

Abstract
Preparation of a novel c(RGDyK) targeted SN38 prodrug incorporating an indolequinone structure for bioreductively triggered drug release is described. This design yields a prodrug that targets surface molecules on tumor cells (αvβ3 integrins) and releases drug under bioreductive conditions. There are three moieties in the prodrug design, namely a therapeutic drug SN38, an indolequinone structure serving as a drug releasing trigger, and an αvβ3 integrin targeting peptide c(RGDyK). Preliminary studies showed that SN38 is released in the presence of a bioreductive enzyme (DT-diaphorase).
There is a tremendous interest in using targeted drug delivery for cancer therapies.1 Targeted delivery can minimize the undesirable drug side effects while retaining desirable therapeutic activities. Advances in molecular biology have facilitated the identification of tumor markers that can function as potential therapeutic targets.2, 3 An example of this is the identification of molecular markers that can differentiate newly formed capillaries from their mature counterparts, providing a strategy for targeted delivery of cytotoxic agents to the tumor vasculature.4, 5 The αvβ3 integrin is one of the most specific markers of tumor-associated vasculature. This marker can be recognized by targeting agents that are restricted to the vascular space during angiogenesis. Cyclic pentapeptide containing RGD is highly selective for αvβ3 and the D amino acid following Asp in the pentapeptide is essential for the 1,000 times higher activity in comparison to the linear RGD.6–8 Because of this, there has been growing interest in cyclic RGD targeted gene delivery,9 imaging applications10 and tumor therapy.11
Tissue hypoxia due to inadequate blood supply is a common feature of solid tumors. Unfortunately, hypoxic tumor cells appear to be resistant to both radiotherapy and chemotherapy.12 However, tumor hypoxia provides a unique strategy for cancer therapy. Several therapeutics have been designed to form prodrugs which can be activated by hypoxia under bioreductive conditions.13, 14 In this regard, prodrugs with indolequinone structures have been studied intensively.15, 16 There are two mechanisms through which indolequinone-based prodrugs can induce cytotoxicity. First, the indolequinone structure itself can be converted to a reactive, cytotoxic species following reduction to a hydroquinone.17 Alternatively, the indolequinone moiety can be used to form a prodrug that selectively releases other cytotoxic agents to hypoxic (i.e., bioreductive) tissues.15
SN38 belongs to the the class of 20(s)-camptothecin (CPT) group of compounds that act as potent topoisomerase I inhibitors. Due to its general toxicity and poor solubility, SN38 cannot be systemically administered to cancer patients. Numerous modifications have been made to enhance the drugs solubility including liposomal formulation,18 antibody conjugation,19 and PEG functionalization.20 CPT-11 (Irinotecan) is a SN38 derivative and clinically approved for the treatment of colorectal carcinoma.21 However, only 2–5% of of the injected dose of CPT-11 is converted to active SN38 and the drug has serious side effects including gastrointestinal toxicity and neutropenia.22
Here we report the design, synthesis and preliminary drug release study of a cyclic RGDyK (denoted by c(RGDyK))23 targeted SN38 prodrug possessing an indolequinone structure for bioreductively triggered drug release (compound 1, Scheme 3). There are three moieties in the prodrug design, namely a therapeutic drug SN38, an indolequinone structure serving as a drug releasing trigger, and an αvβ3 integrin targeting peptide c(RGDyK). We envision that this design will impart the following advantages over the free drug. First, the prodrug will be water soluble due to the presence of the targeting peptide and the short polyethylene glycol tether. Second, the prodrug will be specifically delivered to cells that overexpress the αvβ3 integrin.11 Finally, the active drug SN38 will be released under bioreductive conditions presented in tumor tissues.24 This design will enhance drug specificity towards tumor cells that overexpress the αvβ3 integrin and thus reduce side effects.
Scheme 3.

