

Abstract
Phosphorylation-dependent changes in AMPA receptor function have a crucial role in activity-dependent forms of synaptic plasticity such as long-term potentiation (LTP) and long-term depression (LTD). Although three previously identified phosphorylation sites in AMPA receptor glutamate receptor 1 (GluR1) subunits (S818, S831, and S845) appear to have important roles in LTP and LTD, little is known about the role of other putative phosphorylation sites in GluR1. Here, we describe the characterization of a recently identified phosphorylation site in GluR1 at threonine 840. The results of in vivo and in vitro phosphorylation assays suggest that T840 is not a substrate for protein kinases known to phosphorylate GluR1 at previously identified phosphorylation sites, such as protein kinase A, protein kinase C, and calcium/calmodulin-dependent kinase II. Instead, in vitro phosphorylation assays suggest that T840 is a substrate for p70S6 kinase. Although LTP-inducing patterns of synaptic stimulation had no effect on GluR1 phosphorylation at T840 in the hippocampal CA1 region, bath application of NMDA induced a strong, protein phosphatase 1- and/or 2A-mediated decrease in T840 phosphorylation. Moreover, GluR1 phosphorylation at T840 was transiently decreased by a chemical LTD induction protocol that induced a short-term depression of synaptic strength and persistently decreased by a chemical LTD induction protocol that induced a lasting depression of synaptic transmission. Together, our results show that GluR1 phosphorylation at T840 is regulated by NMDA receptor activation and suggest that decreases in GluR1 phosphorylation at T840 may have a role in LTD.
Keywords: AMPA receptor, GluR1, NMDA receptor, phosphorylation, long-term depression, hippocampus
Introduction
Activity-dependent forms of synaptic plasticity, such as long-term potentiation (LTP) and long-term depression (LTD), are thought to have an essential role in the storage of new information during learning. A key feature of current molecular models of LTP and LTD is that changes in the strength of synaptic transmission in these forms of plasticity are mostly attributable to phosphorylation-dependent changes in the activity and/or trafficking of postsynaptic AMPA-type glutamate receptors (AMPARs) (Collingridge et al., 2004). For example, the induction of NMDA receptor (NMDAR)-dependent forms of LTP is associated with a persistent, calcium/calmodulin-dependent kinase II (CaMKII)-mediated increase in phosphorylation of AMPAR glutamate receptor 1 (GluR1) subunits at serine 831 (Barria et al., 1997a; Lee et al., 2000) and phosphorylation of this site produces an increase in AMPAR channel conductance (Barria et al., 1997b; Derkach et al., 1999) like that seen after LTP induction (Benke et al., 1998; Luthi et al., 2004). Conversely, LTD induction is thought to require activation of protein phosphatases that dephosphorylate postsynaptic AMPARs at S845 (Lee et al., 1998, 2000), leading to decreases in channel activity (Banke et al., 2000) and/or endocytosis of AMPA receptors out of the postsynaptic membrane (Ehlers, 2000) (for review, see Winder and Sweatt, 2001; Collingridge et al., 2004).
Although this AMPAR-centric model of LTP and LTD is supported by a wealth of evidence, it seems clear that more complex models will be needed to fully describe how NMDAR activation can lead to persistent changes in AMPAR function. For instance, increases in GluR1 phosphorylation at S831 and S845 cannot fully account for the activity-dependent insertion of new AMPARs into the synapse that occurs during LTP (Boehm and Malinow, 2005). Moreover, although LTD is strongly disrupted in mice with mutations in GluR1 that prevent phosphorylation at S831 and S845, some patterns of synaptic stimulation can still induce significant, albeit reduced, levels of LTP in these mutants (Lee et al., 2003). It thus seems likely that LTP induction involves phosphorylation at other sites within GluR1 that regulate AMPAR function and/or trafficking, such as S818 (Boehm et al., 2006). Alternatively, LTP and LTD may require changes in phosphorylation of other proteins that can interact with AMPARs and facilitate their movement into and out of the postsynaptic membrane (Nicoll et al., 2006).
Here, we report characterization of a recently identified phosphorylation site in the C terminus of AMPAR GluR1 subunits at threonine 840 (T840) (Lee et al., 2007; Munton et al., 2007) that is an in vitro substrate for p70S6 kinase. Although LTP induction in the hippocampal CA1 region was not associated with an increase in GluR1 phosphorylation at T840, NMDAR activation induced a strong, protein phosphatase 1/2A (PP1/2A)-dependent dephosphorylation at T840. Using different pharmacological protocols to induce either short- or long-term synaptic depression, we find a striking correlation between changes in synaptic strength and GluR1 phosphorylation at T840 suggesting that decreases in GluR1 phosphorylation at T840 may have a role in hippocampal LTD.
Materials and Methods
Slice preparation and electrophysiology.
Standard techniques approved by the University of California, Los Angeles (UCLA) Institutional Animal Care and Use Committee were used to prepare 400-μm-thick slices from hippocampi obtained from 2- to 3-month-old C57BL/6 mice. For some experiments, “mini-slices” containing just the hippocampal CA1 region were prepared by removing the dentate gyrus, CA3 region, and subiculum from freshly cut slices. In all experiments, slices were maintained at 30°C in an interface-type chamber (Fine Science Tools, Foster City, CA) and continuously perfused with an oxygenated (95% O2/5% CO2) artificial CSF (ACSF) consisting of 124 mm NaCl, 4.4 mm KCl, 25 mm Na2HCO3, 1 mm NaH2PO4, 1.2 mm MgSO4, 2 mm CaCl2, and 10 mm glucose. Slices were allowed to recover for at least 2 h before the start of an experiment. Schaffer collateral/commissural fiber synapses onto CA1 pyramidal cells were activated using a bipolar, nichrome wire electrode placed in stratum radiatum of the CA1 region of the slice, and the resulting synaptic potentials were recorded using an ACSF-filled glass microelectrode (5–10 MΩ) placed in stratum radiatum. Single pulses of presynaptic fiber stimulation were delivered at 0.02 Hz using a stimulation intensity that evoked field EPSPs that were 50% of the maximum amplitude that could be evoked using strong stimulation intensities. To examine the effects of synaptic stimulation on GluR1 phosphorylation at T840, we used CA1 mini-slices maintained in interface-slice chambers and used larger bipolar stimulation electrodes fabricated from 66-μm-diameter, Formvar-coated nichrome wire (A-M Systems, Carlsborg, WA). The tip separation of the stimulation electrode was adjusted such that the electrode spanned the width of stratum radiatum and the stimulating electrode was placed at one end of the slice while an extracellular recording electrode was placed in stratum radiatum at the opposite end of the slice.
Western immunoblotting.
Slices were prepared and maintained using techniques identical with those used for electrophysiological recordings. In general, slices obtained from the same animal were placed into up to four separate chambers (three slices per chamber). One chamber was exposed to ACSF alone to provide control, untreated tissue while the remaining chambers were treated with various pharmacological reagents. This allowed us to use a within-subjects design and, by pooling multiple slices per condition, provided sufficient amounts of protein for several immunoblots. Thus, different blots could be used to measure phospho- and total GluR1 levels from the same samples. Pharmacological treatments and tissue homogenization were performed using previously described methods (Delgado and O'Dell, 2005). Synaptoneurosomes were prepared using a previously described protocol (Ho et al., 2004). Proteins (20 μg/lane) were resolved on 12% SDS-PAGE gels, transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes, and incubated overnight with primary antibodies. After a 2–4 h incubation with HRP-conjugated secondary antibodies (1:2000), immunoreactive bands were visualized using enhanced chemiluminesence (Immun-Star; Bio-Rad, Hercules, CA). Image acquisition and analysis were done using a cooled CCD camera and the Quantity One software package from Bio-Rad. To control for potential variations in loading, all blots were reprobed with anti-tubulin or anti-actin antibodies and the optical density values for each band of interest were normalized to the density values obtained for these loading controls in the same lane.
Antibodies.
The rabbit polyclonal antibody against phospho-T840 GluR1 was made by Abcam (Cambridge, UK), using a synthetic peptide (conjugated to KLH) derived from the C terminus of mouse GluR1 phosphorylated at T840. The antibody was subsequently immunogen affinity purified. Phosphospecific antibodies to GluR1 phosphorylated at S845 and S831 (both used at 1:1000) as well as total GluR1 (1:2000) were obtained from Upstate Biotechnology (Lake Placid, NY). Monoclonal antibodies to actin (1:2000) and βIII tubulin (1:20,000) were obtained from Upstate Biotechnology and Sigma (St. Louis, MO), respectively.
Immunoprecipitation.
Hippocampal slices were maintained at 30°C in submerged-slice type chambers and perfused (at 1–2 ml/min) with oxygenated (95% O2/5% CO2) ACSF. Slices were allowed to recover for at least 3 h and then homogenized in 20 mm 4-morpholinepropanesulfonic acid (MOPS), pH 7.4, 2 mm EDTA, 5 mm EGTA, 1% Triton X-100, 0.5% DOC (sodium deoxycholate), 30 mm NaF, 40 mm β-glycerophosphate, 20 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and Roche (Basel, Switzerland) Complete protease inhibitor mixture. Protein concentrations were determined by Micro BCA Protein Assay kit (Pierce, Rockford, IL) and 500 μg of total protein was immunoprecipitated overnight at 4°C with 2 μg of anti-GluR1 (Chemicon, Temecula, CA). After a 2 h incubation with protein G-Sepharose, samples were washed three times with 20 mm MOPS, pH 7.4, 1% Triton X-100, and 1 mm sodium orthovanadate. Proteins were then separated by SDS-PAGE and procedures described above were used for Western blotting.
