appears that not only does inorganic arsenic function as a transforming agent, but also the arsenic metabolites MMAIII and DMAIII contribute to cancers caused by arsenic exposure.
Acknowledgments
The authors would like to thank Dr. Donald Sens and Dr. Mary Ann Sens for the use of the UROtsa and URO-ASSC cell lines. Additionally, the authors appreciate D. Ray Nagle for evaluating the histology of tumors produced in SCID mouse xenograft studies. Immunohistochemical and histological data generated by the TACMASS Core (Tissue Acquisition and Cellular/Molecular Analysis Shared Service) at the Arizona Cancer Center are supported by a grant from the NCI (2P30CA023074). The authors also thank Dr. Bernard Futscher for his guidance with the transformation studies. In addition, the authors thank Dr. John Regan for the use of his microscope that was used to collect the images presented in this document. The research herein was made possible by the NIEHS Superfund Basic Research Program (ES 04940) and the Southwest Environmental Health Sciences Center (ES 06694). T.B. is funded by an NIEHS Training Grant (ES 07091).
References
- Ahmad S, Kitchin KT, Cullen WR. Plasmid DNA damage caused by methylated arsenicals, ascorbic acid and human liver ferritin. Toxicol. Lett. 2002;133:47–57. doi: 10.1016/s0378-4274(02)00079-6. [DOI] [PubMed] [Google Scholar]
- Aposhian HV. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Annu. Rev. Pharmacol. Toxicol. 1997;37:397–419. doi: 10.1146/annurev.pharmtox.37.1.397. [DOI] [PubMed] [Google Scholar]
- Aposhian HV, Gurzau ES, Le XC, Gurzau A, Healy SM, Lu X, Ma M, Yip Li., Zakharyan RA, Maiorina RM, Dart RC, Tircus MG, Gonzalez-Ramirez D, Morgan DL, Avram D, Aposhian MM. Occurrence of monomethylarsonous acid in urine of humans exposed to norganic arsenic. Chem. Res. Toxicol. 2000a;13:693–697. doi: 10.1021/tx000114o. [DOI] [PubMed] [Google Scholar]
- Aposhian HV, Zheng B, Aposhian MM, Le XC, Cebrian ME, Cullen W, Zakharyan RA, Ma M, Dart RC, Cheng Z, Andrews P, Yip Li., ’Malley GF, Maiorino RM, Van Voorhies W, Healy SM, Titcomb A. DMPA-arsenic challenge test. II. Modulation of arsenic species, including monomethylarsonous acid (MMA(III)), excreted in human urine. Toxicol. Appl. Pharmacol. 2000b;165:74–83. doi: 10.1006/taap.2000.8922. [DOI] [PubMed] [Google Scholar]
- Basu A, Mahata J, Gupta S, Giri AK. Genetic toxicology of a paradoxical human carcinogen, arsenic: a review. Mutat. Res. 2001;488:171–194. doi: 10.1016/s1383-5742(01)00056-4. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Kopplin MJ, Gandolfi AJ. Effects of arsenite on UROtsa cells: low-level arsenite causes accumulation of ubiquitinated proteins that is enhanced by reduction in cellular glutathione levels. Toxicol. Appl. Pharmacol. 2004;198:412–418. doi: 10.1016/j.taap.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Chien CW, Chiang MC, Ho IC, Lee TC. Association of chromosomal alteration with arsenite-induced tumorigenicity of human HaCaT keritinocytes in nude mice. Environ. Health Perspect. 2004;112:1704–1710. doi: 10.1289/ehp.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Arnold LL, Uzvolgyi E, Cano M, St. John M, Yamamoto S, Lu X, Le C. Possible role of dimethylarsinous acid in dimethylarsinic acid-induced urothelial toxicity and regeneration in the rat. Chem. Res. Toxicol. 2002;15:1150–1157. doi: 10.1021/tx020026z. [DOI] [PubMed] [Google Scholar]
- Cullen WR, Reimer KJ. Arsenic speciation in the environment. Chem. Rev. 1989;89:713–764. [Google Scholar]