The synthesis of c(RGDyK) targeted SN38 prodrug
The synthesis of the non-targeted SN38 prodrug 12 is demonstrated in Scheme 1. The N-1 methyl analogue of the indolequinone structure has been synthesized by Naylor et. al. Extensive structure-activity studies have been performed.15 We use an ethoxycarbonyl methyl functional group to replace the methyl group at the N-1 position. This synthetic design allows the N-1 site to serve as an open reaction position to facilitate the coupling of a long chain tether with an azido functional group. The azido group is then reacted with an alkyne modified targeting moiety via click chemistry. Commercially available 5-methoxy-2-methylindole was first treated with Vilsmeier reagents to give 3-formyl compound 2 with excellent yield. Alkylation of position 1 was then carried out with sodium hydride and ethyl bromo-acetate. Nitration at the desired position 4 could be achieved at a low reaction concentration and with a large excess of nitric acid. However, a 6-nitro isomer was present at a low ratio (about 15%). The mixture of 6-nitro and 4-nitro had a very poor solubility in organic solvents, making it impossible to purify through chromatography. The mixture was reduced with Sn/HCl to give a mixture of 4- and 6-amine products. The desired 4-amino compound 5 was then purified using chromatography.
Scheme 1.

The synthesis of non-targeted SN38 prodrug
Fremy’s salt was used to covert the aromatic ring to a quinone structure with quantitative yield. The crucial step in this process is the reduction of the 3-carboxaldehyde to a hydroxyl group because several other sites in the structure can also be reduced, particularily the ethyl ester. Selective reduction of the aldehyde rather than the ester could only be achieved by using 5 equivalents of NaBH4 at 0°C over 8 minutes and the yield of this reduction was 67%. Higher temperatures and prolonged reaction times decrease the yield drastically by reducing other structures. Ethyl ester 7 was then hydrolyzed with 1 equivalent of LiOH to give carboxylic acid compound 8. Compound 8 was then coupled with 11-azido-3,6,9-trioxaundecan-1-amine with HATU to give compound 9. First we activated the 3-methylhydoxyl of compound 9 with 4-nitrophenyl chloroformate and let it reacted with the phenolic hydroxyl of SN38 directly via a carbonate bond. However, a preliminary stability study showed that the carbonate bond is not stable in PBS buffer, releasing the drug molecule within 2 hours (data not shown). To enhance the stability of the prodrug, a small diamine spacer was incorporated between the indolequinone structure and the drug molecule that forms two, more stable carbamate bonds. Moreover, the spacer can potentially reduce steric interference from the drug on the linker, thus enhancing the release rate when hypoxia is present.25 To achieve this, the activated 4-nitrophenyl carbonate of compound 9 was condensed with an excess of N, N′-dimethylethylene diamine to give compound 10. Remarkably, only 3 equivalents of diamine was used in the reaction and there was minimal dimerization. The secondary amine of compound 10 was then converted to a carbamoyl chloride with triphosgene, which was subsequently reacted with SN38 to form the non-targeted prodrug 12 with good yield.
The synthesis of alkyne modified c(RGDyK) is shown in Scheme 2. A bifunctional small linker 13 was prepared from propargyl amine and glutaric anhydride. 13 was then activated with TSTU and reacted with the c(RGDyK). The resulting mixture was purified with semi-preparative HPLC. The collected fractions were lyophilized to give 14 (66% yield) with a retention time of 17.4 min on analytical HPLC.
Scheme 2.