Expression of wild-type and T840A GluR1.
Mutagenesis using the QuikChange Site-Directed Mutagenesis kit (Stratagene, Cedar Creek, TX) was performed according to the manufacturer's instructions to produce the mutant GluR1 T840A with the following forward and reverse primers: 5′-GCCATACGGACATCGGCCCTCCCCCGG-3′ and 5′-CCGGGGGAGGGCCGATGTCCGTATGGC-3′. The fidelity of the mutagenesis reaction was verified by DNA sequencing. Transfection of HEK 293T cells was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells were plated on 6 cm dishes and transfected with 16 μg of DNA and 40 μl of Lipofectamine at ∼70–75% confluence. To enhance GluR1 phosphorylation at T840, the protein phosphatase 1 and 2A inhibitor Cantharidin (10 μm; Tocris, Ellisville, MO) was added 24 h after transfection, and cells were incubated for 1 h at 37°C. Cells were then washed three times with ice-cold PBS and lysed in 600 μl of RIPA buffer [50 mm Tris, pH 7.5, 150 mm NaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS plus Complete protease inhibitor mixture (Roche) and phosphatase inhibitor mixtures 1 and 2 (Sigma)] on ice for 30 min. GluR1 was then immunoprecipitated (at 4°C) by first preclearing lysates for 3 h with normal rabbit serum. Each lysate was then incubated overnight with 6 μl of anti-GluR1 antibody (Upstate Biotechnology). Immune complexes were then bound to protein A-Sepharose beads (Zymed, South San Francisco, CA) during a 1 h incubation. The beads were then washed four times in wash buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, 0.1% Triton X-100, 10% glycerol), and proteins were eluted in Laemmli buffer.
Cell culture and immunocytochemistry.
At UCLA, primary cultures of hippocampal neurons were grown on Matrigel (Becton Dickinson Labware, Bedford, MA)-coated coverslips in MEM media (Invitrogen) containing 5% FBS (HyClone, Logan, UT), 2% B-27 supplement (Invitrogen), 2 mm Glutamax (Invitrogen), 24 mg/ml insulin (Sigma), 0.1 mg/ml transferrin (Calbiochem, La Jolla, CA), 28 mm glucose, and 4 mm AraC (Sigma). After 14–21 d in vitro, cultures were fixed with 4% paraformaldehyde for 10 min and cells were permeabilized for 5 min with 0.1% Triton X-100. After quenching free aldehyde groups with 50 mm NH4Cl for 10–15 min, nonspecific antibody binding was blocked by incubation in 10% goat serum for 30 min. Cells were then incubated with primary antibodies in 10% goat serum for 1–3 h at room temperature or overnight at 4°C. Primary antibodies were detected by incubation with 1:2000 Alexa Fluor 488- or 546-conjugated secondary antibodies in 10% goat serum for 1 h at room temperature. Confocal fluorescence images were obtained using a Zeiss (Oberkochen, Germany) Pascal scanning laser microscope using 63× (1.2 numerical aperture) water immersion objective.
At The Sanger Institute, cultures grown on polylysine/laminin-coated coverslips for 14–24 d in vitro were fixed with ice-cold methanol for 7 min, blocked in PBS containing 3% BSA and 0.2% Triton X-100 for 1 h (blocking solution was also used in both antibody steps); primary antibodies were applied together for 1 h at room temperature. Secondary antibodies were applied together for 20 min at room temperature. Coverslips were mounted with Prolong Anti-Fade Gold (Invitrogen). Primary antibodies used were as follows: mouse anti-GluR1 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-phospho T840 GluR1, and chicken anti-microtubule-associated protein 2 (MAP2) (Abcam, Cambridge, UK). Secondary antibodies used were as follows: chicken IgY specific-Cy2 and mouse IgG specific-Rhodamine (Abcam, Cambridge, UK), and rabbit IgG specific-Alexa Fluor 633 (Invitrogen). All images were taken on a Zeiss 510 META confocal microscope using a 63× Plan-apochromat objective.
Peptide array phosphorylation.
The 15-aa-long peptides that encompassed the GluR1 threonine 840 (NEAIRTSTLPRNSGA) were synthesized on cellulose membranes in a parallel manner using SPOT technology (Reineke et al., 2001) and covalently immobilized to glass slides. Each peptide was present in triplicate and a negative control peptide for the phosphorylation site was included, replacing threonine with valine (NEAIRTSVLPRNSGA). As a positive control of kinase activity, consensus phosphorylation peptides were attached. These peptide sequences were included in a broader screen of synaptic phosphorylation sites to be reported elsewhere. Peptide arrays were sealed with Gene-Frame incubation chambers (Abgene House, Surrey, UK), and the chambers were filled with 330 μl of kinase buffer, 20 mm MOPS, pH 7.2, 25 mm β-glycerol phosphate, 5 mm EGTA, 1 mm sodium orthovanadate, 1 mm DTT, and 2 μg of p70 S6 kinase (T412E). The reaction was initiated by the addition of ATP/MgCl2 [final concentrations: 100 μm ATP, 10 μCi of [γ-32P]ATP (GE Healthcare, Piscataway, NJ), 15 mm MgCl2]. After a 40 min incubation at 32°C, the peptide microarrays were washed six times, alternating between 0.1 m phosphoric acid and distilled water. γ-32P incorporation in the immobilized peptide spots was detected on a Typhoon 8600 PhosphorImager (50 μm resolution; Amersham Biosciences). Image analysis and signal quantification were performed using ImageQuant TL (GE Healthcare), and positive signals were defined after background subtraction. The following recombinant kinases were used (all obtained from Upstate Biotechnology): protein kinase A (PKA), catalytic subunit, Akt1/(ΔPH, S473D), protein kinase Cα (PKCα), PKCδ, PKCζ, phosphoinositide-dependent kinase 1 (PDK-1), Cdk5, Raf-1, p38α, mitogen-activated protein kinase kinase 1 (MEK1), extracellular signal-regulated kinase 1 (Erk1), Erk2, Rsk2, c-Jun N-terminal kinase 3 (JNK3), glycogen synthase kinase 3β (GSK3β), CaMKII, casein kinase 1, casein kinase 2, p70 S6 kinase (T412E), Rho GTPase-Rho kinase II (ROCK-II), TANK binding kinase 1 (TBK1), Src, Fyn, Pyk2, and Fes.
Statistical analysis.
Paired and unpaired t tests or one-way ANOVAs followed by Student–Newman–Keuls tests for multiple pairwise comparisons were used to assess statistical significance. Nonparametric versions of these tests (Mann–Whitney rank sum tests and Friedman repeated-measures ANOVAs on ranks) were used where appropriate. Statistical tests were performed using SigmaStat (Systat Software, Richmond, CA). All results are reported as mean ± SEM.
Results
In recent years, with the advent of proteomic technologies, the discovery of phosphorylation sites has increased exponentially with several approaches identifying large numbers of in vivo phosphorylated synaptic proteins (Jaffe et al., 2004; Collins et al., 2005; Trinidad et al., 2006). We previously found that many of these synaptic phosphoproteins exhibit multiple phosphorylation sites, with 331 identified sites distributed among only 79 proteins (Collins et al., 2005). Importantly, most of these new phosphorylation sites were clustered in very short peptide sequences. We thus analyzed the C-terminal region of the mouse AMPA receptor GluR1 subunit between R820 and S855 to search for a putative cluster of phosphorylation sites using two different prediction algorithms, Scansite (Obenauer et al., 2003) and NetPhosK (Blom et al., 1999). The Scansite algorithm identified T840 as a putative phosphorylation site under low and medium levels of stringency, which are conditions that fail to predict any other possible serine/threonine phosphorylation sites, including the well characterized S831 and S845 sites. Similar results were obtained using the NetPhos algorithm in which only T840 and S845 were identified as potential phosphorylation sites. Although the failure of both Scansite and NetPhosK to correctly identify known GluR1 phosphorylation sites (S831 and S845) suggests that the results of the programs should be interpreted with caution, we noted that T840 in GluR1 is conserved across several species including humans, rodents, marsupials, fish, and amphibians (Fig. 1A). We thus decided to examine whether GluR1 is phosphorylated at T840 in vivo and whether phosphorylation at this site might be regulated by NMDAR activation. Importantly, after we began our experiments, a study using mass spectrometry demonstrated that GluR1 is phosphorylated at T840 in vivo (Munton et al., 2007).
Figure 1.