- Delker D, Ouyang M, Welsh W, Roop B, Geter D, Chen Y, O’Brien T, Kitchin K. Gene expression profiling of mouse skin and papillomas following chronic exposure to monomethylarsonous acid in K6/ODC transgenic mice. The Toxicologist CD—An official Journal of the Society of Toxicology. 2006 March;90(S1) Abstract # 54. [Google Scholar]
- Drobona Z, Jaspers I, Thomas DJ, Styblo M. Differential activation of AP-1 in human bladder epithelial cells by inorganic and methylated arsenicals. FASEB. 2003;17:67–69. doi: 10.1096/fj.02-0287fje. [DOI] [PubMed] [Google Scholar]
- Enterline PE, Day R, Marsh GM. Cancers related to exposure to arsenic at a copper smelter. Occup. Environ. Med. 1995;52:28–32. doi: 10.1136/oem.52.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Lu X, Cullen WR, Le C. Unstable trivalent arsenic metabolites, monomethylarsonous acid and dimethylarsinous acid. J. Anal. At. Spectrom. 2001;16:1409–1413. [Google Scholar]
- Guo HR, Wang NS, Hu H, Monson RR. Cell type specificity of lung cancer associated with arsenic ingestion. Cancer Epidemiol., Biomarkers Prev. 2004;13:638–643. [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer) IARC Monographs on the evaluation of carcinogenic risk of chemicals to humans. Vol. 23. World Health Organization; Lyon, France: 1980. Some metals and metallic compounds. [Google Scholar]
- Johansson SL, Cohen SM. Epidemiology and etiology of bladder cancer. Semin. Surg. Oncol. 1997;13:291–298. doi: 10.1002/(sici)1098-2388(199709/10)13:5<291::aid-ssu2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- Kligerman AD, Doerr CL, Tennant AH, Harrington-Brock K, Allen JW, Winkfield E, Poorman-Allen P, Kundu B, Funasaka K, Roop BC, Mass MJ, DeMarini DM. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: induction of chromosomal utations but not gene mutations. Environ. Mol. Mutagen. 2003;42:192–205. doi: 10.1002/em.10192. [DOI] [PubMed] [Google Scholar]
- Krishnamohan M, Seawright AA, Ng JC. Monomethylarsonous acid (MMAIII) is carcinogenic in mice. The Toxicologist CD—An official Journal of the Society of Toxicology. 2006 March;9(S1) Abstract # 2086. [Google Scholar]
- Le XC, Ma M, Cullen WR, Aposhian HV, Lu X, Zheng B. Determination of monomethylarsonous acid, a key arsenic methylation intermediate, in human urine. Environ. Health Perspect. 2000;108:1015–1018. doi: 10.1289/ehp.001081015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TC, Oshimura M, Barrett JC. Comparison of arsenic-induced cell transformation, cytotoxicity, mutation and cytogenetic effects in Syrian hamster embryo cells in culture. Carcinogenesis. 1985;6:1421–1426. doi: 10.1093/carcin/6.10.1421. [DOI] [PubMed] [Google Scholar]
- Lin S, Cullen WR, Thomas DJ. Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem. Res. Toxicol. 1999;12:924–930. doi: 10.1021/tx9900775. [DOI] [PubMed] [Google Scholar]
- Loveland BE, Johns TG, Mackay IR, Vailland F, Wang ZX, Hertzog PJ. Validation of the MTT dye assay for enumeration of cells in proliferative and antiproliferative assays. Biochem. Int. 1992;27:501–510. [PubMed] [Google Scholar]
- Mandal BK, Ogra Y, Suzuki KT. Identification of dimethylarsinous and monomethylarsonous acids in human urine of the arsenic-affected areas in West Bengal, India. Chem. Res. Toxicol. 2001;14:371–378. doi: 10.1021/tx000246h. [DOI] [PubMed] [Google Scholar]
- Mass MJ, Tennant A, Roop BC, Cullen WR, Styblo M, Thomas DJ, Kligerman AD. Methylated trivalent arsenic species are genotoxic. Chem. Res. Toxicol. 2001;14:355–361. doi: 10.1021/tx000251l. [DOI] [PubMed] [Google Scholar]