The modification of c(RGDyK) peptide
The synthesis of c(RGDyK) targeted SN38 prodrug is demonstrated in Scheme 3. After the click reaction, the desired product 1 was purified using a semi-preparative HPLC. Compound 1 was lyophilized as a brownish red fluffy solid (48% yield). The retention time of 1 is 25.1 min on an analytical HPLC. Electrospray mass spectrum gave peaks at 1782.1(M+H) and 902.5 (M+H+Na). (see Supporting Information)
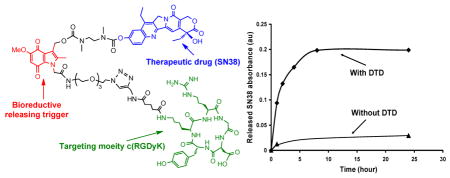
The release of SN38 from prodrug 1 in the presence of DT-diaphorase (DTD) was tested. DTD is an obligate two electron quinine reductase that uses either NADH or NADPH as electron donors. DTD has received great deal of attention as a target for anticancer prodrug development and has been implicated in the bioreductive activation of quinone-containing compounds.26 Upon reduction, the electron density at the indole nitrogen increases drastically, triggering expulsion of the exiting group in a reverse-Michael-like process, releasing the drug coupled to a small spacer. The ability of DTD to activate certain quinones, in conjunction with the fact that certain tumor types contain elevated levels of DTD, suggests that it could mediate hypoxic induced, enzyme-directed bioreductive drug delivery.27 SN38 release in the presence of DTD was characterized and monitored using HPLC. The results showed that in the presence of DTD, SN38 was efficiently released from the prodrug. (Figure 1) The prodrug hydrolysis over the same time frame in PBS without the enzyme was very limited. We further tested if 12 acts as a prodrug (minimally active) and if the the released SN38 is still active. For this, we tested the cytotoxicity of 12 in the human cervical carcinoma KB cells before and after treatment with DTD. We incubated 12 with recombinant human DTD (2mU/μL) added into the cell growth medium, in presence of different concentration of 12 for three days and cytotoxicity was determined by XTT assay. As shown in Figure 2, in the absence of DTD, the compound was essentially non-cytotoxic to up to ~ 300 nM concentration., whereas there was ~50–70% decrease in cell growth in the presence of DTD.
Figure 1.

The release of SN38 from prodrug 1 under DTD.
Figure 2.

Cytotoxicity of 12 in the presence of DTD.
To summarize, we have synthesized a novel c(RGDyK) targeted SN38 prodrug with an indolequinone structure for bioreductive drug release. Preliminary studies showed that free drug can be released from prodrug 1 in the presence of DTD. This indicates that prodrug 1 can be used to deliver anticancer drugs specifically under bioreductive conditions. The in vitro toxicity studies of prodrug 1 towards cancer cell lines expressing the αvβ3 integrin receptor under hypoxia conditions are under investigation.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under award 1 R01 CA119409.
Footnotes
Supporting Information Available: Detailed preparetive procedures, analytical data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Han HK, Amidon GL. AAPS Pharmsci. 2000;2(1):48. doi: 10.1208/ps020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esteva FJ, Hayes DF. Monoclonal Antibody-Based Therapy of Cancer. 1998;15:309. [Google Scholar]
- 3.Atkins JH, Gershell LJ. Nat Rev Cancer. 2002;2:645. [Google Scholar]
