

Abstract
Amino acid, polyamine, and organocation (APC) transporters are secondary transporters that play essential roles in nutrient uptake, neurotransmitter recycling, ionic homeostasis, and regulation of cell volume. Here we present the crystal structure of apo-ApcT, a proton-coupled broad-specificity amino acid transporter, at 2.35 Å resolution. The structure contains 12 transmembrane helices, with the first 10 consisting of an inverted structural repeat of 5 transmembrane helices like LeuT. The ApcT structure reveals an inward-facing, apo state and an amine moiety of Lys158 located in a position equivalent to the Na2 ion of LeuT. We propose that Lys158 is central to proton coupled transport and that the amine group serves the same functional role as the Na2 ion in LeuT, thus demonstrating common principles amongst proton and sodium coupled transporters.
Amino acid, polyamine and organocation (APC) transporters are members of a large family of secondary transport proteins that catalyze the uniport, symport and antiport of a broad range of substrates across the membrane bilayer (1). Present throughout the kingdom of life, APC transporters are integral to cellular physiology and in humans include multiple solute carrier (SLC) families (Fig. 1A; table S1) (2). Canonical APC transporters include the SLC7 glutamate/cystine antiporter (xCT), a sodium independent antiporter that exchanges extracellular cystine for intracellular glutamate(3, 4). Cationic amino acid transporters (CATs) are also members of the SLC7 family and they supply arginine for nitric oxide synthesis, a major contributor to asthma pathogenesis (5). In cancer, the APC l-type amino acid transporter 1 provides amino acids required for tumor growth, is upregulated in tumor cells (6) and mediates the uptake of melphalin, a chemotherapy drug (7). Both cystinuria, the most common primary inherited aminoaciduria, and lysinuric protein intolerance are the consequence of mutations in SLC7 genes (8-10).
Fig. 1. ApcT is a broad specificity amino acid transporter.

(A) Dendrogram of human and selected bacterial APC transporters. Branches corresponding to prokaryotic APCs, CATs, CCCs, VIATT, SNATs, PATs and HATs are defined in table S1. (B) [3H]-Ala uptake is pH dependent. ApcT and no protein control proteoliposomes were loaded with buffer at pH 7 (blue and black, respectively) or pH 4 (red and grey, respectively). Uptake experiment was performed by diluting these liposomes in a pH 4 buffer supplemented with 500nM [3H]Ala. Error bars represent SEM of triplicate measurements. (C) Effect of FCCP and valinomycin treatment on [3H]-Ala uptake. Proteoliposomes were loaded with 100mM KCl, and either pH7 or pH4 20mM citrate buffer. Uptake experiment was performed at 30°C in 20mM citrate buffer pH4, 100mM KCl, 750nM L-[3H] Ala and in the presence or absence of 4μM FCCP and/or 100nM valinomycin. Time points were taken at 20 min. (D) Ala and Glu are the preferred substrates of ApcT in counterflow experiments. Proteoliposomes were loaded with 4mM Ala, pH 4 and uptake of the particular [3H] amino acid, at 500 nM concentration, was measured. Estimates of uptake together with non specific transport or binding are defined by the experiments at pH4 (external; black bars) whereas estimates of non specific transport or binding are defined by experiments at pH 7 (external; gray bars).
The SLC12 family cation chloride cotransporters (CCCs) are subsumed under the APC mantle, are targets for several classes of therapeutic diuretics and are involved in active Cl- absorption in the kidney, blood pressure maintenance, and cell volume homeostasis (11); in the central nervous system, these transporters play an essential role in setting the resting chloride concentration and in γ-amino butyric acid (GABA)- and glycine-mediated neurotransmission (12). Other APC transporters participate in manifold biochemical processes in the nervous system, including the packaging of inhibitory neurotransmitters into synaptic vesicles (13) and the sodium or proton dependent symport of glutamine, a crucial step in the recycling of glutamate and GABA (14). In epithelial cells, SLC36 transporters drive the absorption of amino acids at the apical membrane of intestinal epithelial cells through coupling to a proton gradient (15). Remarkably, some APC transporters serve as ‘transceptors’, transporter-substrate complexes that transduce signals to the inside of the cell in a receptor-like fashion (16).