- Millar IT, Heany H, Heinekey DM, Fernelius WC. Methyliiodoarsine. Inorg. Synth. 1960;6:113–115. [Google Scholar]
- Mure K, Uddin AN, Lopez LC, Styblo M, Rossman TG. Arsenite induces delayed mutagenesis and transformation in human osteosarcoma cells at extremely low concentrations. Environ. Mol. Mutagen. 2003;41:322–331. doi: 10.1002/em.10164. [DOI] [PubMed] [Google Scholar]
- NCR (National Research Council) Arsenic in Drinking Water. National Academy Press; Washington, D.C.: 2000. [Google Scholar]
- Nesnow S, Roop BC, Lambert G, Kadiiska M, Mason RP, Cullen WP, Mass MJ. DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species. 2002;15:1627–1634. doi: 10.1021/tx025598y. [DOI] [PubMed] [Google Scholar]
- Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Aposhian HV. Monomethylarsonous acid (MMAIII) is more toxic than arsenite in change human hepatocytes. Toxicol. Appl. Pharmacol. 2000;163:203–207. doi: 10.1006/taap.1999.8872. [DOI] [PubMed] [Google Scholar]
- Petrick JS, Jagadish B, Mash EA, Aposhian HV. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem. Res. Toxicol. 2001;14:651–656. doi: 10.1021/tx000264z. [DOI] [PubMed] [Google Scholar]
- Petzoldt L, Leigh IM, Duffy PG, Sexton C, Masters RW. Immortalisation of human urothelial cells. Urol. Res. 1995;23:377–380. doi: 10.1007/BF00698738. [DOI] [PubMed] [Google Scholar]
- Qu W, Bortner CD, Sakurai T, Hobson MJ, Waalkes MP. Acquisition of apoptotic resistance in arsenic-induced malignant transformation: role of the JNK signal transduction pathway. Carcinogenesis. 2002;23:151–159. doi: 10.1093/carcin/23.1.151. [DOI] [PubMed] [Google Scholar]
- Rossi MR, Masters JRW, Park S, Todd JH, Garrett SH, Sens MA, Somji S, Nath J, Sens DA. The immortalized UROtsa cell line as a potential cell culture model of human urothelium. Environ. Health Perspect. 2001;109:801–808. doi: 10.1289/ehp.01109801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FJ, Bosland MC. Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin: an animal model for arsenic carcinogenesis. Toxicol. Appl. Pharmacol. 2001;176:64–71. doi: 10.1006/taap.2001.9277. [DOI] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FJ, Bosland MC. Arsenite cocarcinogenesis: an animal model derived from genetic toxicology studies. Environ. Health Perspect. 2002;110:749–752. doi: 10.1289/ehp.02110s5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim EI, Wanibuchi H, Morimura K, Wei M, Mitsuhashi M, Yoshida K, Endo G, Fukushima S. Carcinogenicity of dimethylarsinic acid in p53 heterozygous knockout and wild-type C57BL/6J mice. Carcinogenesis. 2003;24:335–342. doi: 10.1093/carcin/24.2.335. [DOI] [PubMed] [Google Scholar]
- Schwerdtle T, Walter I, Mackiw I, Hartwig A. Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA. Carcinogenesis. 2003;24:967–974. doi: 10.1093/carcin/bgg018. [DOI] [PubMed] [Google Scholar]
- Sens DA, Park S, Gurel V, Sens MA, Garrett SH, Somji S. Inorganic cadmium- and arsenite-induced malignant transformation of human bladder urothelial cells. Toxicol. Sci. 2004;79:56–63. doi: 10.1093/toxsci/kfh086. [DOI] [PubMed] [Google Scholar]
- Shen J, Wanibuchi H, Waalkes MP, Salim EI, Kinoshita A, Yoshida K, Endo G, Fukushima S. A comparative study of the sub-chronic toxic effects of three organic arsenical compounds on the urothelium in F344 rats; gender-based differences in response. Toxicol. Appl. Pharmacol. 2006;210:171–180. doi: 10.1016/j.taap.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Styblo M, Serves SV, Cullen WR, Thomas DJ. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem. Res. Toxicol. 1997;10:27–33. doi: 10.1021/tx960139g. [DOI] [PubMed] [Google Scholar]
- Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 2000;74:289–299. doi: 10.1007/s002040000134. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Barrett JC, Tsutsui T. Transformation by inorganic arsenic compounds of normal Syrian hamster embryo cells into a neoplastic state in which they become anchorage-independent and cause tumors in newborn hamsters. Int. J. Cancer. 2002;99:629–634. doi: 10.1002/ijc.10407. [DOI] [PubMed] [Google Scholar]
- Vahter M. Species differences in the metabolism of arsenic compounds. Appl. Organomet. Chem. 1994;8:175–182. [Google Scholar]
- Valenzuela OL, Borja-Aburto VH, Garcia-Vargas GG, Cruz-Gonzalez MB, Garcia-Montalvo EA, Calderon-Aranda ES, Del Razo LM. Urinary trivalent methylated arsenic species in a population chronically exposed to inorganic arsenic. Environ. Health Perspect. 2005;113:250–254. doi: 10.1289/ehp.7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega L, Styblo M, Patterson R, Cullen W, Wang C, Germolec D. Differential effects of trivalent and pentavalent arsenical son cell proliferation and cytokine secretion in normal human epidermal keratinocytes. Toxicol. Appl. Pharmacol. 2001;172:225–232. doi: 10.1006/taap.2001.9152. [DOI] [PubMed] [Google Scholar]
- Walen KH. Spontaneous cell transformation: karyoplasts derived from multinucleated cells produce new cell growth in senescent human epithelial cultures. In Vitro Cell Dev. Biol., Anim. 2004;40:150–158. doi: 10.1290/1543-706X(2004)40<150:SCTKDF>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Wang TS, Chung CH, Wang AS, Bau DT, Samikkannu T, Jan KY, Cheng YM, Lee TC. Endonuclease III, formamidopyrimidine-DNA glycosylase and proteinase K additively enhance arsenic-induced DNA strand breaks in human cells. Chem. Res. Toxicol. 2002;15:1254–1258. doi: 10.1021/tx025535f. [DOI] [PubMed] [Google Scholar]
- Wei M, Wanibuchi H, Yamamoto S, Li W, Fukushima S. Urinary bladder carcinogenicity of dimethylarinic acid in male F344 rats. 1999;20:1873–1876. doi: 10.1093/carcin/20.9.1873. [DOI] [PubMed] [Google Scholar]
- Xie Y, Trouba KJ, Liu J, Waalkes MP, Germolec DR. Biokinetics and subchronic toxic effects of oral arsenite, arsenate, monomethylarsonic acid, and dimethylarsinic acid in v-Ha-ras transgenic (Tg,AC) mice. Environ. Health Perspect. 2004;112:1255–1263. doi: 10.1289/txg.7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Konishi Y, Matsuda T, Murai T, Shibata MA, Matsui-Yuasa I, Otani S, Kuroda K, Endo G, Fukushima S. Cancer induction by an organic arsenic compound, dimethylarsinic acid (cacodylic acid), in F344/DuCrj rats after pretreatment with five carcinogens. Cancer Res. 1995;55:1271–1276. [PubMed] [Google Scholar]
- Zakharyan R, Aposhian HV. Enzymatic reduction of arsenic compounds in mammalian systems: the rate limiting enzyme of rabbit liver arsenic biotransformation is MMAV reductase. Chem. Res. Toxicol. 1999;12:1278–1283. doi: 10.1021/tx9901231. [DOI] [PubMed] [Google Scholar]
- Zakharyan R, Wu Y, Bogdan GM, Aposhian HV. Enzymatic methylation of arsenic compounds: assay, partial purification, and properties of arsenite methyltransferase and monomethylarsonic acid methyltransferase of rabbit liver. Chem. Res. Toxicol. 1995;8:1029–1038. doi: 10.1021/tx00050a006. [DOI] [PubMed] [Google Scholar]