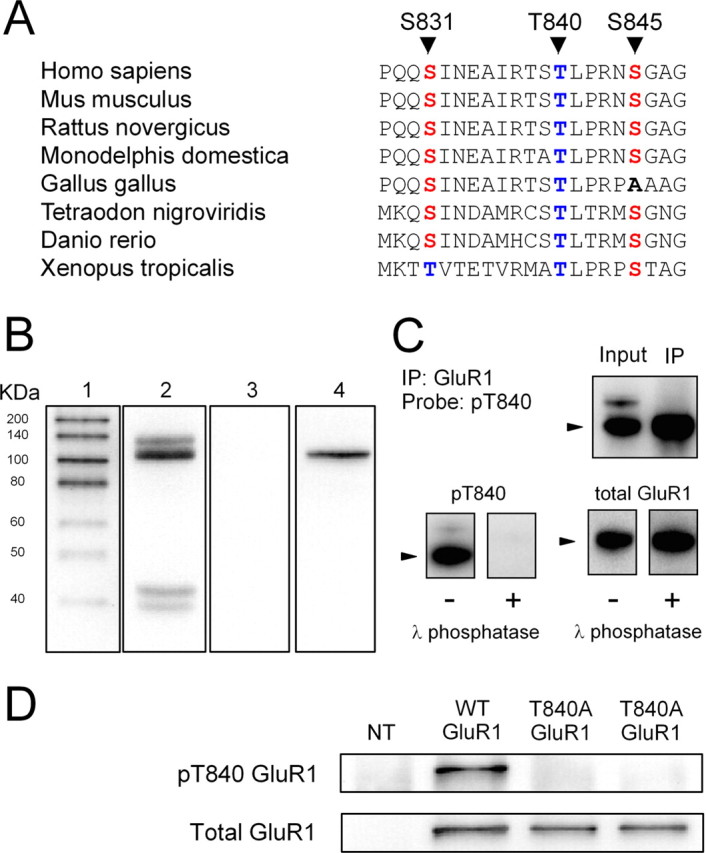
Characterization of phospho-T840 antibody. A, Sequence alignment for amino acids surrounding S831, T840, and S845 in GluR1 across several different vertebrate species. B, Proteins in homogenates prepared from mouse hippocampi were resolved by SDS-PAGE, transferred to PVDF membranes, and probed with the anti-phospho-T840 GluR1 antibody (lanes 2 and 3). Four distinct immunoreactive bands were detected in blots probed with the anti-phospho-T840 GluR1 antibody (antibody concentration, 0.12 μg/ml; lane 2), whereas no signal was detected in blots in which the peptide antigen used to generated the phospho-T840 GluR1 antibody was present during the primary antibody incubation (peptide concentration, 0.3 μg/ml; lane 3). Lane 1 shows biotinylated molecular weight markers, whereas lane 4 shows the same blot as lane 3 after reprobing with anti-total GluR1 antibody. C, Top, Immunoblots show bands detected by phospho-T840 GluR1 antibody before and after immunoprecipitating GluR1 from hippocampal extracts. Bottom, Immunoblots showing the effects of treating blots with λ phosphatase (2000 U for 40 min at 32°C) before incubation in anti-phospho-T840 GluR1 antibody (left set of blots). These blots were subsequently stripped and reprobed with a total GluR1 antibody (right set of blots). The arrowheads indicate the ≈110 kDa band corresponding to GluR1. D, Western blot analysis of antibody specificity for T840 phosphorylated GluR1. GluR1 was immunoprecipitated from lysates prepared from HEK cells expressing wild-type (WT) GluR1 or a mutant form GluR1 in which T840 was changed to an alanine (T840A GluR1). Blots were probed with anti-phospho-T840 GluR1 (top), and then stripped and reprobed with total GluR1 antibodies (bottom). The first lane shows nontransfected (NT) controls.
A phosphorylation site-specific antibody detects T840-phosphorylated GluR1 in vivo
To examine whether GluR1 is phosphorylated at T840 in vivo, we generated a phosphorylation site-specific antibody against T840 phosphorylated GluR1 by injecting rabbits with a 15-aa-long phosphopeptide corresponding to sequence surrounding threonine 840 in GluR1 (NEAIRTSpTLPRNSGA). As shown in Figure 1B, this antibody recognized four distinct bands with molecular weights of ∼135, 110, 43, and 39 kDa in immunoblots of homogenates prepared from whole mouse hippocampus. The most prominent band at 110 kDa is near the expected molecular weight for GluR1 (≈106 kDa) and comigrated with total GluR1 in our gels (Fig. 1B, lanes 2 and 4), suggesting that this band corresponds to T840 phosphorylated GluR1. Consistent with this, a band of the same molecular weight was readily detected by the anti-phospho-T840 GluR1 antibody in experiments in which GluR1 subunits were immunoprecipitated from hippocampal extracts before Western blotting (Fig. 1C). Detection of all four bands was completely blocked when the phosphopeptide used to generate the anti-phospho-T840 GluR1 antibody was present during the primary antibody incubation step (Fig. 1B, lane 3), whereas a nonphosphorylated form of the same peptide had no effect (data not shown). Moreover, the anti-phospho-T840 GluR1 antibody also failed to detect bands on blots pretreated with λ phosphatase before primary antibody incubation (Fig. 1C).
The results shown in Figure 1, B and C, indicate that the phospho-T840 GluR1 antibody specifically recognizes phosphorylated but not nonphosphorylated forms of GluR1 and suggest that GluR1 is phosphorylated at T840 in the hippocampus. To confirm that our antibody specifically recognizes GluR1 phosphorylated at T840, as opposed to other potential sites in GluR1, we performed two different experiments. First, we compared the ability of the phospho-T840 GluR1 antibody to detect GluR1 immunoprecipitated from lysates obtained from transfected HEK cells expressing either wild-type GluR1 or GluR1 in which T840 was mutated to an alanine (T840A GluR1). As shown in Figure 1D, the anti-phospho-T840 antibody readily detected GluR1 immunoprecipitated from cells expressing wild-type GluR1, whereas no bands were detected in lysates obtained from cells expressing T840A GluR1. Along with the results shown in Figure 1C, this indicates that the phospho-T840 GluR1 antibody only recognizes GluR1 phosphorylated at T840. Second, we also examined whether phosphorylation of GluR1 detected with this antibody was altered in Western blots of homogenates prepared from hippocampal slices treated with activators of protein kinases known to increase GluR1 phosphorylation at S845 or S831. In these experiments, we compared levels of GluR1 phosphorylation at S845, S831, and T840 in homogenates prepared from either untreated slices or slices bathed in activators of PKA or PKC. As shown in Figure 2A, activation of PKA, either by direct activation of adenylyl cyclase with forskolin (FSK) (10–20 μm for 15 min) or by activation of Gs-coupled β-adrenergic receptors with isoproterenol (ISO) (1 μm for 5 min) produced a more than twofold increase in GluR1 phosphorylation at S845, a site known to be phosphorylated by PKA (Roche et al., 1998; Oh et al., 2006), but had no effect on levels of T840 phosphorylated GluR1 (phospho-T840 GluR1 levels were 109 ± 10 and 101 ± 12% of untreated controls in FSK- and ISO-treated slices, respectively; n = 6 for both). Similarly, activation of PKC with the phorbol ester phorbol 12,13-dibutyrate (10 μm for 15 min) had no effect on GluR1 phosphorylation at T840 (phospho-T840 GluR1 levels were 102 ± 6% of untreated controls; n = 4), although it induced a nearly threefold increase in GluR1 phosphorylation at S831, a site phosphorylated by both PKC and CaMKII (Barria et al., 1997a; Mammen et al., 1997) (Fig. 2B). Activation of Gq-coupled, metabotropic glutamate receptors (mGluRs) with the agonist (1S,3R)-aminocyclopentane-1,3-dicarboxylic acid (100 μm for 10 min) also significantly increased GluR1 phosphorylation at S831 but had no effect on T840 phosphorylation (Fig. 2B). These results indicate that the phospho-T840 GluR1 antibody does not recognize GluR1 subunits phosphorylated at S845 or S831. In addition, they also suggest that neither PKA nor PKC phosphorylate GluR1 at T840 in vivo. Notably, the pharmacological manipulations used in these experiments also induced robust increases in levels of the dually phosphorylated, active form of Erk2 (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Moreover, mGluR activation also induced a significant increase in levels of threonine 286 phosphorylated, persistently active αCaMKII (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). The fact that T840 phosphorylation is unchanged in slices in which these kinases are strongly activated suggests that Erk2 and αCaMKII also do not phosphorylate GluR1 at T840 in vivo. Consistent with these findings, we also observed that prolonged incubation of slices in ACSF containing PKA, PKC, or MEK inhibitors had no effect on basal levels of GluR1 phosphorylation at T840 (supplemental Fig. 1C–E, available at www.jneurosci.org as supplemental material).
Figure 2.