- 4.Baillie CT, Winslet MC, Bradley NJ. Brit J Cancer. 1995;72:257. doi: 10.1038/bjc.1995.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruoslahti E. Nat Rev Cancer. 2002;2:83. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- 6.Aumailley M, Gurrath M, Muller G, Calvete J, Timpl R, Kessler H. Febs Letters. 1991;291:50. doi: 10.1016/0014-5793(91)81101-d. [DOI] [PubMed] [Google Scholar]
- 7.Haubner R, Gratias R, Diefenbach B, Goodman SL, Jonczyk A, Kessler H. J Am Chem Soc. 1996;118:7461. [Google Scholar]
- 8.Pfaff M, Tangemann K, Mueller B, Gurrath M, Mueller G, Kessler H, Timpl R, Engel J. J Biol Chem. 1994;269:20233. [PubMed] [Google Scholar]
- 9.Kunath K, Merdan T, Hegener O, Haberlein H, Kissel T. J Gene Med. 2003;5:588. doi: 10.1002/jgm.382. [DOI] [PubMed] [Google Scholar]
- 10.Haubner R, Bruchertseifer F, Bock M, Kessler H, Schwaiger M, Wester H. Nuklearmedizin-Nuclear Med. 2004;43:26. doi: 10.1267/nukl04010026. [DOI] [PubMed] [Google Scholar]
- 11.Shukla R, Thomas TP, Peters J, Kotlyar A, Myc A, Baker JR. Chem Commun. 2005;46:5739. doi: 10.1039/b507350b. [DOI] [PubMed] [Google Scholar]
- 12.Brown JM, William WR. Nat Rev Cancer. 2004;4:437. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 13.Lee HH, Wilson WR, Ferry DM, vanZijl P, Pullen SM, Denny WA. J Med Chem. 1996;39:2508. doi: 10.1021/jm9600104. [DOI] [PubMed] [Google Scholar]
- 14.Priyadarsini KI, Naylor MA, Stratford MRL, Wardman P. Free Radical Res. 1996;25:99. doi: 10.3109/10715769609149914. [DOI] [PubMed] [Google Scholar]
- 15.Naylor MA, Jaffar M, Nolan J, Stephens MA, Butler S, Patel KB, Everett SA, Adams GE, Stratford IJ. J Med Chem. 1997;40:2335. doi: 10.1021/jm9608422. [DOI] [PubMed] [Google Scholar]
- 16.Hernick M, Flader C, Borch RF. J Med Chem. 2002;45:3540. doi: 10.1021/jm020191b. [DOI] [PubMed] [Google Scholar]
- 17.Walton MI, Smith PJ, Workman P. Cancer Commun. 1991;3:199. doi: 10.3727/095535491820873164. [DOI] [PubMed] [Google Scholar]
- 18.Sadzuka Y, Takabe H, Sonobe T. J Control Release. 2005;108:453. doi: 10.1016/j.jconrel.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 19.Moon SJ, Govindan SV, Cardillo TM, D’Souza CA, Hansen HJ, Goldenberg DM. J Med Chem. 2008;51:6916. doi: 10.1021/jm800719t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao H, Rubio B, Sapra P, Wu DC, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C, Greenberger LM, Horak ID. Bioconjugate Chem. 2008;19:849. doi: 10.1021/bc700333s. [DOI] [PubMed] [Google Scholar]
- 21.Vanhoefer U, Harstrick A, Achterrath W, Cao SS, Seeber S, Rustum YM. J Clin Oncol. 2001;19:1501. doi: 10.1200/JCO.2001.19.5.1501. [DOI] [PubMed] [Google Scholar]
- 22.Oreilly S, Rowinsky EK. Crit Rev Oncol Hemat. 1996;24:47. doi: 10.1016/1040-8428(96)00211-9. [DOI] [PubMed] [Google Scholar]
- 23.Auernheimer J, Haubner R, Schottelius M, Wester HJ, Kessler H. Helv Chim Acta. 2006;89:833. [Google Scholar]
- 24.Everett SA, Swann E, Naylor MA, Stratford MRL, Patel KB, Tian N, Newman RG, Vojnovic B, Moody CJ, Wardman P. Biochem Pharmacol. 2002;63:1629. doi: 10.1016/s0006-2952(02)00885-7. [DOI] [PubMed] [Google Scholar]
- 25.Saari WS, Schwering JE, Lyle PA, Smith SJ, Engelhardt EL. J Med Chem. 1990;33:97. doi: 10.1021/jm00163a016. [DOI] [PubMed] [Google Scholar]
- 26.Phillips RM, Naylor MA, Jaffar M, Doughty SW, Everett SA, Breen AG, Choudry GA, Stratford IJ. J Med Chem. 1999;42:4071. doi: 10.1021/jm991063z. [DOI] [PubMed] [Google Scholar]
- 27.Workman P. Oncol Res. 1994;6:461. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.