To deepen our understanding of APC transporters as a whole and to understand the molecular commonalities and distinctions between proton coupled and sodium coupled transporters, we have studied the atomic structure and mechanism of a proton dependent APC transporter. We initiated this effort by precrystallization screening of bacterial orthologs of the glutamate/cysteine antiporter via fluorescence detection, size exclusion chromatography (FSEC) (17). Analysis of several thermophilic orthologs revealed that a 435 amino acid integral membrane protein from Methanocaldococcus jannaschii (ApcT; GI:1591319; Fig. 1A) possessed a sharp symmetrical elution profile, both alone and in complex with an anti-ApcT Fab fragment (fig. S1), and was thus a promising APC transporter for studies of function and structure.
We discovered the function of ApcT by examining the uptake of four 3H labeled amino acids - l-Ser, l-Gln, l-Ala, and Gly - using purified ApcT reconstituted into lipid vesicles. Several APC transporters are exchangers that are active over a range of pH values (3, 18, 19), thus we loaded vesicles with cold substrate and explored various internal and external proton concentrations. At low external pH, transport of l-[3H]-Ser, l-[3H]Gln, l-[3H]-Ala, and [3H]-Gly is robust; at neutral pH, however, transport is negligible (fig. S2). Because ApcT efficiently transports [3H]Ala (fig. S3), we chose alanine for further experiments. Substrate uptake does not require internal Ala, although uptake activity is trans-stimulated by ~3-fold in the presence of internal substrate (fig. S4). A time course of [3H]-Ala uptake under conditions of a proton gradient and in the absence of internal Ala is demonstrated (Fig. 1B). Ala exchange is dependent on acidic pH values (fig. S5), yet uptake does not require sodium or a sodium gradient (fig. S6). Inclusion of proton and potassium ionophores, which collapse the pH gradient, dramatically diminish transport activity to approximately the level measured in the absence of a proton gradient (Fig. 1C and fig. S7).
To probe the substrate specificity of ApcT, we employed the canonical 20 l-amino acids, d-Ala, β-Ala, and GABA as cold competitive inhibitors in a [3H]-Ala uptake assay. Uptake of [3H]Ala is inhibited by almost all l-amino acids except Lys, Arg and Pro (fig. S8). Transport is stereo-specific, as d-Ala does not effectively compete for l-Ala transport. β-Ala does inhibit transport, demonstrating that an α-amino group is not required for inhibition. On the basis of the cold competition experiment, we selected a subset of amino acids to test as bona fide substrates, finding that ApcT transports a range of amino acids (l-Glu, l-Ala, > l-Ser, l-Gln >l-Phe,) but transports the smallest (Gly) or largest (Trp) amino acids very slowly, if at all (Fig. 1D). Taken together, our data demonstrates that ApcT is a sodium independent, broad specificity amino acid transporter active at acidic pH. We suggest that ApcT couples substrate uptake to one or more protons, although conclusive determination of this coupling, as well as the substrate to proton stoichiometry, requires further study.
ApcT solublized in n-octyl-β-d-thioglucoside forms crystals that diffract to high resolution both alone and in complex with the 7F11 Fab fragment. We solved the crystal structure of ApcT by a combination of molecular replacement, using a known Fab structure as a search probe (tables S2 and S3) and single wavelength anomalous scattering, using selenomethionine labeled ApcT. Because the crystals of ApcT alone, grown at pH ~9, diffract to higher resolution than those of the Fab complex, and the conformation of ApcT is similar in both crystal forms, we employed the former structure for analysis. The well resolved electron density maps of ApcT allowed for a complete tracing of the polypeptide chain, the accurate positioning of almost all side chains and the faithful fitting of ~180 water molecules. We found no evidence for bound ions or substrate molecules and have not yet grown crystals of a substrate complex. The present structure therefore represents a high pH, apo state.
The structure of ApcT reveals an overall cylindrical shape, intracellular amino and carboxyl termini, short cytoplasmic and extracellular loops and 12 transmembrane (TM) helices (Figs. 2A, 2B, and fig. S9). Within the first 10 TM helices, segments 1-5 and 6-10 share a similar conformation and are related by a pseudo 2-fold axis of symmetry oriented parallel to the membrane and positioned between TMs 1 and 6, about halfway across the putative bilayer (Fig. 2A, 2B, S10). The two 5 helix repeats superimpose, with not only the ‘scaffold’ helices 3-5 and 8- 10 superimposing well, but also the ‘bundle’ helices 1-2 and 6-7 and the intracellular and extracellular loops, IL1 and EL4 (Fig. 2A, 2B). Together, the two penta-helical repeats form a cylindrical barrel on the outside of which reside TMs 11 and 12. The protein fold and pseudo symmetry of ApcT is similar to that of LeuT (20), a sodium coupled amino acid transporter, and superposition confirmed that the two transport proteins harbor a remarkable kinship, in agreement with hydrophathy profile analysis (21). Other LeuT- like sodium coupled transporters, including vSGLT (22), Mph1 (23) and BetP (24), do not superimpose on ApcT as well as LeuT (supporting online text).