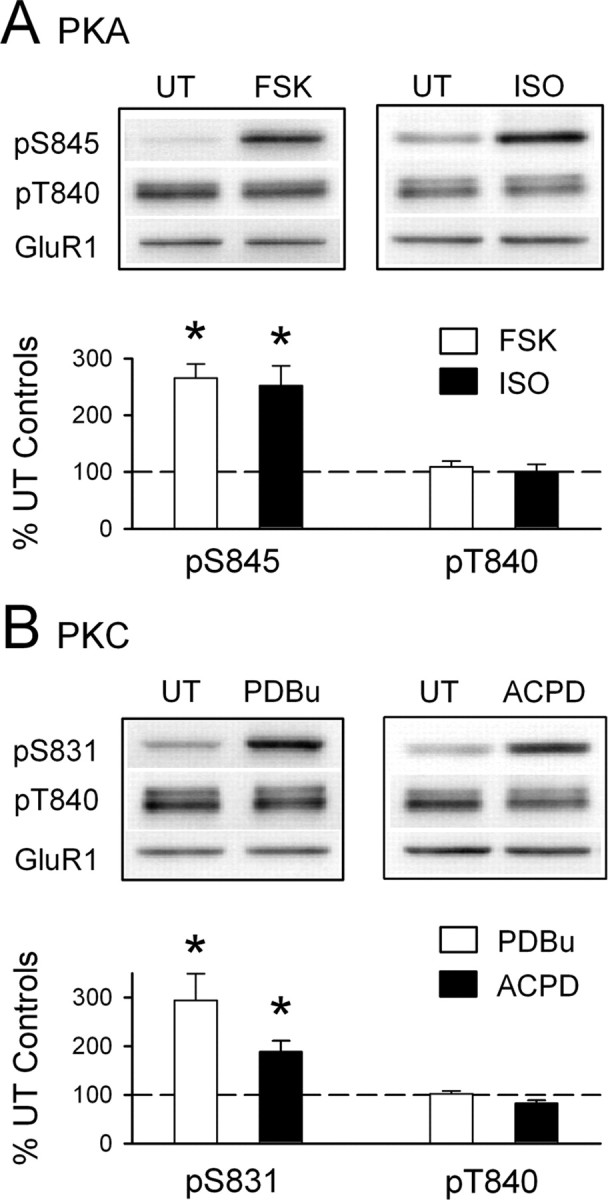
The anti-phospho-T840 GluR1 antibody does not recognize S831 and S845 phosphorylated GluR1. A, Hippocampal slices obtained from the same animal were untreated (UT) or exposed to the PKA activators FSK (10–20 μm for 10 min; n = 5) or the β-adrenergic receptor agonist ISO (1 μm for 5 min; n = 4). Both FSK and ISO induced a significant increase in GluR1 phosphorylation at S845 (*p < 0.05 compared with untreated control slices) but had no effect on GluR1 phosphorylation at T840. B, Bath application of the PKC activator PDBu (10 μm for 15 min) significantly increased GluR1 phosphorylation at S831 (*p < 0.05 compared with untreated control slices; n = 4) but had no effect on GluR1 phosphorylation at T840. A 10 min bath application of the mGluR agonist ACPD (1-aminocyclopentane-1,3-dicarboxylic acid) (100 μm) also induced a significant increase in phospho-S831 GluR1 levels (*p < 0.05 compared with untreated controls; n = 5) but had no effect on GluR1 phosphorylation at T840. The immunoblots above each histogram in A and B show representative blots probed with the indicated phospho-specific antibodies. None of the protein kinase activators had effects on total GluR1 levels. Error bars indicate SEM.
Finally, we also examined GluR1 phosphorylation at T840 in vivo by using the phospho-T840 GluR1 antibody for immunofluorescent staining of cultured hippocampal neurons. As shown in Figure 3A, prominent phospho-T840 GluR1-like immunoreactivity is present in both the cell soma and in MAP2-positive dendritic processes. At higher levels of magnification, phospho-T840 GluR1-like immunostaining showed a discrete, punctate-like pattern of staining in dendrites that colocalized with the postsynaptic marker PSD-95 but showed little colocalization with the presynaptic marker synaptophysin (Fig. 3B). This suggests that that the phospho-T840 GluR1-like immunostaining is attributable, in part, to phosphorylation of postsynaptic AMPARs. Indeed, triple immunofluorescent staining of cultured hippocampal neurons with anti-GluR1, anti-phospho-T840 GluR1, and anti-MAP2 antibodies revealed strong colocalization of GluR1 and phospho-T840 immunofluorescence within MAP2-positive processes (Fig. 3C). Although by immunoblotting the pT840 antibody recognized three bands in addition to the GluR1 band in hippocampal homogenates, immunocytochemistry with the antibody showed very low cell body/dendrite staining in hippocampal cultures prepared from GluR1 null mutant mice (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Of particular note was the complete absence of any puncta colocalizing with synaptic markers in the GluR1 knock-out cells (Fig. 3D; supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Thus, by immunocytochemistry the antibody primarily detects the pT840 epitope, with no background staining of synaptic proteins.
Figure 3.
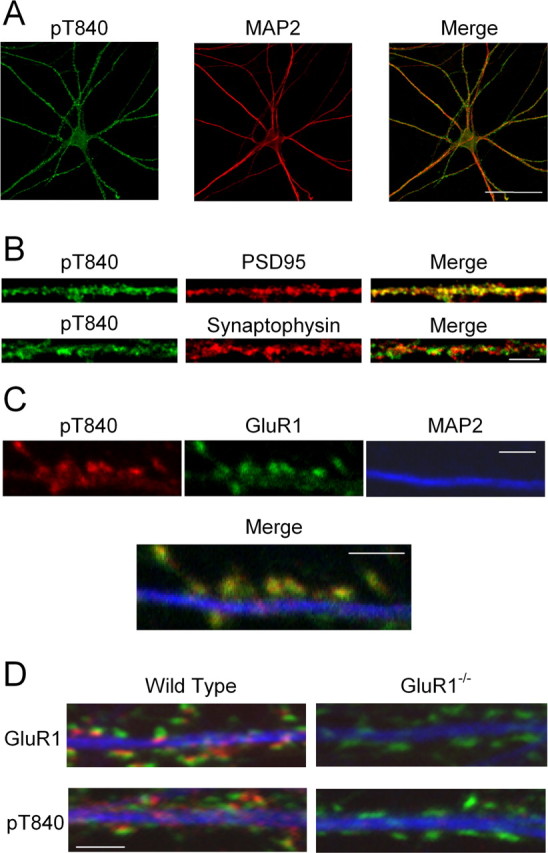
Immunofluorescent staining of cultured mouse hippocampal neurons with the phospho-T840 GluR1 antibody. A, Confocal images showing that phospho-T840 GluR1-like immunofluorescence is present throughout the soma and MAP2-positive dendrites of hippocampal neurons. Scale bar, 50 μm. B, Higher magnification images of processes in cultured neurons reveal extensive colocalization of phospho-T840 GluR1-like immunofluorescence with the postsynaptic marker PSD-95 (top) but little overlap with the presynaptic marker synaptophysin (bottom). Scale bar, 5 μm. C, Triple staining of cultured hippocampal neurons with anti-phospho-T840 (green), anti-GluR1 (red), and anti-MAP2 (blue) antibodies reveals strong colocalization of phospho-T840 GluR1-like immunofluorescence with total GluR1 staining. Scale bar, 2 μm. D, Cultures of hippocampal neurons obtained from wild-type and GluR1-null mutant mice were stained with anti-MAP2 (blue) and anti-synaptophysin (green) antibodies. Immunostaining for both GluR1 (red; top two panels) and phospho-T840 GluR1 (red; bottom two panels) was strongly diminished in GluR1−/− neurons. Scale bar, 2 μm.
LTP induction is not associated with increases in GluR1 phosphorylation at T840
To determine whether changes in GluR1 phosphorylation at T840 might be associated with LTP induction in the hippocampal CA1 region, we next examined the effects of theta pulse stimulation (TPS) on GluR1 phosphorylation in CA1 mini-slices. As shown in Figure 4A, CA1 mini-slices exhibit robust LTP after a brief (30 s) train of TPS. Thus, in a separate set of experiments, we used Western immunoblotting to compare levels of GluR1 phosphorylation at T840 in TPS-stimulated slices harvested 5 min after a 30 s train of TPS to unstimulated control slices obtained from the same animal. As shown in Figure 4B, TPS had no effect on GluR1 phosphorylation at T840 (5 min post-TPS phospho-T840 GluR1 levels were 106 ± 2% of unstimulated controls; n = 8). As a positive control, we also examined GluR1 phosphorylation at S831 in these same samples and observed a statistically significant increase in GluR1 phosphorylation at S831 in stimulated slices (phospho-S831 GluR1 levels were increased to 153 ± 6% of unstimulated controls; n = 8; p < 0.01) as reported by others (Barria et al., 1997a; Lee et al., 2000). These findings suggest that increases in GluR1 phosphorylation at T840 are unlikely to be involved in TPS-induced LTP.
Figure 4.
