

Summary
Inappropriate drug delivery, secondary toxicities and persistent chemo- and immuno-resistance have traditionally compromised treatment response in melanoma. Using cellular systems and genetically engineered mouse models, we show that melanoma cells retain an innate ability to recognize cytosolic dsRNA and mount persistent stress response programs able to block tumor growth, even in highly immunosuppressed backgrounds. The dsRNA mimic polyinosine-polycytidylic acid (pIC), coadministered with polyethyleneimine (PEI) as a carrier, was identified as an unanticipated inducer of autophagy downstream of an exacerbated endosomal maturation program. A concurrent activity of the dsRNA helicase MDA-5 driving the proapoptotic protein NOXA resulted in an efficient autodigestion of melanoma cells. These results reveal tractable links for therapeutic intervention among dsRNA helicases, endo/lysosomes and apoptotic factors.
Significance
Here we show that despite the multiple genetic and epigenetic alterations that melanoma cells accumulate, they still remain highly sensitive to dsRNA mimics, shown here for the synthetic molecule pIC. However, the delivery vehicle is critical. PEI, a polycation that favors endosomal uptake and cytosolic release, was able to shift the mode of action of pIC from a transient innocuous transcriptional program, to persistent cycles of fusion events involving a sequential recruitment of Rab7 (a small GTPase), LC3 (autophagosome marker) and lysosomes. A convergent mechanism of cellular stress was found driven by MDA-5 and involving an efficient NOXA-dependent caspase activation. Selective antitumor activity of [pIC]PEI in vivo, further supports cytosolic dsRNA sensors as viable targets for drug development in melanoma.
Introduction
Melanoma remains a prototype of solid cancers with increasing incidence and extremely poor prognosis at advanced stages (Jemal et al., 2008). Considerable effort has been devoted to the identification of molecular determinants underlying melanoma chemo- and immuno-resistance (Chin et al., 2006; Gray-Schopfer et al., 2007). Still, the average survival of patients with inoperable disseminated metastases is less than 10 months (Tawbi and Kirkwood, 2007). High throughput histogenetic and functional studies have revealed complex mechanisms associated with treatment failure (Fecher et al., 2008). These range from increased expression of drug pumps and detoxification enzymes, to a pleiotropic potentiation of key survival pathways (Fecher et al., 2008; Gray-Schopfer et al., 2007; Soengas and Lowe, 2003). In addition, apoptotic programs involving the mitochondria, the endoplasmic reticulum or death receptors are invariably ineffective in vivo (Hersey and Zhang, 2008; Soengas and Lowe, 2003). Consequently, current anticancer drugs either do not reach their target(s) in a productive manner, or have to be administered at dosing schedules that result in unbearable toxicities to normal cellular compartments (Tawbi and Kirkwood, 2007). Similarly, melanomas have an inherent ability to bypass or overcome anti-tumoral activities of immunomodulators (Ilkovitch and Lopez, 2008; Tormo et al., 2006; Verma et al., 2008)
Autophagy, and in particular macroautophagy, which involves the sequestration of bulk cytosolic components in autophagosomes for subsequent lysosomal degradation (Xie and Klionsky, 2007), is an understudied process in melanoma. The clinical relevance of macroautophagy -herein referred to as autophagy for simplicity- stems from its potential to protect cells against a variety of intracellular and extracellular stress signals and favor tumor development (Mathew et al., 2009; Mizushima et al., 2008). Paradoxically, autophagy has also been associated with cell death (Kroemer et al., 2009). Thus, excessive or persistent autophagy can promote cell killing by depletion of key organelles (e.g. endoplasmic reticulum or mitochondria), re-wiring of survival signals, deregulation of lysosomal enzymes and/or activation of caspase-dependent apoptotic programs (Eisenberg-Lerner et al., 2009; Hoyer-Hansen and Jaattela, 2008; Maiuri et al., 2007; Xie and Klionsky, 2007). Given these pro- and anti-apoptotic roles of autophagy, it is unclear whether this program could be a viable target for drug development (Kroemer and Levine, 2008; Rubinsztein et al., 2007; Scarlatti et al., 2009).
Autophagy genes can also have pleiotropic roles in the immune system (Virgin and Levine, 2009). Thus, autophagy can modulate antigen presentation, inhibit or potentiate interferon responses, and display critical functions in the clearance of intracellular viral and bacterial pathogens (Levine and Deretic, 2007; Sanjuan and Green, 2008). Typically, these responses are engaged to protect infected cells or the host (Virgin and Levine, 2009). The precise mechanisms underlying this immune-autophagy are not well defined. Membrane-bound pattern recognition receptors of the Toll-Like Receptor family (particularly TLR-3, TLR-4, and TLR-7) can favor pathogen sequestration in autophagosomes (Delgado and Deretic, 2009; Levine and Deretic, 2007). These TLRs have a restricted expression pattern, being enriched in cells of the immune system, such as macrophages and dendritic cells (Paulos et al., 2007; Wenzel et al., 2008). Whether melanoma cells have other sensors of viral pathogens that can be engaged to induce autophagy and cell death is unknown.
Here, we have assessed the interplay between autophagy and apoptosis in the context of tumor cell-selective elimination of melanoma cells.
Results
Identification of autophagosome inducers in melanoma cells
Melanoma cells stably expressing the autophagosome marker LC3 fused with GFP (Klionsky et al., 2008) were used to screen for autophagy inducers among commercially available chemotherapeutic drugs and immunomodulators. To improve intracellular delivery, cationic carriers, e.g., polyethyleneimine (PEI), were added to DNA or RNA-based agents (Bieber et al., 2002). The initial screen was performed with the SK-Mel-103 cell line. Subsequent validation studies were performed using a panel of nine human metastatic melanoma cell lines of diverse genetic background (see Supplementary Table 1), as well as the well-known B16 mouse melanoma cells. Primary skin melanocytes, keratinocytes and fibroblasts were included as controls for normal cells. Drug response was also tested in mouse embryonic fibroblasts (MEFs) expressing or deficient for the autophagy factor Atg5, to provide a genetically controlled model to assess classical autophagy programs (Salazar et al., 2009).
Multiple drugs were found to promote focal GFP-LC3 fluorescence emission without significantly affecting cell viability (A.C. and M.S.S. unpublished data). Among pro-death agents, the classical dsRNA mimic polyinosine-polycytidylic acid (pIC, Wenzel et al., 2000) complexed with PEI ([pIC]PEI), induced a potent accumulation of GFP-LC3 foci (Figure 1A). About 50% of cells treated with low doses (0.5-1 μg/ml) of [pIC]PEI showed GFP-LC3 staining within 2-4 h. These results were intriguing as pIC had been linked to autophagy in immune cells (Delgado et al., 2008), but not in the context of tumor cell death. Therefore, we focused on the identification of the cellular machinery that sensed and executed the response of melanoma cells to [pIC]PEI.
Figure 1. Induction of autophagy by [pIC]PEI results in melanoma cell death.

(A) Fluorescence imaging of eGFP-LC3 distribution in SK-Mel-103 melanoma cells treated for 6 h with 1 μg/ml [pIC]PEI. Cells treated with PEI as single agent are shown as reference controls.
(B) Changes in the electrophoretic mobility of the endogenous LC3 in SK-Mel-103 treated for the indicated times with pIC, [pIC]PEI or vehicle control (PEI).
(C) Visualization of eGFP-LC3 distribution in WT or Atg5-/- MEFs treated for 8 h with 25 nM Rapamycin (Rap) or 1 μg/ml pIC]PEI. pIC-treated cells are included as controls (upper two rows). Bottom panels show quantification of the percentage of cells with focal GFP-LC3 at the indicated times post treatment. Error bars correspond to SEM of three independent experiments.