Fig. 2. Architecture of ApcT.
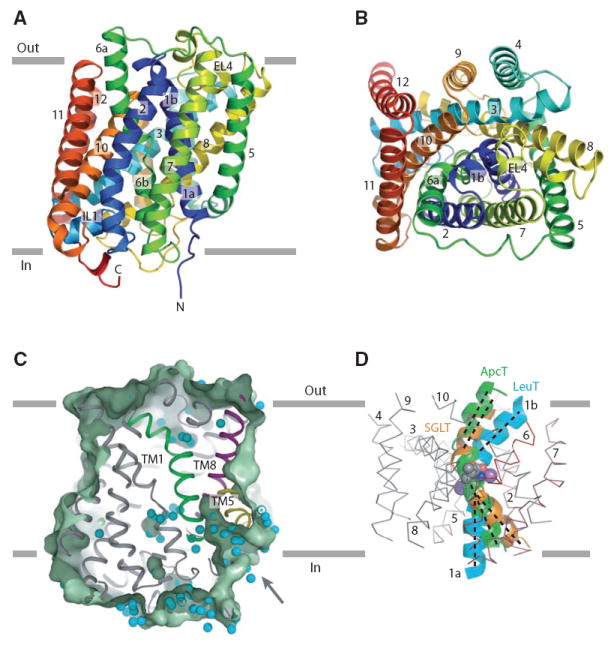
(A) Ribbon diagram of the ApcT structure, viewed parallel to the membrane, along the pseudo 2- fold axis of molecular symmetry. (B) ‘Top’ down view of ApcT from the outside. (C) Slice through a solvent accessible surface of ApcT showing a solvent accessible pathway reaching deep into the transporter. Water molecules are shown as cyan spheres. (D) Superposition of the scaffold helices TMs 3-5 and 8-10 of ApcT and vSGLT on to the equivalent elements of LeuT shows that in ApcT, TM1b is closed to the outside and TM1a is partially open to the inside. TMs 2-10 of LeuT are shown as an α-carbon trace.
The arginine/agmatine antiporter from Escherichia coli (19) (AdiC) is a member of the APC transporter family. Recently Gao et al. reported its crystal structure (25). Because ApcT is related to AdiC in amino acid sequence and in biochemical function, we compared our structure of ApcT, determined at 2.35 Å resolution, to the structure of AdiC, solved at ~3.6 Å resolution (PDB code 3H5M). As anticipated from the relationships in amino acid sequence, the protein folds of ApcT and AdiC are similar. We found significant discrepancies, however, between the superpositions of the ApcT and AdiC structures and the independently aligned amino acid sequences in the transmembrane segments 6, 7 and 8 (figs. S11 and S12). Analysis of the structures and the sequence alignments leads us to conclude that in the AdiC structure, in the regions of TMs 6, 7 and 8 and perhaps to the C-terminus, the amino acid sequence is off register by several amino acid residues relative to ApcT, beginning at the ‘loop’ between TMs 5 and 6. This means that many residues after the end of TM5, including key residues such as Glu208, Tyr239 and Trp293, are ‘frame shifted’ by ~4 residues or about one turn of α-helix(25) (figs. S11-S13). Because of this frame-shift we have not used the AdiC structure in our analysis of APC transporters.
Analysis of the ApcT structure using three different criteria suggests that it adopts a substrate-free, inward-facing yet occluded conformation. First, there is a solvent accessible path from the cytoplasmic solution to Lys158, between TMs 1 and 5 (Fig. 2C). Second, analysis of the conformation of TM 1 from the LeuT, vSGLT and ApcT structures, an element of structure that plays a key role in allowing access to the substrate binding pocket, demonstrates that TM1 adopts a conformation that completely occludes access to the outside but only partially occludes access to the inside (Figs. 2D, S14, and table S4). Third, mapping of bound water molecules onto the transporter clearly shows a belt of solvent at the extracellular face of the transporter, a solvent-free swath about 8 Å thick between the extracellular boundary and the putative substrate binding pocket, and a wide belt of solvent sites within and around the cytoplasmic ‘half’ of the transporter (fig. S15). This apo conformation of ApcT is most similar to that of the substrate bound, occluded conformation of the ~3.3 Å resolution BetP structure (fig. S14) (24), thus demonstrating, by way of our high resolution structure, that formation of this occluded state is not dependent upon the presence of bound substrate.