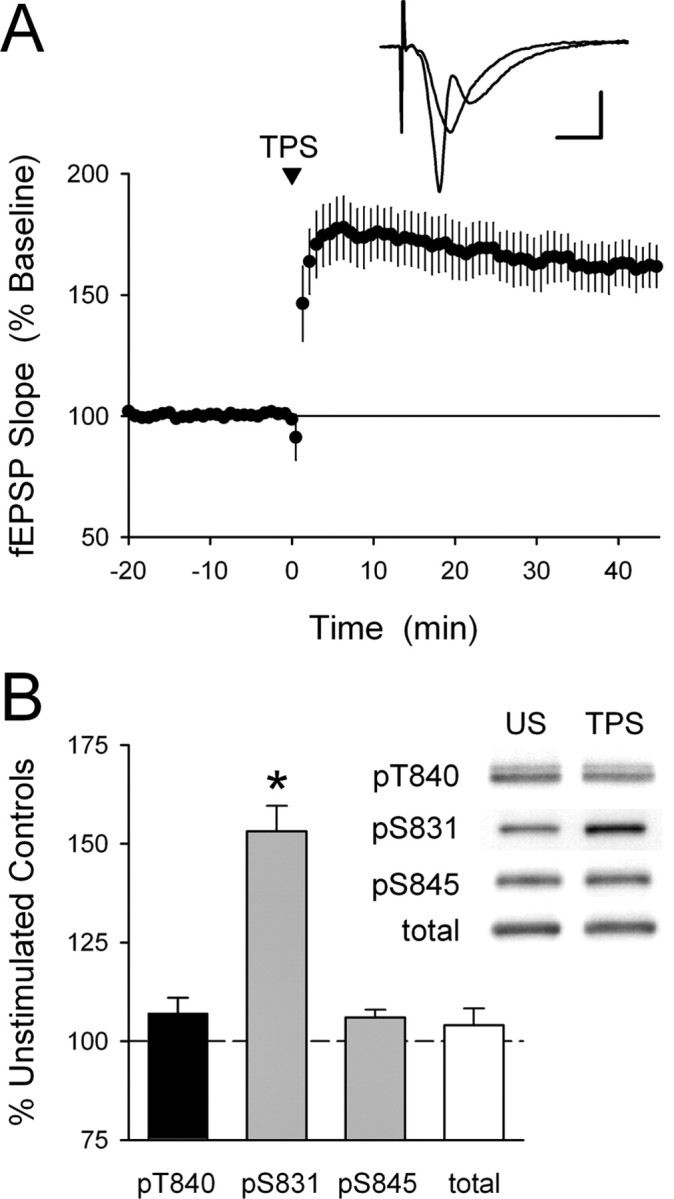
GluR1 phosphorylation at T840 is not enhanced by LTP-inducing patterns of synaptic stimulation. A, A TPS protocol consisting of 150 pulses of presynaptic fiber stimulation delivered at 5 Hz induces significant LTP in hippocampal CA1 mini-slices. Field EPSPs were potentiated to 161 ± 8% of baseline 60 min post-TPS (p < 0.001 compared with baseline; n = 5). The inset shows fEPSPs recorded during baseline and 45 min post-TPS. Calibration: 2 mV, 5 ms. B, LTP inducing patterns of synaptic stimulation have no effect on GluR1 phosphorylation at T840. CA1 mini-slices obtained from the same animal were either unstimulated (US) or snap frozen 5 min after activating presynaptic fibers in stratum radiatum with a TPS protocol (150 pulses at 5 Hz). Whereas TPS induced a significant increase in GluR1 phosphorylation at S831 (*p < 0.01 compared with US controls), it had no effect on GluR1 phosphorylation at T840 or S845 (n = 8 experiments). The immunoblots show phospho-GluR1 and total-GluR1 levels in samples from one experiment. Error bars indicate SEM.
NMDA receptor activation induces a dephosphorylation of AMPAR GluR1 subunits at T840
We next explored the effects of NMDAR activation on GluR1 phosphorylation at T840 by pharmacologically activating NMDARs with bath-applied NMDA. In these experiments, we first measured levels of GluR1 phosphorylation at T840 in hippocampal slices obtained from the same animal that were either bathed in agonist-free ACSF (untreated controls) or exposed to a 3 min bath application of ACSF containing different concentrations of NMDA. As shown in Figure 5A, NMDA induced a significant, dose-dependent decrease in GluR1 phosphorylation at T840 [at the highest concentration tested (100 μm), NMDA reduced phospho-T840 GluR1 levels to 43 ± 5% of untreated controls; n = 6]. Significant decreases in GluR1 phosphorylation at T840 after NMDAR activation could also be readily detected in both NMDA-treated CA1 mini-slices (levels were reduced to 62 ± 5% of untreated control mini-slices; n = 5) (Fig. 5B) and in synaptoneurosome preparations prepared from NMDA-treated slices (levels were reduced to 50 ± 7% of untreated controls; n = 4) (Fig. 5C). Moreover, immunostaining with the anti-phospho-T840 GluR1 antibody revealed a clear, NMDA-induced decrease in phospho-T840 GluR1-like immunoreactivity in stratum radiatum of the hippocampal CA1 region (Fig. 5D). NMDAR activation also induced a significant decrease in dendritic phospho-T840 GluR1-like immunofluorescence in cultured hippocampal neurons (data not shown). Together, these findings suggest that the GluR1 subunits of AMPARs are rapidly dephosphorylated at T840 after activation of NMDARs on CA1 pyramidal cells.
Figure 5.
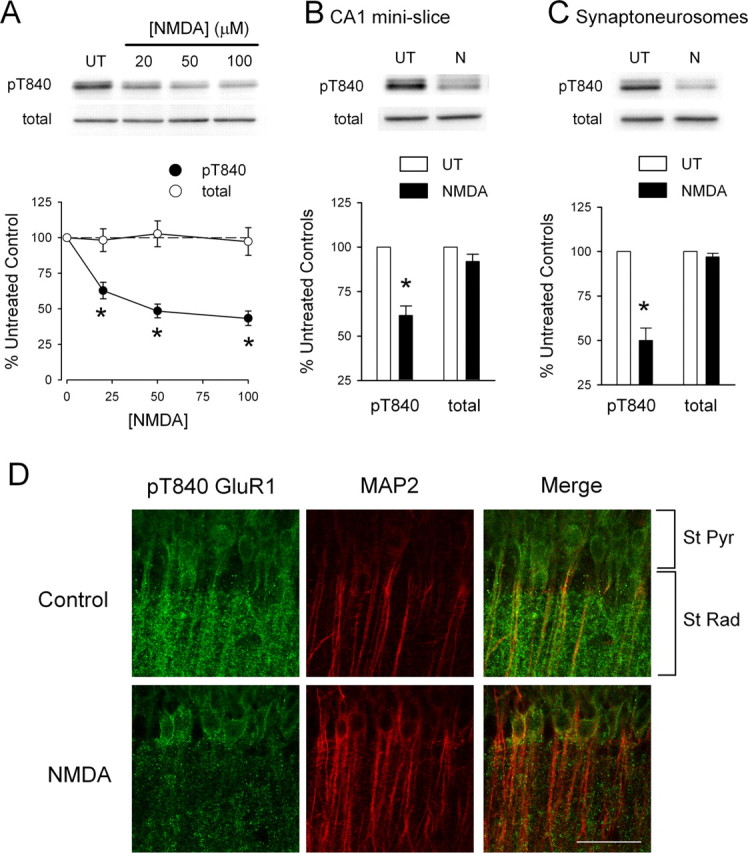
NMDAR activation dephosphorylates GluR1 at T840. A, Hippocampal slices obtained from the same animal were either left untreated (UT) or exposed to 3 min bath applications of the indicated concentrations of NMDA. NMDA induced a significant, dose-dependent decrease in GluR1 phosphorylation at T840 (filled symbols; *p < 0.05 compared with untreated controls; n = 6). NMDAR activation had no effect on total GluR1 levels (open symbols). The immunoblots show changes in GluR1 phosphorylation at T840 as well as total GluR1 levels in samples obtained from one experiment. B, Bath application of NMDA (20 μm for 3 min) induces significant dephosphorylation of GluR1 at T840 in isolated CA1 mini-slices (*p < 0.05 compared with untreated controls; n = 4). C, Dephosphorylation of GluR1 at T840 is readily detected in synaptoneurosomes prepared from NMDA-treated slices (20 μm NMDA for 3 min; *p < 0.01 compared with synaptoneurosomes from untreated control slices; n = 4). Error bars indicate SEM. D, Phospho-T840 GluR1-like immunofluorescence in stratum radiatum of the hippocampal CA1 region is reduced in NMDA-treated slices. Hippocampal slices were fixed immediately after a 3 min application of 20 μm NMDA and stained with phospho-T840 GluR1 and MAP2 antibodies. Confocal images of the hippocampal CA1 region in an untreated control slice (top) and in a slice exposed to NMDA (bottom). Scale bar, 50 μm.
Chem-LTD induction is associated with persistent dephosphorylation of GluR1 subunits at T840
To determine how the temporal profile of NMDA induced changes in GluR1 phosphorylation at T840 might correspond to changes in synaptic strength, we next examined the effects of a chemical stimulation protocol for inducing LTD (chem-LTD) that has proven useful for studying biochemical processes underlying NMDAR-dependent LTD (Kameyama et al., 1998; Lee et al., 1998; Colledge et al., 2003; Brown et al., 2005; Oh et al., 2006). In these experiments, hippocampal slices were maintained under identical conditions and used in either electrophysiological experiments or treated with NMDA (20 μm for 3 min) and collected for Western analysis at different time points after NMDAR activation. Although a 3 min bath application of 20 μm NMDA has been shown to induce robust chem-LTD in the hippocampal CA1 region (Kameyama et al., 1998; Lee et al., 1998), under our experimental conditions synaptic transmission was strongly depressed by NMDA receptor activation but then rapidly recovered to baseline levels after NMDA washout (20 min post-NMDA application fEPSPs were 107 ± 16% of baseline; n = 6) (Fig. 6A). Strikingly, NMDA-induced changes in GluR1 phosphorylation at T840 closely paralleled these changes in synaptic strength. As shown in Figure 6B, GluR1 phosphorylation at T840 was significantly decreased immediately after NMDA application (levels were reduced to 68 ± 7% of untreated controls; p < 0.05; n = 5) but then rapidly recovered after NMDA washout (20 min after NMDA washout, phospho-T840 GluR1 levels recovered to 96 ± 6% of untreated controls). In a separate set of experiments, we found that stronger activation of NMDARs with a longer bath application of NMDA (20 μm for 5 min) also failed to induced a lasting depression of synaptic strength and induced a robust, but transient, decrease in GluR1 phosphorylation at T840 (data not shown).