(D) Differential response of SK-Mel-103 melanoma cells to rapamycin and [pIC]PEI determined by visualization of LC3-GFP fluorescence (upper two rows), and brigthfield microscopy imaging (bottow row). Cells treated with vehicle were included as controls.
(E) Immunoblots of total cell extracts isolated from SK-Mel-103 cells treated as indicated and probed with antibodies specific for phospho-S6 or tubulin.
(F) Transmission electron micrographs of melanoma cells treated with [pIC]PEI. Arrows point to conjugates of pIC and PEI being endocytosed into the treated cells. Note the large multivesicular structures at late time points after [pIC]PEI treatment (panel d).
(G) Murine B16 melanoma cells and the indicated human melanoma cell lines compared for sensitivity to PEI and pIC as single agents or in combination. Cell death was estimated by trypan blue exclusion 24 and 48h after treatment, and presented as means ± SEM of three independent experiments.
Consistent with autophagy, [pIC]PEI induced electrophoretic mobility changes of the endogenous LC3, which are characteristic of lipidation of this protein during autophagy (Figure 1B). [pIC]PEI also lead to LC3-foci formation in oncogenically transformed MEFs, an activity that required Atg5 (Figure 1C, upper panels). In fact, inhibition of LC3 redistribution in Atg5-/- cells treated with [pIC]PEI was as prominent as with rapamycin (Figure 1C), a classical autophagy inducer (Noda and Ohsumi, 1998). However, rapamycin and [pIC]PEI were not equivalent. LC3-foci were transient in melanoma treated with rapamycin, but sustained with [pIC]PEI (Figure 1D). In addition, rapamycin, but not [pIC]PEI inhibited the mTOR pathway as visualized by monitoring the expression of the phosphorylated S6 kinase (Figure 1E).
Electron microscopy (EM) provided independent evidence of autophagosome/autolysosome formation driven by [pIC]PEI. Thus, large mutivesicular structures (>500 nm diameter; Figure 1Fd) were found following uptake of [pIC]PEI nanoparticles in melanoma cells (Figure 1Fb). This uptake was likely facilitated by the known activity of PEI to coat genetic material and favor endocytosis (Boussif et al., 1995; Kopatz et al., 2004). Importantly, neither naked pIC nor PEI alone was able to induce autophagosome formation (see Figure S1 for representative examples). Together, these results suggest an mTOR-independent autophagy driven by pIC that requires an appropriate cellular delivery method.
Selective melanoma cell death following [pIC]PEI -driven autophagy
Notably, in all melanoma cell lines tested in this study, the early activation of autophagy by [pIC]PEI was invariably followed by cell death (Figure 1G and Table SI). In contrast, melanocytes retained their viability and did not display markers of autophagy (Figure S2A-C). Interestingly, although complexed efficiently with PEI, dsDNA, the immunogenic variant ds-BDNA (Ishii et al., 2006), or other dsRNA molecules such as pA:U had no obvious impact on melanoma physiology (Figure S2D and results not shown). These results are consistent with the known superior efficacy of pIC over other dsRNA mimic as an immunomodulator (Alexopoulou et al., 2001).
Inhibition of metastatic melanoma dissemination by [pIC]PEI in immunocompetent and immunosuppressed mice
A detailed characterization of the mechanism of action of [pIC]PEI would not be meaningful without significant activity in vivo. To this end, three tumor models were investigated. First, B16 melanoma cells were tested following subcutaneous or intravenous inoculation in immunocompetent mice. In a second setting, B16 or SK-Mel-103 cells expressing GFP (to allow for non-invasive imaging) were used in a surrogate model of lung metastasis in SCID-beige mice (Figure 2A-D). These animals lack functional B cells, T cells and NK cells (Croy and Chapeau, 1990), representing severely immunocompromised backgrounds, which can be found in melanoma patients. As summarized in Figures 2A-D and S3A-C, [pIC]PEI showed a considerably stronger anti-melanoma activity than naked pIC in all the models tested, even in the highly immunosuppressed SCID-beige mice where pIC had virtually no effect.
Figure 2. Anti-melanoma activity of [pIC]PEI in mice.
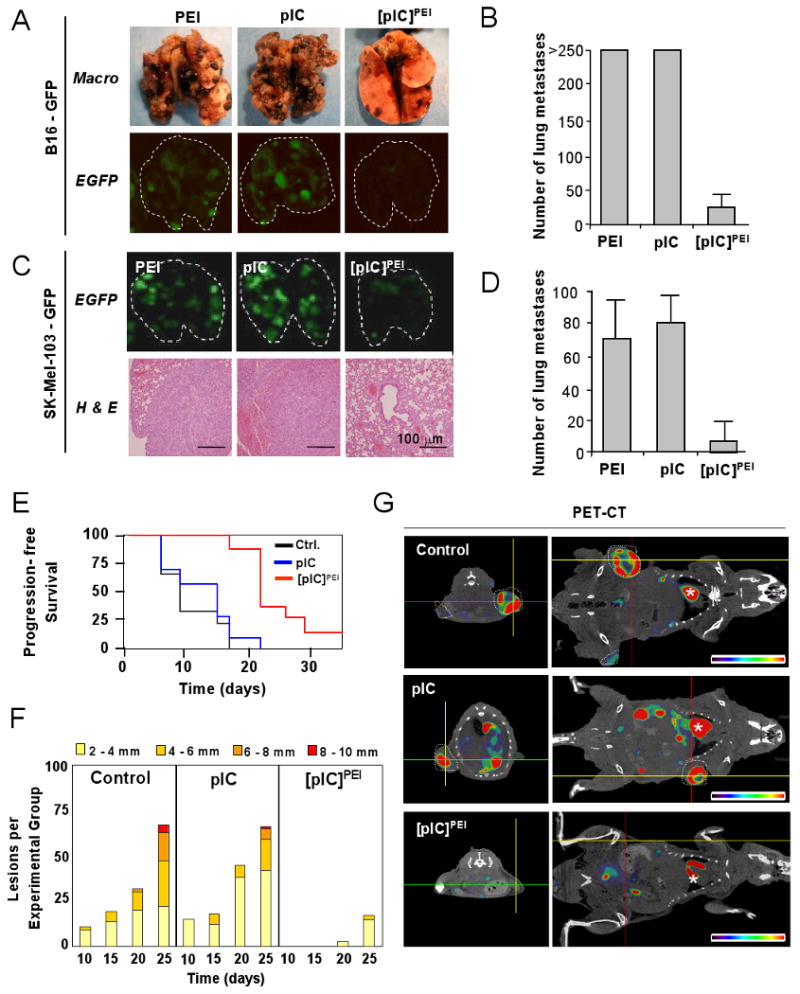
A-D are results from SCID Beige mice; E-G are results from Tyr::RasQ61K × Ink4a/Arf-/- mice.
(A) Representative images of lungs of mice 14 days after i.v. inoculation of B16 melanoma cells and treatment as indicated, and photographed under visible (Macro) or fluorescent (eGFP) light.
(B) Representation of the mean number of metastases (± SEM) induced by B16 as indicated in (A). P*<0.01 between PEI or pIC and [pIC]PEI treatment groups (n=5; generalized Mann–Whitney test).
(C) Lung colonization by eGFP-SK-Mel-103 in SCID-beige mice treated with the indicated agents and assessed by fluorescence imaging and histological analyses.
(D) Quantification of average number of external lung nodules (± SEM) in the indicated treatment groups shown in (C). P*<0.01 between PEI, pIC and [pIC]PEI treatment groups (n=5; generalized Mann–Whitney test).