There is a water-filled pocket in ApcT approximately halfway across the membrane bilayer that overlaps with the substrate site of LeuT (Fig. 3A, 3B).This putative substrate binding site is adjacent to non α-helical regions of TMs1 and 6, residues from TMs 3 and 8, and amino acids from IL1 and EL4 on the ‘bottom’ and ‘top’, respectively (Fig. 3B, fig. S9). The volume of the water-filled pocket is ~195 Å3 and is large enough to accommodate phenylalanine (191.9 Å3), but not tryptophan (228.2 Å3)(26), consistent with our determination that Phe is a better substrate than Trp (Fig. 1D) (27). The pocket is lined by a combination of polar, aromatic and aliphatic amino acids and thus, like LeuT (20), the ionic interactions between the substrate’s amino and carboxyl groups are probably satisfied by hydrogen bonding interactions with polar side chain atoms and with main chain carbonyl and amide moieties.
Fig. 3. Substrate pocket.
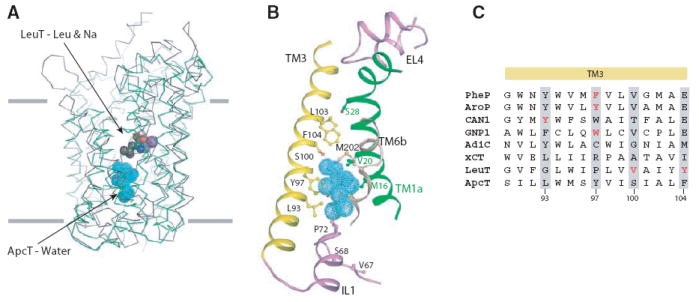
(A) Structural superposition of TMs 1-10 of LeuT (grey) and ApcT (green). TMs 1-10 of LeuT were superposed on that of ApcT using the DaliLite program and their Cα traces are shown. The substrate leucine and two sodium ions in LeuT are shown as spheres. C, N, O and Na atoms are grey, blue, red and violet respectively. The water molecules buried in the middle of the TM regions in ApcT are shown in cyan. (B) A closeup view of the putative solvent pocket in ApcT. TM helices 1, 3 and 6 and the IL1 and EL4 loops surrounding the solvent pocket are shown. Key residues predicted to be involved in substrate binding are shown as ball and stick models. (C) Role of TM3 in substrate binding. The sequence alignment shown is a composite of independent alignments generated by PROMALS 3D of the prokaryotic transporters (PheP, AroP, CAN1, GNP1, AdiC, LeuT and ApcT) and of ApcT alone with xCT orthologues. Residues implicated in substrate binding are highlighted in red and equivalent residues in other orthologs are highlighted by grey shading.
In related APC transporters residues in TMs 3 and 8, as well as IL1, play important roles in substrate binding. In TM3, Phe111 in PheP and Tyr103 in AroP of E. coli and the equivalent Trp239 in yeast Gnp1 mediate substrate specificity (28). The corresponding residue in ApcT, Tyr97 of TM3, lines part of the putative substrate binding site. In the human glutamate/cystine exchanger the residue equivalent to Tyr97 of ApcT is Arg135 and we suggest that this residue interacts with the substrate’s γ-carboxylate entity. Additional residues in TM3 that line the water-filled pocket – Leu93, Ser100 and Phe104- correspond to residue Tyr173 of yeast Can1 and Val104 and Tyr108 of LeuT, amino acids which also play central roles in substrate specificity (Fig. 3C) (20, 28). In TM8, Ala287 and Thr291 of ApcT correspond to substrate binding residues Ser355 and Ile359 in LeuT(20), while Trp293 of AdiC (TM8) is crucial for substrate binding and transport (29). Together these observations substantiate the water-filled pocket as a plausible substrate binding site.