Figure 6.

NMDA-induced changes in GluR1 T840 phosphorylation parallel NMDA-induced changes in synaptic strength. A, A 3 min bath application of 20 μm NMDA has no lasting effect on synaptic transmission. NMDA was applied starting at time = 0 (indicated by the arrow). Although synaptic transmission was strongly depressed in the presence of NMDA, synaptic strength returned to baseline levels after NMDA washout (60 min post-NMDA fEPSPs were 112 ± 6% of baseline; n = 6; p = 0.14 compared with baseline). B, NMDAR activation transiently dephosphorylates GluR1 at T840. Hippocampal slices obtained from the same animal were either left untreated or stimulated with NMDA (20 μm for 3 min) and collected for analysis either immediately after NMDA treatment or after NMDA had been washed from the slice chamber for the indicated times. Although levels of T840 phosphorylation GluR1 (filled symbols) were significantly decreased at the end of a 3 min application of 20 μm NMDA, GluR1 phosphorylation at T840 recovered to basal levels by 20 min after NMDA washout. Total GluR1 levels (open symbols) were unchanged. Results are from seven separate experiments; *p < 0.05 compared with untreated control slices (time = 0 on the plot). The inset shows phospho-T840 GluR1 and total GluR1 levels from one experiment. C, Bath application of NMDA (20 μm for 3 min) induces significant LTD in slices bathed in high-Ca2+ ACSF. Sixty minutes after NMDA washout, fEPSPs were depressed to 48 ± 8% of baseline; p < 0.001 compared with baseline; n = 11). The inset shows fEPSPs recorded during baseline and 60 min after NMDA application in a representative experiment. Calibration bars: 1 mV, 5 ms. D, NMDAR activation induces a persistent dephosphorylation of GluR1 at T840 (filled symbols) in slices bathed in high-Ca2+ ACSF (n = 6; *p < 0.05 compared with untreated controls). Total GluR1 levels (open symbols) were unchanged. Error bars indicate SEM. The immunoblots show levels of T840 phosphorylated GluR1 and total GluR1 in a representative experiment.
To examine how the effects of NMDA receptor activation on GluR1 phosphorylation at T840 might be altered under experimental conditions in which NMDA receptor activation induces LTD, we took advantage of a previous finding showing that elevated levels of external calcium can enhance LTD induction (Norris et al., 1996). Consistent with the findings of Norris et al. (1996), bathing slices in a high-Ca2+ ACSF containing 4.0 mm CaCl2 significantly facilitated the induction of LTD by a 15-min-long train of 1 Hz synaptic stimulation [60 min after 1 Hz stimulation, fEPSPs were depressed to 86 ± 3% of baseline in control slices (n = 6) and were depressed to 61 ± 3% of baseline in slices bathed in high-Ca2+ ACSF; n = 8; p < 0.001]. Similarly, the induction of chem-LTD by bath-applied NMDA was also significantly enhanced in high-Ca2+ ACSF (Fig. 6C). Based on this observation, we reexamined the effects of NMDAR activation on GluR1 phosphorylation using hippocampal slices maintained in high-Ca2+ ACSF to determine how GluR1 phosphorylation at T840 is altered under conditions in which chem-LTD is induced. In these experiments, we maintained hippocampal slices obtained from the same animal in high-Ca2+ ACSF and then harvested slices for Western blot analysis at different time points after transiently activating NMDARs with bath-applied NMDA (20 μm for 3 min). Although high-Ca2+ ACSF by itself had no effect on basal levels of GluR1 phosphorylation at T840 (data not shown), the effects of NMDAR activation on T840 phosphorylation were strongly altered in slices bathed in high-Ca2+ ACSF. As shown in Figure 6D, bath application of NMDA under experimental conditions that induce chem-LTD not only induced a significantly stronger initial decrease in GluR1 phosphorylation at T840 relative to control experiments but also induced a significant, persistent decrease in T840 phosphorylation (GluR1 phosphorylation at T840 was reduced to 67 ± 5% of baseline at 40 min after NMDA washout; p < 0.05 compared with untreated control slices; n = 6). Thus, across three different stimulation protocols, we find that changes in GluR1 phosphorylation at T840 show a striking correlation with both short- and long-term changes in synaptic strength.
To examine whether the induction of LTD by synaptic stimulation is also associated with a decrease in GluR1 phosphorylation at T840, we measured phospho-T840 GluR1 levels 5 min after stimulating CA1 mini-slices hippocampal slices with a conventional LTD stimulation protocol (900 pulses of presynaptic fiber stimulation delivered at 1 Hz). Although we observed robust dephosphorylation of T840 in chem-LTD (Fig. 6D), we were unable to observe significant changes in GluR1 phosphorylation at T840 in slices that received 1 Hz stimulation relative to unstimulated controls (data not shown) (n = 12). Importantly, in these same samples we were also unable to observe low-frequency stimulation-induced changes in GluR1 at S845 (Lee et al., 2000), suggesting that our techniques lack sufficient sensitivity to detect changes in GluR1 phosphorylation that occur during synaptically induced LTD.
Identification of T840 kinases and phosphatases
To begin to identify the protein kinases that might be able to phosphorylated GluR1 at T840, we tested whether GluR1 T840 could serve as a kinase substrate in vitro using peptide arrays and recombinant kinases (Collins et al., 2005). A 15-aa-long peptide that encompassed the GluR1 threonine 840 (NEAIRTSTLPRNSGA; 840 shown in bold) was synthesized and covalently immobilized to glass slides. A control peptide with a valine substitution at 840 (T840V) was used as a negative control. We chose to use a library of 25 recombinant kinases (see Materials and Methods) based on the fact that these kinases were previously identified as components of the postsynaptic proteome (Collins et al., 2005) and could thus potentially phosphorylate postsynaptic substrates such as GluR1. Although Scansite analysis under low-stringency conditions indicates that T840 may be phosphorylated by either PKA or the protein kinase Akt, neither kinase phosphorylated GluR1 at T840 in vitro (Table 1). Indeed, of the 25 protein kinases tested only 1, p70S6 kinase (p70S6K), was able to induce phosphorylation of the GluR1 T840 peptide (Table 1; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This phosphorylation was absent in the T840V control peptide indicating that T840 is an in vitro kinase substrate with specificity for p70S6K.
Table 1.
Results from in vitro peptide array phosphorylation assays examining GluR1 phosphorylation at T840
| Kinase | Protein | Sequence | Site | Level (%) |
|---|---|---|---|---|
| PKA | GluR1 | NEAIRTSTLPRNSGA | T840 | 0 |
| PKA | Control | LRRASLGVLKI | Consensus | 100 |
| Akt | GluR1 | NEAIRTSTLPRNSGA | T840 | 0 |
| Akt | Control | GVGRPRTSSFAEGAG | Consensus | 100 |
| p70S6K | GluR1 | NEAIRTSTLPRNSGA | T840 | 38 ± 10 |
| p70S6K | GluR1 | NEAIRTSVLPRNSGA | T840V | 0 |
| p70S6K | Control | GGKKRNRTLTKKRGA | Consensus | 100 |
Although PKA and Akt failed to phosphorylate the GluR1 T840 peptide, significant phosphorylation was detected in assays with p70S6K. Levels show optical density measurements normalized to signals from positive control peptides in the same array containing consensus sites for PKA, Akt, or p70S6K. Note that p70S6K phosphorylation of the peptide is abolished when T840 is replaced with a valine (T840V).
The protein phosphatases PP1 and PP2A are thought to have a crucial role in the induction of NMDAR-dependent LTD in the hippocampal CA1 region (Mulkey et al., 1993; for review, see Winder and Sweatt, 2001). Thus, to identify the protein phosphatases responsible for dephosphorylating GluR1 at T840, we examined whether cantharidin, a potent inhibitor of PP1 and PP2A (Honkanen, 1993), could prevent the NMDA-induced dephosphorylation of GluR1 at T840 in hippocampal slices bathed in high-Ca2+ ACSF. In these experiments, slices obtained from the same animal were bathed in either high-Ca2+ ACSF or pretreated for 1 h in high-Ca2+ ACSF containing 10 μm cantharidin. In each condition, one set of slices was left untreated, whereas the remaining slices were exposed to NMDA (20 μm for 3 min) and collected for analysis either immediately after NMDA application or 40 min after NMDA washout. Consistent with the notion that PP1 and/or PP2A regulate GluR1 phosphorylation at T840, basal levels of GluR1 phosphorylation at T840 were significantly elevated in cantharidin-treated slices (Fig. 7A). Moreover, although NMDA induced a significant, persistent dephosphorylation of T840 in control slices bathed in high-Ca2+ ACSF alone, it had no effect on phospho-T840 GluR1 levels in cantharidin-treated slices (Fig. 7A). As shown in Figure 7B, blocking PP1 and PP2A with cantharidin also significantly inhibited chem-LTD.