(E) Cohorts of Tyr::RasQ61K × Ink4a/Arf-/- mice were treated topically with a single dose of 200 μg DMBA at 8 weeks of age. Upon appearance of pigmented lesions of >1 mm diameter, mice were treated as indicated. Control groups received PEI in 5% glucose. The fraction of animals with tumors of <1mm diameter (progression free survival) was represented by Kaplan-Meier graphs.
(F) Average number of cutaneous melanocytic neoplasms developing in each of the different treatment groups. Scoring was performed every five days and tumors were grouped by size as indicated.
(G) Representative transverse (left panels) and coronal sections (right panels) of PET /CT fused images to assess metabolic activity (18F-FDG incorporation) of representative examples of mice treated as indicated. Tumors are encircled with white dashed lines. The asterisks mark animal hearts.
The third system analyzed was the Tyr::NRASQ61K; INK4a/ARF-/- mice. These animals develop melanomas with key features similar to the human disease (Ackermann et al., 2005). Here, again, the antitumor activity of [pIC]PEI was significantly higher than that of pIC, as determined by quantification of tumor number, tumor size, metabolic activity, and histological analyses (Figures 2E-G and S3D). Of note, [pIC]PEI prolongs the progression free survival (Figure 2E) at dosing schedules without noticeable secondary toxicities (see below). Together, these results support an efficient and tumor cell-selective antitumor activity of [pIC]PEI.
Requirement of ATG-5 for [pIC]PEI-mediated cell death
Next, we investigated the molecular basis by which [pIC]PEI mediates self killing of melanoma cells. EM (Fig. 3A), real-time fluorescence analyses of GFP-LC3 distribution (Movies S1 and S2) and Nomarski interference contrast microscopy analyses (Figure 3A and Movie S3) indicated that melanoma cell death invariably followed the activation of classical autophagocytic vesicles (see Figure S4 for an example). To confirm that autophagy is a key effector of [pIC]PEI action (and not a byproduct of cellular stress), knockdown of key autophagy genes (Beclin1 and ATG7) was attempted in various melanoma cells. However, transient or stable knockdown of these genes induced either cellular senescence or cell death (A.C. and M.S.S. unpublished findings), as described in other systems (Hoyer-Hansen and Jaattela, 2007; Mathew et al., 2009; Miller et al., 2008). This is consistent with a basal rate of autophagy that can be visualized by monitoring LC3-GFP dynamics in untreated melanoma cells (see Movie S1). Therefore, to avoid confounding results of testing drug response in cells whose proliferative capacity and viability is already severely compromised, [pIC]PEI-driven cell death was determined in transformed wild type and Atg5-/- MEFs. Although oncogenically transformed wild type MEFs were less sensitive to [pIC]PEI-mediated death than melanoma cells, they were significantly more sensitive than their Atg5-/- counterparts (Figure 3B). However, it should be noted that Atg5-/- transformed MEFs died efficiently at high doses of [pIC]PEI (>1 μg/ml; not shown), perhaps reflecting the dual roles of autophagy in cell survival and cell death, as in immune cells (Virgin and Levine, 2009).
Figure 3. [pIC]PEI-triggered melanoma cell death is driven by autophagosome-autolysosome formation.

(A) Representative bright field (left and middle panels) and electron microscope (right panel) micrographs of SK-Mel-103 treated as indicated (30 h).
(B) Transformed wild-type or Atg5-/- MEFs treated with low dose (0.2 μg/ml) of [pIC]PEI or control. Cell death was estimated by trypan blue exclusion 48 h after treatment, and presented as means of three independent experiments.
(C) Confocal fluorescence images of SK-Mel-103 transduced by Cherry-GFP-LC3 to detect autophagosomes (red and green foci) and autolysosomes (red-only foci) formation after treatment with [pIC]PEI, 25 nM Rapamycin (Rap) or solvent control.
(D) Inhibitory effect of 100 μM Bafilomycin (Bafil), 20 μM Chloroquine (Chlor) or 10μg/ml Pepstatin (PEP) on cell death estimated by trypan blue exclusion 20h after treatment with vehicle (white bars) or [pIC]PEI (black bars). Data are indicated as means ± SEM of three independent experiments.
(E) Confocal fluorescence images of SK-Mel-103 cells stably transfected with eGFP-Rab5 WT and incubated with [pIC]PEI labeled with Fluor Red. Shown is the internalization of [pIC]PEI by the melanoma cells in the presence or absence of Chloroquine.
(F) Confocal visualization of lysosomal-dependent proteolysis upon cleavage and release of the fluorescent moiety of DQ-BSA (Green) in control or [pIC]PEI-treated SK-Mel-103. Chloroquine is included to monitor DQ-BSA emission in cells with blocked lysosomal activity. Cells were simultaneously imaged in the presence of lysotracker red (LTR-red) to visualize the lysosomal compartment.
(G) The DQ-BSA-lysotracker colocalization was estimated in a minimum of 150 cells in two independent experiments, and it is expressed (as arbitrary fluorescence units) with respect to control treated cells. Error bars correspond to ± SEM of three independent experiments.
Requirement of autophagosome-lysosome fusion in [pIC]PEI -mediated cell death
As indicated above, a defining ultrastructural feature of [pIC]PEI treatment is the induction of large multivesicular structures (Figure 1Fd). These vesicles could result from halted autophagosomes in which lysosomes are either not recruited or dysfunctional (Amaravadi et al., 2007; Ostenfeld et al., 2008). In this scenario, autophagosomes would grow in size as a consequence of accumulation of improperly degraded material. Alternatively, these vesicles could correspond to homotypic fusions among large endosomes, subsequently recruiting multiple autophagosomes (to generate hybrid structures also referred to as amphisomes). To assess these possibilities, melanoma cells were transduced with Cherry-GFP-LC3, which displays dual red-green fluorescence in autophagosomes but loses GFP signal in the acidic environment of autolysosomes (Tasdemir et al., 2008). We found that [pIC]PEI, similar to rapamycin, induced autolysosomes in melanoma cells (see “red-only” LC3 foci in Figure 3C). Supporting a lysosomal-dependent mode of action of [pIC]PEI, transient treatment with the lysosomotropic agent chloroquine protected melanoma cells against [pIC]PEI-driven cell death (Figure 3D), without affecting the endosomal uptake of this dsRNA mimic (as determined by colocalization of FluoRed labeled [pIC]PEI with GFP-fused early endosomal protein Rab5; Figure 3E). Similar inhibitory effects were observed using the broad spectrum protease inhibitors E64d and pepstatin A, and the vacuolar ATPase blocker bafilomycin (Figure 3D),
To independently monitor lysosomal activity during [pIC]PEI treatment, cells were assayed for the ability to process DQ-BSA (a derivative of BSA whose green fluorescence is quenched unless cleaved by proteolytic enzymes). As shown in Figures 3F and 3G, DQ-BSA was efficiently cleaved in the presence of [pIC]PEI (note that DQ-BSA emission was detected at the lysosomes, as indicated by colocalization with lysotracker-Red).