ApcT is a proton coupled amino transporter and thus we asked if there were any titratable groups in functionally conspicuous regions. Remarkably, the well ordered, primary amine group of Lys158 (TM5) is situated between TMs 1 and 8 (Fig. 4A; fig. S19) and forms hydrogen bonds to the main chain carbonyl oxygen atom of G19 (TM1) and hydroxyl of S283 (TM8) (Fig. 4B). On the basis of a structural alignment with LeuT, the amine of Lys158 (ApcT) superimposes on the Na2 ion (LeuT; fig. S16). Consistent with the buried location of the Lys158 amine group, calculations predict a pKa that is 3-4 pH units below that of an unperturbed lysine residue (30). To test the hypothesis that Lys158 is important for ApcT transport activity we made the Lys to Ala substitution (K158A) and found that the mutant had no measurable 3H-Ala transport activity (fig. S17).
Fig. 4. Lys158 is essential to ApcT transport function.

(A) View of Lys158 bound between TMs1 and 8. (B) The amine of Lys158 forms hydrogen bonds with the carbonyl and hydroxyl oxygens of Gly19 and S283, respectively. (C) We propose that ApcT adopts an inward facing, occluded conformation when the amine group of K158 is neutral (bottom-left panel). Acidic pH promotes protonation of K158 and isomerization to an outward facing conformation (top-left). Upon substrate binding (top-right), ApcT isomerizes to an open-to-in state (bottom-right). The release of substrate and proton to the cytoplasm precedes formation of the inward facing yet occluded conformation observed in the present crystal structure (bottom-left panel).
The wild-type ApcT crystal structure was determined at alkaline pH, where the transporter is not active, and thus we reasoned that if Lys158 undergoes reversible protonation and deprotonation during the transport cycle, the present structure may represent the neutral, non protonated state of the amine group, i.e. the functional state of the transporter following unbinding of substrate and proton(s) to the cytoplasm. To assess the structural consequences of removing the lysine residue, we solved the ApcT K158A crystal structure. In agreement with our hypothesis that the wild-type structure represented the neutral, non protonated state of Lys 158, the ApcT K158A crystal structure showed neither significant rearrangement to the overall fold of the protein nor significant changes in the positions of residues surrounding residue 158 (fig. S18).
Structural alignments of ApcT, LeuT, vSGLT (SLC5), Mhp1 (NCS-1) and BetP show the ammonium group of the lysine occupies a position very similar to the predicted sodium sites in Mhp1, vSGLT and BetP. Moreover, the crucial oxygen atoms defining these essential sodium or ammonium ion binding sites between TM1 and TM8 are also conserved in sodium-potassiumchloride cotransporters (fig. S16) revealing that this monovalent cation binding site is broadly conserved across many different secondary transporters. Lys158 and its hydrogen bonding partners in TM1 and TM8 of ApcT are also conserved in the glutamate/cystine antiporter (SLC7).
The structure of ApcT provides an atomic resolution view of an APC transporter captured in an inward facing yet occluded, apo state. We suggest that this state may represent the conformation of the transporter after substrate and proton have unbound to the intracellular solution, via an open-to-the-inside state, and the intracellular gate has partially closed. Upon protonation of the Lys158 amine group, we speculate that ApcT will isomerize from this inward facing state to an open-to-the-outside conformation, ready to bind substrate and return to an inward facing conformation.
How might protonation and deprotonation of Lys158 in ApcT, or sodium binding and unbinding to the Na2 site in LeuT modulate the conformation of the transporter? We suggest that proton binding and unbinding to Lys158 toggles the conformation of TM1 and changes the position of the kink in the helix, opening and closing the extracellular gate, respectively (Fig. 4C). How might this happen? If we compare the structures of LeuT and ApcT (fig. S16), we see that the Na2 ion is fully coordinated by 5 ligands from TMs1 and 8. By contrast, the primary amine group of Lys158, which we assert is neutral in the present structure, is only coordinated by two hydrogen bond donors, one each in TM1 and TM8. Because ApcT and LeuT are closely related in structure and in the sequences of their TM1 helices, we suggest that when Lys158 of ApcT binds a proton, the coordination of the now positively charged amine group changes, with TM1 forming a kink at a position similar to the kink in LeuT, recruiting the carbonyl oxygen of Ile22 (TM1), and the hydroxyl groups of Ser283 and Ser286 (TM8). Proton binding and unbinding to the amine group of Lys158 may not only modulate the local conformation of TM1, but in doing so may also promote the movement of TMs 1a, 1b, 6a and 6b, thus facilitating the opening and closing of the extracellular and intracellular gates.