Figure 7.
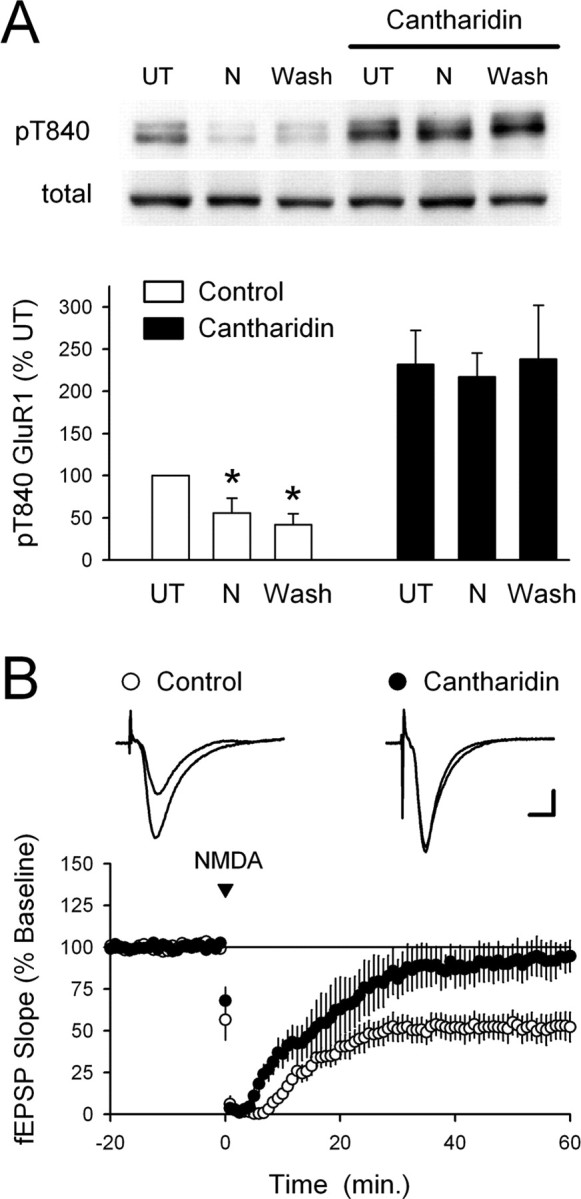
Blocking PP1 and PP2A inhibits NMDA-induced dephosphorylation of GluR1 at T840. A, Slices from the same animal were maintained in high Ca2+-ACSF either alone or in the presence of the PP1 and PP2A inhibitor cantharidin (10 μm). Under control conditions, GluR1 phosphorylation at T840 was significantly decreased both immediately after NMDA application (20 μm for 3 min; N) and 40 min after NMDA washout (Wash; *p < 0.05 compared with untreated controls; n = 5). Basal levels of T840 phosphorylated GluR1 were significantly increased in cantharidin-treated slices (p < 0.01 compared with UT slices bathed in high-Ca2+ ACSF alone), and both the initial and persistent NMDA-induced dephosphorylation of T840 were completely blocked. The immunoblots show phospho-T840 GluR1 and total GluR1 levels from a representative experiment. Error bars indicate SEM. B, Chem-LTD is inhibited in cantharidin-treated slices. Sixty minutes after NMDA application (20 μm for 3 min), fEPSPs were depressed to 52 ± 8% of baseline in vehicle control experiments (open symbols; 0.1% DMSO; n = 6) and were 93 ± 10% of baseline in cantharidin-treated slices (filled symbols; n = 5; p = 0.01 compared with control). Slices were continuously bathed in ACSF containing 10 μm cantharidin for at least 1 h before an experiment.
Discussion
Although the notion that phosphorylation-dependent changes in AMPAR function underlie activity-dependent changes in synaptic strength seems well established, a number of key steps in the signaling pathways that link NMDAR activation to persistent changes in AMPAR number and/or activity remain to be discovered. Importantly, proteomic profiling of the postsynaptic density (PSD) of excitatory synapses has revealed that the PSD is composed of >1000 different proteins (Grant, 2006) and recent studies of in vivo phosphorylation sites within these proteins has identified hundreds of previously unrecognized phosphorylation sites (Collins et al., 2005; Trinidad et al., 2006; Munton et al., 2007). Given this surprisingly high degree of complexity of the synaptic proteome and phosphoproteome, it seems likely that numerous key proteins and phosphorylation events are missing from our current models of LTP and LTD. Indeed, a recent study has found that phosphorylation of AMPAR GluR1 subunits at a newly identified site (serine 818) may be involved in LTP (Boehm et al., 2006). Based on these studies, as well as recent work suggesting that phosphorylation sites of postsynaptic proteins are not randomly distributed but instead are often found in clusters (Collins et al., 2005), we reasoned that there might be hitherto-unknown phosphorylation sites adjacent to those previously described in AMPA receptors. Using two phosphorylation prediction programs (Scansite and NetPhosK), we identified T840 as a potential phosphorylation site in GluR1. Although computational methods have proven to be useful for identifying phosphorylation sites and protein interaction domains (Park et al., 2006; Poolos et al., 2006), in silico approaches are still in their early stages of development and both programs failed to identify known phosphorylation sites (S831 and/or S845) in the C terminus of GluR1. We noted, however, the T840 is highly conserved in vertebrates and found that a peptide corresponding to a 15-aa-long stretch of the C terminus of GluR1 containing T840 was phosphorylated at T840 by p70S6K in vitro. This indicates that GluR1 may indeed be phosphorylated at T840. Consistent with this, recent studies using immobilized metal affinity chromatograph and mass spectrometry (Munton et al., 2007) and phospho-peptide mapping (Lee et al., 2007) have demonstrated that AMPAR GluR1 subunits are phosphorylated at T840.
GluR1 phosphorylation at T840 and LTP
Using the anti-phospho-T840 antibody, we could readily detect T840-phosphorylated GluR1 in immunoblots run using homogenates prepared from both intact hippocampus and hippocampal slices maintained in vitro, suggesting that AMPAR GluR1 subunits are basally phosphorylated at T840 in hippocampal neurons. Both in situ (slices) and in vitro kinase assays indicate, however, that none of four serine/threonine protein kinases that have a crucial role in NMDA receptor-dependent LTP (PKA, PKC, CaMKII, and ERK2) phosphorylate GluR1 at T840. Consistent with this, we found that LTP induction had no effect on GluR1 phosphorylation at T840. Although it is possible that high basal levels of phosphorylation at T840 might make it difficult to detect increases in phosphorylation in slices treated with various protein kinase activators, the approximately twofold increase in T840 phosphorylation induced by the protein phosphatase inhibitor cantharidin (Fig. 7) suggests that this is unlikely. The lack of a potential role for PKA, PKC, and ERK is also supported by our observation that inhibitors of these kinases had no effect on basal levels of GluR1 phosphorylation at T840 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Surprisingly, only 1 protein kinase of 25 tested in our in vitro assays, p70S6K, was able to phosphorylate GluR1 at T840. Although the lack of specific p70S6K inhibitors prevents direct validation of this finding in hippocampal slices, the presence of p70S6K in the dendrites of hippocampal neurons (Cammalleri et al., 2003; Tsokas et al., 2005) is consistent with a potential role for this kinase in regulating GluR1 phosphorylation at T840. Interestingly, p70S6K is best known as a downstream target for mammalian target of rapamycin (mTOR) and has been implicated as an important component of the signaling pathways underlying NMDAR-dependent changes in protein synthesis in LTP (Tang et al., 2002; Cammalleri et al., 2003; Klann and Dever, 2004; Tsokas et al., 2005). Thus, although our results indicate that LTP induction is not associated with a rapid increase in GluR1 phosphorylation at T840, additional experiments will be required to determine whether increases in GluR1 phosphorylation at T840 might occur during the later, protein synthesis-dependent phases of LTP (often referred to as L-LTP). If so, this could not only explain our failure to see increases in GluR1 phosphorylation at T840 soon after relatively weak TPS protocols but would also suggest the intriguing possibility that increases in GluR1 phosphorylation at T840 might have a unique role in regulating AMPAR function at synapses that have undergone L-LTP. Notably, p70S6K associates with PP2A and the activity of this phosphatase is inhibited by mTOR, one of the upstream activators of p70S6K (Peterson et al., 1999). Thus, the candidate T840 kinases and phosphatases identified in our experiments may act in a highly coordinated manner to regulate GluR1 phosphorylation at T840. It is important to note, however, that although our in vitro kinase assays suggest that T840 is uniquely phosphorylated by p70S6K, some kinase/substrate interactions are notoriously weak in vitro. For instance, although PKA readily phosphorylates GluR1 at S845 in intact hippocampal neurons, PKA phosphorylation of S845 is difficult to demonstrate using in vitro kinase assays (Oh et al., 2006). In addition, recent evidence suggests that T840 may also be phosphorylated by PKC (Lee et al., 2007). Thus, additional work is needed to determine whether other protein kinases can phosphorylate GluR1 at T840 in vivo and to determine whether T840 is indeed an in vivo substrate for p70S6 kinase.