Dynamic endosome mobilization by [pIC]PEI
Having determined that autophagosomes fuse to active lysosomes in response to [pIC]PEI, we assessed whether these organelles interacted with or were recruited to endosomes. First, endosomal dynamics were assessed in melanoma cells expressing a GFP-fused to the late endosomal marker Rab7 (Luzio et al., 2007). Basal endosome generation and resolution (i.e., progressive reduction in size) was detected in untreated melanoma cells (Figure S5, right panels, Movie S4). [pIC]PEI treatment markedly enhanced endosomal activity, inducing a sustained and multi-wave generation of endosomes (Figure S5, left panels, and Movie S5). These endosomes were found to be filled with lysosomes, as determined by dual imaging of GFP-Rab7 and Lysotracker-Red (Figure 4A). Moreover, time-lapse microscopy revealed fast kinetics of multiple recruitments of lysosomes to GFP-Rab7 decorated endosomes (Movie S7; see also a sequential series of fusion events in Figure 4B). Importantly, as shown in Figure 4A (right panels), endosome-lysosome fusion was significantly inhibited if cells overexpressed Rab7-T22N, a known dominant negative mutant of this protein (Gutierrez et al., 2004). In total, these results uncovered mobilization of endo/lysosomal compartments in tumor cells treated with [pIC]PEI.
Figure 4. Generation and resolution of amphisomes upon [pIC]PEI treatment.

(A) SK-Mel-103 cells stably transfected with eGFP-Rab7 WT or eGFP-Rab7 T22N were incubated with [pIC]PEI for visualization of Rab7 (green fluorescence) and Lysotracker-Red. Microphotographs were captured by confocal microscopy 10 h after treatment with [pIC]PEI. Values in insets correspond to the average area contained in Rab7-decorated vesicles.
(B) Sequence of confocal microphotographs taken at the indicated time intervals (in seconds) to illustrate the fusion to and incorporation of lysosomes to Rab7-positive vesicles upon [pIC]PEI treatment.
(C) Real-time triple-imaging fluorescence microscopy of GFP-Rab7 WT, Cherry-LC3 and Lysotracker Blue (green, red and blue fluorescence, respectively) in SK-Mel-103 cells treated with [pIC]PEI. Images were taken at the indicated time intervals (in minutes), 1h after treatment initiation. Arrows mark the first sequence in which the indicated markers appear visible.
(D) Incorporation of LC3 to the surface of Rab7 endosomal vesicles prior to internalization and subsequent degradation. These endosome/LC3 hybrid structures (amphisomes) were visualized by fluorescence real-time microscopy of SK-Mel-103 cells expressing eGFP-Rab7 WT and Cherry-LC3.
Generation and resolution of [pIC]PEI-driven amphisome formation
To determine whether and when autophagosomes are recruited to endosomes melanoma cells were imaged simultaneously for GFP-Rab7, Cherry-LC3 and Lysotracker -Blue at different time points after [pIC]PEI addition. As shown in Figures 4C and S6A, the first event detected was generally the incorporation of Rab7 to the membrane of preexisting “empty” (early endosome) vesicles. LC3 was recruited afterwards usually starting from an initial nucleation event, to ultimately decorate the entire Rab7-positive vesicles. Lysotracker-Blue incorporation into these endosome/autophagosome hybrids (amphisomes) was found to be the last to occur. At this point, the corresponding vesicle progressively diminished in size to ultimately become undetectable, indicating a bona fide autodegradative process (Figures 4C and S6; see arrows marking Rab7>LC3>Lysotracker sequence). The presence of lysotracker did not interfere with endosomal dynamics, as similar progressive recruitment and internalization of LC3 to Rab7 endosomes was found in its absence (Figure 4D).
[pIC]PEI links autophagy to apoptotic caspases
Notably, the sequential fusion events mentioned above for a single vesicle were found to involve large numbers of endosomes and occur in a persistent manner throughout the entire cytosol of treated cells (Figure S6). This was independent of the imaging reagents employed (see examples of cells imaged with Lysotracker-Blue in S6A or Lysotracker-Red in Figure S6B). Still, we found a considerable delay from the starting point of endosome mobilization (2-5 h), until final cellular collapse (24-48 h). Thus, we hypothesized that cell demise required an additional, later events. Interestingly, cleaved regulatory and effector caspases (casp-8, 9, 7 and 3) were detected 15-20h after [pIC]PEI treatment (Figure S7A). Accordingly, caspase inhibitors (zVAD-fmk, and to a lesser extent DEVD-CHO) reduced [pIC]PEI-driven cell death (Figure S7B). Similarly, the sensitivity of MEFs to [pIC]PEI was significantly reduced in cells deficient for the apoptotic factors Bax and Bak (Figure S7C, left panels). Importantly, neither caspase inactivation in melanoma cells nor BAX/BAK deficiency in MEFs prevented the generation of GFP-LC3 foci (Figure S7B,C). Therefore, [pIC]PEI is able to induce an early but persistent autophagy and a late apoptotic program. This is in contrast to other systems where caspases are the initiating death signals and autophagy is induced to favor the clearance of partially degraded cytosolic components (Kroemer and Levine, 2008a).
Differential gene expression induced by naked and PEI-complexed pIC in melanoma cells
Next, we investigated the molecular sensor(s) of [pIC]PEI that might link autophagy to apoptosis in a tumor cell-selective manner. Using cDNA arrays, melanocytes were found to undergo minimal changes in gene expression at early or late time points after treatment with pIC or [pIC]PEI, In contrast, melanoma cells responded to both agents with substantial alterations of their transcriptome (Figure S8A). Interestingly, while pIC induced expected changes in interferon response genes, this response was largely transient. On the other hand, the effect of [pIC]PEI was sustained and extended to additional transcripts (Figure S8A,B). These results reveal key idiosyncratic differences between melanocytes and melanoma cells regarding the uptake and signaling to dsRNA mimics.
Qualitatively different activation of MDA-5 by pIC in the absence and presence of PEI
The best known innate programs that control the sensing of long dsRNA are (i) the Toll-Like Receptor 3 (TLR3), (ii) the Melanoma Differentiation-Associated gene-5 (MDA-5)/Interferon Induced with Helicase C domain 1 protein (IFIH1), and (iii) the dsRNA-activated protein kinase (PKR, also known as eIF2AK2) (Takeuchi and Akira, 2008). Of these genes, MDA-5 showed the strongest and most sustained induction by [pIC]PEI (Figure 5A). Moreover, knockdown of TLR3 with independent siRNA or shRNA had no impact on [pIC]PEI-driven cell death (Figure 5B). Therefore, we focused on MDA-5 for further analyses. Protein immunoblotting demonstrated a sustained activation of full length MDA-5 by [pIC]PEI (Figure 5C). In addition, an apoptosis-associated processing of MDA-5 (Barral et al., 2007; Kovacsovics et al., 2002) was found preferentially (and in a more sustained manner) in [pIC]PEI-treated cells (Figure 5C). Of note, this induction and processing of MDA-5 was not simply a consequence of activated apoptotic programs. Treatment with bortezomib, a proteasome inhibitor which engages intrinsic apoptotic pathways in melanoma (Fernandez et al., 2005; Wolter et al., 2007), had no effect on MDA-5 levels or processing (Figure 5C).
Figure 5. MDA-5 is a sensor and driver of [pIC]PEI cytotoxicity in melanoma cells.

(A) Fold-induction of the transcript levels of the indicated pathogen-associated receptors relative to non-treated cells. The IFIT-1 gene was used as reference for a classical IFN-responsive factor.
(B) Inhibition of TLR-3 expression by transient transduction of siRNA (left panels) or stable infection with lentivirus coding for shTLR3 (right panels). Cell death was assessed by trypan blue exclusion after treatment with vehicle (white bars) or [pIC]PEI (black bars). Insets correspond to mRNA levels detected by RT-PCR. Data are shown as means ± SEM of three independent experiments.