Supplementary Material
Footnotes
Supplementary materials include Supplemental Experimental Methods, 18 figures and 4 tables.
References
- 1.Jack DL, Paulsen IT, Saier MHJ. Microbiology. 2000;146:1797. doi: 10.1099/00221287-146-8-1797. [DOI] [PubMed] [Google Scholar]
- 2.Hediger MA, et al. Pflugers Arch. 2004;447:465. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 3.Sato H, Tamba M, Ishii T, Bannai S. J Biol Chem. 1999;274:11455. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 4.Bannai S, Kitamuri E. J Biol Chem. 1980;255:2372. [PubMed] [Google Scholar]
- 5.Zimmermann N, et al. J Clin Invest. 2003;111:1815. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yanagida O, et al. Biochim Biophys Acta. 2001;1514:291. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 7.Harada N, et al. Acta Haematol. 2000;103:144. doi: 10.1159/000041037. [DOI] [PubMed] [Google Scholar]
- 8.Calonge MJ, et al. Nat Genet. 1994;6:420. doi: 10.1038/ng0494-420. [DOI] [PubMed] [Google Scholar]
- 9.Torrents D, et al. Nat Genet. 1999;21:293. doi: 10.1038/6809. [DOI] [PubMed] [Google Scholar]
- 10.Borsani G, et al. Nat Genet. 1999;21:297. doi: 10.1038/6815. [DOI] [PubMed] [Google Scholar]
- 11.Hebert SC, Mount DB, Gamba G. Pflugers Arch - Eur J Physiol. 2004;447:580. doi: 10.1007/s00424-003-1066-3. [DOI] [PubMed] [Google Scholar]
- 12.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Neuron. 2009;61:820. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 13.McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Nature. 1997;389:747. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- 14.Chaudhry FA, Reimer RJ, Edwards RH. J Cell Biol. 2002;157:349. doi: 10.1083/jcb.200201070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boll M, Daniel H, Gasnier B. Pflugers Arch - Eur J Physiol. 2004;447:776. doi: 10.1007/s00424-003-1073-4. [DOI] [PubMed] [Google Scholar]
- 16.Taylor PM. Biochem Soc Trans. 2009;37:237. doi: 10.1042/BST0370237. [DOI] [PubMed] [Google Scholar]
- 17.Kawate T, Gouaux E. Structure. 2006;14:673. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Reig N, et al. J Biol Chem. 2007;282:13270. doi: 10.1074/jbc.M610695200. [DOI] [PubMed] [Google Scholar]
- 19.Iyer R, Williams C, Miller C. J Bacteriol. 2003;185:6556. doi: 10.1128/JB.185.22.6556-6561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Nature. 2005;437:215. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 21.Lolkema JS, Slotboom D-J. Mol Membr Biol. 2008;25:567. doi: 10.1080/09687680802541177. [DOI] [PubMed] [Google Scholar]
- 22.Faham S, et al. Science. 2008;321:810. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weyand S, et al. Science. 2008;322:709. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Nature. 2009;458:47. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 25.Gao X, et al. 2009 doi: 10.1126/science.1173654. [DOI] [Google Scholar]
- 26.Tsai J, Taylor R, Chothia C, Gerstein M. J Mol Biol. 1999;290:253. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
- 27.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. Science. 2008;322:1655. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cosgriff AJ, et al. J Bacteriol. 2000;182:2207. doi: 10.1128/jb.182.8.2207-2217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casagrande F, et al. J Biol Chem. 2008;283:33240. doi: 10.1074/jbc.M806917200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Robertson AD, Jensen JH. Proteins. 2005;61:704. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 31.We appreciate the beamtime and support of the staffs at beamlines 8.2.1, 8.2.2 and 5.0.2 at the Advanced Light Source. We also thank Jennifer Michel for cloning of the 7F11 gene, Lori Vaskalis for assistance with preparation of figures and Chayne Piscitelli for analysis of the AdiC structure. P.L.S. was supported by an individual NIH NRSA postdoctoral fellowship. This work was supported by the New York Consortium for Membrane Protein Structure, a NIH supported Protein Structure Initiative (E.G.). E.G. is an investigator with the Howard Hughes Medical Institute. Coordinates and structure factors of the apo ApcT, ApcT-7F11 Fab complex, and ApcT K158A-7F11 Fab complex have been deposited in the Protein Data Bank with accession codes 3GIA, 3GI9 and 3GI8, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.