GluR1 phosphorylation at T840 and LTD
NMDAR activation induced strong dephosphorylation of GluR1 subunits at T840 indicating that NMDAR-activated protein phosphatases regulate GluR1 phosphorylation at this site. Indeed, using a number of different chem-LTD induction protocols, we find that NMDA-induced changes in GluR1 phosphorylation at T840 parallel NMDA-induced changes in synaptic strength and that GluR1 is persistently dephosphorylated at T840 only when significant chem-LTD is induced. These results suggest that dephosphorylation of GluR1 at T840 may have a role in LTD, and, consistent with this, we found that inhibitors of protein phosphatases known to be activated during LTD block NMDA-induced dephosphorylation of T840.
Chem-LTD induction protocols provide a way to induce a LTD-like depression of synaptic transmission at large numbers of synapses in slice preparations and have proven to be useful for studying changes in phosphorylation of synaptic proteins. The induction of chem-LTD occludes the induction of LTD by synaptic stimulation (Lee et al., 1998), suggesting that common molecular mechanisms underlie the both forms of LTD. It seems unlikely, however, that the mechanisms underlying the persistent depression of synaptic transmission induced by pharmacological activation of NMDARs are identical with those responsible for synaptically induced forms of LTD (Kameyama et al., 1998). Moreover, we were unable to detect decreases in GluR1 phosphorylation at T840 after low-frequency synaptic stimulation. Although it seems likely that our failure to detect changes in GluR1 phosphorylation at T840 with synaptic stimulation may be attributable to the relatively few synapses activated by the low stimulation strengths needed to induce LTD, we cannot rule out the possibility that persistent decreases in GluR1 phosphorylation at T840 only occur in response to excessive and/or prolonged bouts of NMDAR activation. Thus, rather than having an important role in LTD, persistent decreases in GluR1 phosphorylation at T840 may provide a mechanism for modulating AMPARs in response to pathological levels of NMDAR activity.
An important question not addressed by our experiments is how dephosphorylation of AMPAR GluR1 receptors at T840 might be involved in regulating synaptic transmission. One possibility is that phosphorylation of GluR1 at T840 regulates AMPAR ion channel activity in a manner analogous to that seen after phosphorylation of S831 and S845 (Barria et al., 1997b; Derkach et al., 1999; Banke et al., 2000). Interestingly, high-stringency Scansite analysis of the C terminus of GluR1 indicates that T840 lies within a predicted 14-3-3 protein-binding domain. 14-3-3 proteins interact with target proteins in a serine/threonine phosphorylation-dependent manner and are thought to act as adaptor or scaffolding proteins that regulate a large number of different cellular processes including ion channel gating (Li et al., 2006) as well as protein kinase and protein phosphatase signaling (Fu et al., 2000; Berg et al., 2003). Moreover, 14-3-3 proteins have been shown to have a crucial role in the trafficking of some types of potassium channels to the plasma membrane (Rajan et al., 2002). Thus, another possibility is that GluR1/14-3-3 interactions dependent on phosphorylation of GluR1 at T840 have an important role in AMPAR trafficking, perhaps by stabilizing AMPARs at the synapse or enabling reinsertion of AMPARs back to the synapses after endocytosis. If so, dephosphorylation of T840 and subsequent dissociation of 14-3-3 proteins from GluR1 might promote removal of AMPARs from the postsynaptic membrane. Although additional experiments will be required to address these issues, our results suggest that future studies examining the effects of GluR1 phosphorylation at T840 on AMPAR function may yield important insights into mechanisms underlying NMDAR-dependent modulation of AMPARs.
Footnotes
This work was supported by the National Institute of Mental Health (T.J.O.), the National Science Foundation (T.J.O., K.R.T.), The W. M. Keck Foundation and EJLB Foundation (K.C.M.), and The Wellcome Trust Genes to Cognition Programme and The Wellcome Trust Sanger Institute (S.G.N.G.). M.C. was supported by the Human Frontiers Science Program. C.L.H. was supported by a National Alliance for Research on Schizophrenia and Depression Award. K.C.M. is a member of The Semel Institute for Neuroscience and Human Behavior. T.J.O. and K.C.M. are members of the University of California, Los Angeles, Brain Research Institute. GluR1 mutant mice were provided by Drs. P. Seeburg and R. Sprengel. We thank M. O. Collins and J. Choudhary for mass spectrometry experiments, and A. Morton and S. Syed Salim for assistance with primary cultures.
References
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling T. Regulatory phosphorylation of AMPA-type glutamate receptors by CaMKII during long-term potentiation. Science. 1997a;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate type glutamate receptor. J Biol Chem. 1997b;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Benke TA, Luthi A, Isaac JT, Collingridge GL. Modulation of AMPA receptor unitary conductance by synaptic activity. Nature. 1998;393:793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat Rev Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;2945:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Boehm J, Malinow R. AMPA receptor phosphorylation during synaptic plasticity. Biochem Soc Trans. 2005;33:1354–1356. doi: 10.1042/BST0331354. [DOI] [PubMed] [Google Scholar]
- Boehm J, Kang M-G, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron. 2005;45:81–94. doi: 10.1016/j.neuron.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Lutjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci USA. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JTR, Wang YT. Receptor trafficking and synaptic plasticity. Trends Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Collins MO, Yu L, Coba MP, Husi H, Campuzano I, Blackstock WP, Choudhary JS, Grant SG. Proteomic analysis of in vivo phosphorylated synaptic proteins. J Biol Chem. 2005;280:5972–5982. doi: 10.1074/jbc.M411220200. [DOI] [PubMed] [Google Scholar]
- Delgado JY, O'Dell TJ. Long-term potentiation persists in an occult state following mGluR-dependent depotentiation. Neuropharmacol. 2005;48:936–948. doi: 10.1016/j.neuropharm.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- Grant SGN. The synapse proteome and phosphoproteome: a new paradigm for synapse biology. Biochem Soc Trans. 2006;34:59–63. doi: 10.1042/BST0340059. [DOI] [PubMed] [Google Scholar]
- Ho OH, Delgado JY, O'Dell TJ. Phosphorylation of proteins involved in activity-dependent forms of synaptic plasticity is altered in hippocampal slices maintained in-vitro. J Neurochem. 2004;91:1344–1357. doi: 10.1111/j.1471-4159.2004.02815.x. [DOI] [PubMed] [Google Scholar]
- Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett. 1993;330:283–286. doi: 10.1016/0014-5793(93)80889-3. [DOI] [PubMed] [Google Scholar]
- Jaffe H, Vinade L, Dosemeci A. Identification of novel phosphorylation sites on postsynaptic density proteins. Biochem Biophys Res Commun. 2004;321:210–218. doi: 10.1016/j.bbrc.2004.06.122. [DOI] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bi-directional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Kameyama K, Kaiwen He, Yu S, Rossetti L, Wilen D, Huganir RL. Identification and characterization of a novel phosphorylation site on the GluR1 subunit of AMPA receptors. Mol Cell Neurosci. 2007;36:86–94. doi: 10.1016/j.mcn.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wu Y, Zhou Y. Modulation of inactivation properties of CaV2.2 channels by 14-3-3 proteins. Neuron. 2006;51:755–771. doi: 10.1016/j.neuron.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Luthi A, Wikstrom MA, Palmer MJ, Matthew P, Benke TA, Isaac JTR, Collingridge GL. Bi-directional modulation of AMPA receptor unitary conductance by synaptic activity. BMC Neurosci. 2004;5:44. doi: 10.1186/1471-2202-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Munton RP, Tweedie-Cullen R, Livingstone-Zatchej M, Weinandy F, Waidlich M, Longo D, Gehrig P, Potthast F, Rutishauser D, Gerrits B, Panse C, Schlapback R, Mansuy IM. Qualitative and quantitative analyses of protein phosphorylation in naïve and stimulated mouse synaptosomal preparations. Mol Cell Proteomics. 2007;6:283–293. doi: 10.1074/mcp.M600046-MCP200. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- Norris CM, Korol DL, Foster TC. Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J Neurosci. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Park DJ, Freitas TA, Wallick CJ, Guyette CV, Warn-Cramer BJ. Molecular dynamics and in vitro analysis of connexin-43: a new 14-4-4 mode-1 interacting protein. Protein Sci. 2006;15:2344–2355. doi: 10.1110/ps.062172506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson PT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proc Natl Acad Sci USA. 1999;96:4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Bullis JB, Roth MK. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J Neurosci. 2006;26:7995–8003. doi: 10.1523/JNEUROSCI.2069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Preisig-Muller R, Wischmeyer E, Nehring R, Hanley PJ, Renigunta V, Musset B, Schlichthorl G, Derst C, Karschin A, Daut J. Interaction with 14-3-3 proteins promotes functional expression of the potassium channels TASK-1 and TASK-3. J Physiol (Lond) 2002;545:13–26. doi: 10.1113/jphysiol.2002.027052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke U, Volkmer-Engert R, Schneider-Mergener J. Applications of peptide arrays prepared by the SPOT-technology. Curr Opin Biotechnol. 2001;12:59–64. doi: 10.1016/s0958-1669(00)00178-6. [DOI] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1998;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad JC, Specht CH, Thalhammer A, Shoepfer R, Burlingame AL. Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol Cell Proteomics. 2006;5:914–922. doi: 10.1074/mcp.T500041-MCP200. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan PM, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25:5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]