(C) MDA-5 induction visualized by immunoblotting in SK-Mel-28 and SK-Mel-147 treated with PEI, pIC or [pIC]PEI. Bortezomib was used as a control for an effective death inducer. Full length and cleaved MDA-5 (MDA-5FL and MDA-5C, respectively) are indicated with arrows. Asterisk corresponds to a non-specific band (not blocked by MDA-5 shRNA; not shown).
(D) MDA-5 mRNA levels (left panel) and cell death (right panels) in SK-Mel-103 infected with the pLKO lentivirus expressing scrambled or MDA-5 shRNAs, and treated as indicated. Data are shown as means ± SEM of three independent experiments. P<0.05 between control and shMDA-5 cells in the presence of [pIC]PEI (two-tailed Student's t-test)
(E) Inhibition of MDA-5 expression by transient transduction of a second set of control and MDA-5 siRNAs driven by the psi-vector frame (see Materials and Methods). Cell death in control and [pIC]PEI-treated SK-Mel-103 (24h) were determined by trypan blue exclusion, and plotted as means ± SEM of three independent experiments. In the [pIC]PEI treatment group, P<0.05 between control shRNA and shMDA-5 transduced cells (two-tailed Student's t-test).
(F) Confocal visualization of GFP-LC3 in MeWo melanoma cells infected with Adenovirus-control or Adenovirus-MDA-5. [pIC]PEI was included as a reference for autophagosome formation. Numbers represent fraction of cells with eGFP-LC3 foci.
(G) Cell death resulting from adenovirus-driven expression of MDA-5 in MeWo cells. The viral concentration (in plate forming units, pfu) is indicated. Data correspond to means ± SEM of three independent experiments.
Downregulation of MDA-5 expression by siRNA (Figure 5D) or shRNA (Figure 5E) reduced [pIC]PEI-driven cell death by 50-60%, providing evidence for a critical role of MDA-5 as sensor and effector of this dsRNA mimic. Interestingly, although MDA-5 needs interaction with dsRNA for full activation (Takeuchi and Akira, 2008), ectopic expression of this protein is sufficient to promote cell death in a subset of melanoma cells (e.g., MeWo; (Kang et al., 2004; Kang et al., 2002)). In these cells, MDA-5 was also found to engage focal aggregation of LC3 (Figure 5F) prior to cell killing (Figure 5G and results not shown). These results uncover a pro-autophagy role of MDA-5. However, although MDA-5 may favor autophagy, it is unlikely that this is the sole contribution of this protein to [pIC]PEI-mediated cell death. Thus, MDA5-shRNA or Mda-5-/- MEFs could still engage autophagosome formation after sustained treatment with [pIC]PEI (not shown).
MDA-5 driven NOXA-dependent cell death
A main known role of endogenous MDA-5 is to elicit IFN-responses to blunt viral infection (Takeuchi and Akira, 2008). IFN-α was secreted in [pIC]PEI-treated melanoma cells (results not shown). However, IFN-α was a poor inducer of melanoma cell death, even at doses 100 times higher than those secreted by [pIC]PEI -treated cells (Figure S8C).
cDNA microarray data generated above was analyzed to identify the mediators of [pIC]PEI/MDA5-driven melanoma cell death. Among known pro-apoptotic factors, a strong and sustained upregulation (up to 16 fold) of NOXA mRNA and protein levels was found (Figure 6A-D). Moreover, [pIC]PEI-mediated death induction was reduced by 50-60% when the NOXA level was reduced using a shRNA (Figure 6E, F). Importantly, [pIC]PEI-driven NOXA induction was significantly abrogated by MDA-5 shRNA (Figure 6G). Therefore, these results uncover functional links between dsRNA sensors and the apoptosis program via NOXA.
Figure 6. [pIC]PEI engages apoptosis via NOXA independently of the status of p53 or without inducing compensatory MCL-1 activation.
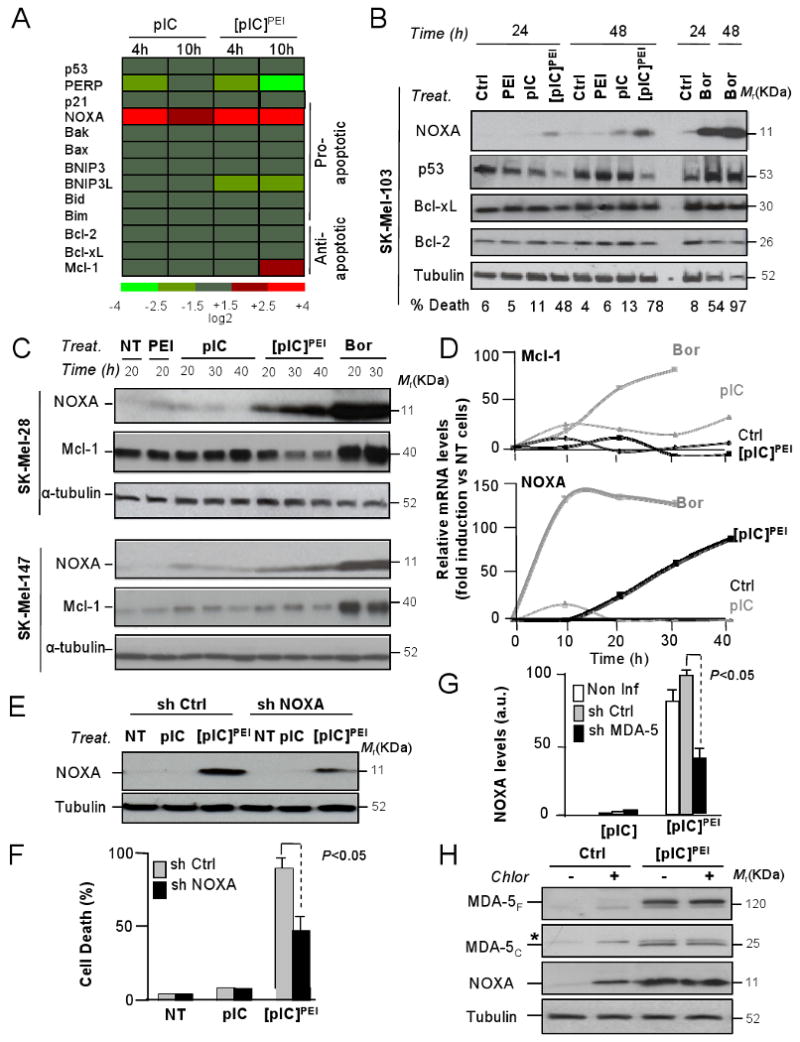
(A) Heat map of log2-ratio values of the indicated genes after pIC or [pIC]PEI treatment of SK-Mel-103, and calculated with respect to control (PEI)-incubated cells.
(B) Immunoblots of total cell extracts isolated from SK-Mel-103 treated for 24 and 48h with pIC, [pIC]PEI or Bortezomib for the relative levels of NOXA, p53 and anti-apoptotic Bcl-2 and Bcl-xL proteins.
(C) SK-Mel-28 and SK-Mel-147 (expressing p53 L145A and p53 wild type, respectively) were treated with PEI, pIC, [pIC]PEI or 25 nM Bortezomib. Shown are the relative levels of NOXA and MCL-1 protein at different time points after treatment.
(D) Quantification of the levels of Mcl-1 and NOXA in SK-Mel-28 after each treatment and represented with respect to control untreated cells.
(E) NOXA protein expression in melanoma SK-Mel-103 treated for 24 h with pIC or [pIC]PEI two days after infection with a lentiviral vector expressing inactive shRNA (sh Ctrl) or shRNA for NOXA.
(F) Death rates (± SEM) of control and NOXA shRNA transduced SK-Mel-103 melanoma cells and incubated with pIC or [pIC]PEI for 24 h.
(G) Inhibitory effect of MDA-5 downregulation on NOXA induction by [pIC]PEI determined in SK-Mel transduced with control or MDA5 shRNAs. Data is represented as means ± SEM of four independent experiments. Levels of NOXA were quantified by densitometry of the corresponding immunoblots and plotted with respect to untreated controls.
(H) Immunoblots of total cell extracts isolated from SK-Mel-103 treated for 24h with vehicle or [pIC]PEI in the presence or absence of Chloroquine, and probed for the detection of MDA-5 (full length and cleaved forms) and NOXA.
Other chemotherapeutic agents such as the proteasome inhibitor bortezomib can also upregulate NOXA and kill melanoma cells deficient for Apaf-1 or other apoptotic modulators (Fernandez et al., 2005; Qin et al., 2005). However, a main disadvantage of bortezomib is that concomitantly induces various antiapoptotic factors, including MCL-1, a NOXA antagonist (Qin et al., 2006; Wolter et al., 2007). In contrast, no compensatory accumulation of MCL-1 protein was found in the response to [pIC]PEI (Figure 6C,D).
Importantly, the induction of MDA-5 and NOXA by [pIC]PEI was found to occur even when lysosome activity was blocked by chloroquine or by cathepsin inhibitors (Figures 6H and S7D and results not shown). Thus, NOXA can be activated independently of the autophagy program. From a therapeutic perspective, it was interesting to note that cDNA arrays failed to identify changes in NOXA expression in melanocytes (not shown). Similarly, MDA-5 and NOXA were not upregulated in skin fibroblasts (Figure S8D), which also showed increased resistance to [pIC]PEI in culture (Figure S8E) and no obvious signs of toxicity in biopsies from treated animals (Figure S8F; note also lack of toxicity to other organs).
[pIC]PEI-mediated therapeutic effects in vivo: MDA-5 requirement, LC3 foci formation and NOXA induction
Next, we investigated the dependency on MDA-5 and the dual activation of autophagosome and apoptotic markers identified above by [pIC]PEI in vivo. Available antibodies against MDA-5 cannot distinguish full length from processed forms of this protein. In the absence of current Mda-5-deficient mouse melanoma models, we decided to take advantage of homozygous knockout MEFs for the Mda-5 gene. As shown in Figure 7A, transformed Mda-5-/- MEFs were significantly more resistant to [pIC]PEI than their wild type counterparts, both in tissue culture systems, and when grown as xenografts in mice.
Figure 7. Defining the mode of action of [pIC]PEI in vivo.

(A) Response of oncogenically transformed wild type (+/+) or Mda-5-/- (-/-) MEFs, measured as time-dependent changes in tumor size (± SEM) upon treatment with naked pIC (V) or [pIC]PEI. Shown are P values for comparative analyses between the indicated treatment groups (n=10, generalized Mann-Witney test). Inset bar graphs corresponds to death rates (± SEM; n=3).of the indicated cell populations 24 h after treatment in culture with 1 μg/ml [pIC]PEI.
(B) SK-Mel-103 GFP-LC3 melanoma cells implanted s.c. into SCID Beige mice were left to grow to 6-8 mm diameter and then treated with PEI or [pIC]PEI. Paraffin-embedded sections were stained for LC3 or NOXA antibodies. For LC3, high magnification photographs are also included to show the formation of foci characteristic of autophagosomes.
(C) Model summarizing main results of this study. A transient activation of IFN and other antiviral stress response factors by naked pIC (1) can be shifted into a sustained stress program when this dsRNA mimic is delivered to cells with PEI (2). PEI packs pIC into nanoparticles and favors both its endosomal uptake and delivery to the cytosol for subsequent activation of the helicase MDA-5 (3). In addition to favor endosomal swelling, PEI has been described to favor endosome-endosome fusion. Within 2-3h after incubation with [pIC]PEI melanoma cells undergo massive ultrastructural changes in the endosomal compartment (4). These changes involve a sustained and cyclic Rab7>LC3>Lysotraker sequence of fusion events, revealing an active generation and resolution of endosome-autophagosome-lysosome hybrids (5). Although MDA-5 can facilitate autophagosome formation, a main function of this protein described here is the induction of the pro-apoptotic factor NOXA (6). NOXA is per se a poor apoptotic inducer, but can lower the threshold for caspase processing. Sustained lysosomal-dependent degradative process together with the activation of apoptotic caspases can ultimately converge in efficient tumor cell death (7). Importantly, we demonstrate a potent anti-melanoma activity of [pIC]PEI in various animal models, at concentrations with no obvious toxicity to normal cellular compartments.
Selective antibodies for LC3 and NOXA were used to visualize autophagosome formation and activation of NOXA by [pIC]PEI in human melanoma xenografts. Although sporadic stainings for LC3 or NOXA were detected in vehicle-treated SK-Mel-103 xenografts, the total number of positive cells and the corresponding signal intensity of NOXA or LC3 per individual cell were noticeably stronger in the [pIC]PEI-treated specimens (Figure 7B). The animal model used (SCID beige) is highly immunodeficient. Therefore, these results serve as the proof-of-principle for the ability of [pIC]PEI to bypass the requirement for immune effectors and drive tumor cell death by a coordinated autophagy/apoptosis mechanism.
Discussion
Melanomas accumulate a plethora of genetic and epigenetic alterations that contribute to the limited efficacy of current anticancer treatments (Chin et al., 2006; Soengas and Lowe, 2003). However, here we have shown that melanoma cells retain the ability to sense and respond to mimetics of viral dsRNA, and that this feature can be exploited therapeutically. Specifically, our results revealed tractable points of crosstalk between innate sensors of dsRNA, endo/lysosomal compartments and tumor cell death.
Given the ability of melanoma cells to deactivate death programs (Gray-Schopfer et al., 2007), it is intriguing that they remain sensitive to lysosomal-dependent autodegradative mechanisms. This is particularly relevant because autophagy has been abundantly linked to cytoprotection in innate and acquired immune responses (Levine and Deretic, 2007; Sanjuan and Green, 2008; Virgin and Levine, 2009). Activation of autophagosomes by TLR3 has also been reported for naked dsRNA in macrophages, but in this case, for protection of the host at both the cellular and organismal levels (Delgado et al., 2008). In the melanoma cells studied here, however, TLR3 was found dispensable for [pIC]PEI-driven autophagy or apoptotic induction, perhaps reflecting inherent differences in TLR3 expression between professional immune cells and melanomas.
An intriguing feature of the response of melanoma cells to [pIC]PEI was a considerable time lag from the detection of the first series of endosome-autophagosome-lysosome fusions (within an hour of treatment) to the final cellular collapse (24-48 h). It is therefore conceivable that autophagy is engaged in response to [pIC]PEI as an initial mechanism of protection, which is later shifted into a pro-death program (see model in Figure 7C). Thus, autophagy could be engaged to resolve an exacerbated endocytosis driven by pIC complexed to PEI (Boussif et al., 1995; Kopatz et al., 2004). Endocytic recruitment of autophagosomes could initially help tumor cells to maintain a “clean” cytosol. Intra-endocytic degradation would prevent leakage or the accumulation of partially degraded autophagocytic structures, and/or damaged lysosomes, which could be potentially lethal to cells (Kroemer and Jaattela, 2005). However, the physical concentration of autophagosomes and lysosomes within endocytic vesicles may also provide a point of vulnerability of melanoma cells. Recurrent cycles of endo/lysosome generation-degradation could lower the threshold for the activation of death programs (i.e. by depleting ATP and/or key proteins or organelles required for cell maintenance) as described in other systems (Eisenberg-Lerner et al., 2009). In fact, a main difference between [pIC]PEI (which kills melanoma cells) and rapamycin (innocuous to melanoma cells), is the sustainability of the endolysosome-autophagosome fusion events. An additional defining feature of [pIC]PEI not shared by rapamycin or by other chemotherapeutic agents, is the ability to engage a potent wave of pro-apoptotic events driven, at least in part, by the MDA-5 dsRNA sensor (see Figure 7C).
MDA-5, a helicase now considered as a first line of defense against viral dsRNA (Kato et al., 2006), was first described as a melanoma differentiation associated gene (Kang et al., 2002). It was noted that MDA-5 had pro-apoptotic activities when expressed ectopically at high levels (Kang et al., 2004; Kovacsovics et al., 2002). However, even forced expression of MDA-5 was rather inefficient in tumor cells with hyperactivated RAS/MEK/ERK pathway (Lin et al., 2006), as is the case of melanomas (Gray-Schopfer et al., 2007). Therefore, it was unclear how to drive a sustained activation of the endogenous MDA-5 protein. Moreover, the identity of death-inducing targets of MDA-5 remains unclear (Takeuchi and Akira, 2008). We showed that endogeous MDA-5 can in fact be induced to kill melanoma cells (with pIC delivered appropriately to the cytosol). Further, we uncovered roles of MDA-5 beyond the activation of IFN-driven immune responses. Specifically, we showed the ability of MDA-5 to drive autophagosome formation and NOXA activation. Still, our data also suggest additional death inducers activated by [pIC]PEI are likely to act in parallel or in concert with MDA-5 and NOXA (i.e., depletion of these proteins do not completely abrogate the antitumoral activity of [pIC]PEI). In this context, the cDNA arrays performed here indicate that [pIC]PEI can selectively induce a variety of stress kinases with pro-autophagic and pro apoptotic activity (D.T. and M.S.S. unpublished results).
From a therapeutic perspective, perhaps one the most unexpected findings in this study is the cell-autonomous anti-tumoral activity of [pIC]PEI. pIC has been used for more than 4 decades as a synthetic dsRNA mimic to boost the immune system in an IFN-dependent manner (Field et al., 1967). Unfortunately, clinical trials with naked pIC showed poor pIC stability and IFN induction, and no detectable antitumor effect in melanoma (Robinson et al., 1976). Complexes with low molecular weight poly-L-lysine, carboxymethylcellulose, liposomes or PEI have been reported to favor pIC's therapeutic activity via IFN-dependent immune responses (Fujimura et al., 2006; Levine et al., 1979). Our results show that PEI significantly enhances the ability of pIC to induce melanoma cells to express classical IFN-α targets. However, we also show that IFN, per se, does not recapitulate the anti-melanoma activity of [pIC]PEI in cultured cells. Moreover, [pIC]PEI can display an efficient anti-melanoma activity in animals with defective NK, T or B cell signaling, a condition common of melanoma patients (Kirkwood et al., 2008; Wenzel et al., 2008). In addition to the xenografts in immunosuppressed and immunodeficient mice, our results in two additional in vivo models (MDA-5 deficient cells and autochthonous cutaneous melanomas generated in the Tyr::NRASQ61K; INK4a/ARF-/- mice), further emphasized the physiological relevance of our data. Altogether, our results provide the proof of principle for dsRNA sensors as therapeutic targets to overcome the inherent resistance of melanoma cells to current anticancer treatments.
Experimental Procedures
Treatment, imaging, and expression analyses in cultured cells
The human and mouse metastatic melanoma cell lines used in this study are described in the Supplementary Information. Normal human melanocytes and fibroblasts were isolated from anonymous discarded foreskins as previously reported (Wolter et al., 2007), using protocols approved by the University of Michigan and the Instituto Carlos III-CNIO ethics committees. Wild type and Atg 5 or Bax/Bak deficient mouse embryonic fibroblasts have been described before (Kuma et al., 2004; Lindsten et al., 2000). Oncogenically transformed derivatives of these cells, expressing HRASG12V and the SV40 large T antigen were a kind gift of G. Velasco (Universidad Complutense, Madrid). Information on PEI-complexed pIC, pharmacological inhibitors of apoptosis and autophagy, and other reagents and techniques used here to assess cell death and cell viability are described in Supplementary Information. Details on transmission electron microscopy, confocal and fluorescence microscopy, protein immunoblotting, lentiviral mediated RNA interference and cDNA microarray expression profiling are also described in the Supplementary Information. Full datasets corresponding to the cDNA arrays reported in this study in melanocytes and SK-Mel-103 cells were deposited to the Gene Expression Omnibus database (with accession numbers GSE16337 and GSE14445, respectively).
Treatment response in vivo
All experiments in mouse models were performed in accordance with institutionally approved protocols by the University of Michigan Unit for Laboratory Animal Medicine (ULAM) Committee, and the Instituto Carlos III-CNIO ethics committee. Generation and treatment of autochthonous melanomas in the Tyr:: N-RasQ61K/°; Ink4a/Arf-/- mice and the analysis of melanoma xenografts in C57BL/6 and in the immunosuppressed SCID beige mice are summarized in Supplementary Materials.
Histopathological and Immunohistochemical Analyses
Tumors or tissue sections from the indicated treatment groups were fixed in 10% buffered formalin, embedded in paraffin, and routinely stained with hematoxylin and eosin. Alternatively, tumors were immunostained with the NOXA antibody (Calbiochem, San Diego, CA, USA) and LC3 antibody (Cell Signaling, Danvers, MA, USA). Slides were digitalized using Dotslide (Olimpus, Watford, UK).
Statistical analyses
Viability data are expressed as means +/- s.e.m, and statistical analysis of the differences was determined by the two-tailed Student's t-test. P < 0.05 was considered significant. For statistical evaluation of tumor growth and metastasis in vivo, the generalized Mann-Whitney test was used to compare the values of continuous variables between two groups. P values of <0.05 were considered significant.
Supplementary Material
Acknowledgments
This study was initiated at Univ. of Michigan Department of Dermatology (USA), and the authors would like to thank all our colleagues there, particularly, Monique Verhaegen, MaryBeth Ribblet, Keith Wolter and Anj Dlugosz for their help and support. We also thank José Esteban (Center for Molecular Biology, Spain) for critical reading of this manuscript, Gabriel Núñez (Univ. Michigan, USA), Marino Zerial (Max Planck Institute, Germany), and Terje Johansen (Univ of Tromso, Norway) for eGFP-LC3, eGFP-Rab7 (WT and T22N) and Cherry-GFP-LC3, respectively. We also thank Friedrich Beermann (ISREC, Switzerland) for the Tyr::NRASQ61K mice, and Guillermo Velasco (Univ Complutense, Spain) and Patricia Boya (CIB, Spain) for Bax/Bak and Atg5 deficient MEFs. This work was supported by grants NIH R01 CA107237 and Spanish Ministry of Science and Innovation SAF2008-1950 (M.S.S); R01 GM068448 (P.B.F); Tu90-6/1 and DKH 10741 (T.T), and institutional grants from the Spanish Association Against Cancer and Spanish National Cancer Research Centre (M.S.S). P.B.F. holds the Thelma Newmeyer Corman Chair in Cancer Research at the VCU Massey Cancer Center. D.T. and D.A-C are recipients of a Juan de la Cierva Postdoctoral Fellowship and a Scientists in Training Predoctoral Fellowship, respectively, from the Spanish Ministry of Science and Innovation. E. R-F is the recipient of a post-residency training program from “Obra Social de Caja Navarra”.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005;65:4005–4011. doi: 10.1158/0008-5472.CAN-04-2970. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, Racaniello VR. MDA-5 is cleaved in poliovirus-infected cells. J Virol. 2007;81:3677–3684. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieber T, Meissner W, Kostin S, Niemann A, Elsasser HP. Intracellular route and transcriptional competence of polyethylenimine-DNA complexes. J Control Release. 2002;82:441–454. doi: 10.1016/s0168-3659(02)00129-3. [DOI] [PubMed] [Google Scholar]
- Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–2182. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- Croy BA, Chapeau C. Evaluation of the pregnancy immunotrophism hypothesis by assessment of the reproductive performance of young adult mice of genotype scid/scid.bg/bg. J Reprod Fertil. 1990;88:231–239. doi: 10.1530/jrf.0.0880231. [DOI] [PubMed] [Google Scholar]
- Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. Embo J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- Fecher LA, Amaravadi RK, Flaherty KT. The MAPK pathway in melanoma. Curr Opin Oncol. 2008;20:183–189. doi: 10.1097/CCO.0b013e3282f5271c. [DOI] [PubMed] [Google Scholar]
- Fernandez Y, Verhaegen M, Miller TP, Rush JL, Steiner P, Opipari AW, Jr, Lowe SW, Soengas MS. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 2005;65:6294–6304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- Field AK, Tytell AA, Lampson GP, Hilleman MR. Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc Natl Acad Sci U S A. 1967;58:1004–1010. doi: 10.1073/pnas.58.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura T, Nakagawa S, Ohtani T, Ito Y, Aiba S. Inhibitory effect of the polyinosinic-polycytidylic acid/cationic liposome on the progression of murine B16F10 melanoma. Eur J Immunol. 2006;36:3371–3380. doi: 10.1002/eji.200636053. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- Hersey P, Zhang XD. Adaptation to ER stress as a driver of malignancy and resistance to therapy in human melanoma. Pigment Cell Melanoma Res. 2008;21:358–367. doi: 10.1111/j.1755-148X.2008.00467.x. [DOI] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576–1582. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Jaattela M. Autophagy: an emerging target for cancer therapy. Autophagy. 2008;4:574–580. doi: 10.4161/auto.5921. [DOI] [PubMed] [Google Scholar]
- Ilkovitch D, Lopez DM. Immune modulation by melanoma-derived factors. Exp Dermatol. 2008 doi: 10.1111/j.1600-0625.2008.00779.x. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Lin L, Randolph A, Valerie K, Pestka S, Fisher PB. Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: a novel type I interferon-responsive apoptosis-inducing gene. Oncogene. 2004;23:1789–1800. doi: 10.1038/sj.onc.1207300. [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002;99:637–642. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kirkwood JM, Tarhini AA, Panelli MC, Moschos SJ, Zarour HM, Butterfield LH, Gogas HJ. Next generation of immunotherapy for melanoma. J Clin Oncol. 2008;26:3445–3455. doi: 10.1200/JCO.2007.14.6423. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopatz I, Remy JS, Behr JP. A model for non-viral gene delivery: through syndecan adhesion molecules and powered by actin. J Gene Med. 2004;6:769–776. doi: 10.1002/jgm.558. [DOI] [PubMed] [Google Scholar]
- Kovacsovics M, Martinon F, Micheau O, Bodmer JL, Hofmann K, Tschopp J. Overexpression of Helicard, a CARD-containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr Biol. 2002;12:838–843. doi: 10.1016/s0960-9822(02)00842-4. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008 doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Levine AS, Sivulich M, Wiernik PH, Levy HB. Initial clinical trials in cancer patients of polyriboinosinic-polyribocytidylic acid stabilized with poly-L-lysine, in carboxymethylcellulose [poly(ICLC)], a highly effective interferon inducer. Cancer Res. 1979;39:1645–1650. [PubMed] [Google Scholar]
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Su Z, Lebedeva IV, Gupta P, Boukerche H, Rai T, Barber GN, Dent P, Sarkar D, Fisher PB. Activation of Ras/Raf protects cells from melanoma differentiation-associated gene-5-induced apoptosis. Cell Death Differ. 2006;13:1982–1993. doi: 10.1038/sj.cdd.4401899. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, Mizushima NN, Iwasaki A, He YW, Swat W, Virgin HWt. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostenfeld MS, Hoyer-Hansen M, Bastholm L, Fehrenbacher N, Olsen OD, Groth-Pedersen L, Puustinen P, Kirkegaard-Sorensen T, Nylandsted J, Farkas T, Jaattela M. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy. 2008;4:487–499. doi: 10.4161/auto.5774. [DOI] [PubMed] [Google Scholar]
- Paulos CM, Kaiser A, Wrzesinski C, Hinrichs CS, Cassard L, Boni A, Muranski P, Sanchez-Perez L, Palmer DC, Yu Z, et al. Toll-like receptors in tumor immunotherapy. Clin Cancer Res. 2007;13:5280–5289. doi: 10.1158/1078-0432.CCR-07-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin JZ, Xin H, Sitailo LA, Denning MF, Nickoloff BJ. Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Res. 2006;66:9636–9645. doi: 10.1158/0008-5472.CAN-06-0747. [DOI] [PubMed] [Google Scholar]
- Robinson RA, DeVita VT, Levy HB, Baron S, Hubbard SP, Levine AS. A phase I-II trial of multiple-dose polyriboinosic-polyribocytidylic acid in patieonts with leukemia or solid tumors. J Natl Cancer Inst. 1976;57:599–602. doi: 10.1093/jnci/57.3.599. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M, Egia A, Vazquez P, Blazquez C, Torres S, Garcia S, et al. Cannabinoid action induces autophagymediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359–1372. doi: 10.1172/JCI37948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan MA, Green DR. Eating for good health: linking autophagy and phagocytosis in host defense. Autophagy. 2008;4:607–611. doi: 10.4161/auto.6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009;16:12–20. doi: 10.1038/cdd.2008.101. [DOI] [PubMed] [Google Scholar]
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–3151. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, Modjtahedi N, Kroemer G. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008;445:29–76. doi: 10.1007/978-1-59745-157-4_3. [DOI] [PubMed] [Google Scholar]
- Tawbi HA, Kirkwood JM. Management of metastatic melanoma. Semin Oncol. 2007;34:532–545. doi: 10.1053/j.seminoncol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Tormo D, Ferrer A, Bosch P, Gaffal E, Basner-Tschakarjan E, Wenzel J, Tuting T. Therapeutic efficacy of antigen-specific vaccination and toll-like receptor stimulation against established transplanted and autochthonous melanoma in mice. Cancer Res. 2006;66:5427–5435. doi: 10.1158/0008-5472.CAN-06-0399. [DOI] [PubMed] [Google Scholar]
- Verma S, Petrella T, Hamm C, Bak K, Charette M. Biochemotherapy for the treatment of metastatic malignant melanoma: a clinical practice guideline. Curr Oncol. 2008;15:85–89. [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J, Tormo D, Tuting T. Toll-like receptor-agonists in the treatment of skin cancer: history, current developments and future prospects. Handb Exp Pharmacol. 2008:201–220. doi: 10.1007/978-3-540-72167-3_10. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Verhaegen M, Fernandez Y, Nikolovska-Coleska Z, Riblett M, de la Vega CM, Wang S, Soengas MS. Therapeutic window for melanoma treatment provided by selective effects of the proteasome on Bcl-2 proteins. Cell Death Differ. 2007;14:1605–1616. doi: 10.1038/sj.cdd.4402163. [DOI] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.