

Summary
Axoplasm from the squid giant axon contains a soluble protein translocator that induces movement of microtubules on glass, latex beads on microtubules, and axoplasmic organelles on microtubules. We now report the partial purification of a protein from squid giant axons and optic lobes that induces these microtubule-based movements and show that there is a homologous protein in bovine brain. The purification of the translocator protein depends primarily on its unusual property of forming a high affinity complex with microtubules in the presence of a nonhydrolyzable ATP analog, adenylyl imidodiphosphate. The protein, once released from microtubules with ATP, migrates on gel filtration columns with an apparent molecular weight of 600 kilodaltons and contains 110–120 and 60–70 kilodalton polypeptides. This protein is distinct in molecular weight and enzymatic behavior from myosin or dynein, which suggests that it belongs to a novel class of force-generating molecules, for which we propose the name kinesin.
Introduction
Most animal cells transport organelles within their cytoplasm (Schliwa, 1984). In neurons, bidirectional organelle transport within axons is thought to be the basis of fast axonal transport (Smith, 1980; Tsukita and Ishikawa, 1980). The squid giant axon, because of its large size, has been particularly useful for studying organelle movement (Allen et al., 1982; Brady et al., 1982). Axoplasm from the giant axon also can be dissociated, allowing bidirectional organelle movements to be visualized along individual cytoplasmic filaments with video-enhanced differential interference contrast microscopy (Vale et al., 1985a; Allen et al., 1985). These cytoplasmic filaments were identified as single microtubules by electron microscopy and immunofluorescence using antitubulin antibodies (Schnapp et al., 1985). Microtubules also support organelle movements in a variety of nonneuronal cells (Schliwa, 1984; Hayden et al., 1983).
Directed organelle movement within cells depends on ATP (Forman et al., 1984; Schliwa, 1984), and it has been generally assumed that movement is powered by an ATPase. Since all organelles move at the same velocity along isolated microtubules, the same type of molecular motor, or “translocator” may interact with a variety of organelles (Vale et al., 1985a). However, it is not known which proteins comprise the translocator. Dynein is the only molecule known to interact with microtubules to generate motive force (Gibbons, 1981), but attempts to identify dynein in neural tissue, where organelle movements are common, have been unsuccessful (Murphy et al., 1983).
Biochemical identification of the proteins involved in organelle movement requires an assay system for studying this phenomenon in vitro. Recently, we showed that isolated axoplasmic organelles move on purified squid optic lobe microtubules and that movement is enhanced by a soluble protein or proteins in axoplasm (Vale et al., 1985b). This experiment suggests that the translocator is a protein that binds reversibly to organelles and to microtubules. Organelle movement in the reconstituted system is similar to that in dissociated axoplasm except that movement along single microtubules in the reconstituted system is primarily unidirectional (Vale et al., 1985a; Schnapp et al., 1985).
The soluble fraction from axoplasm also induces movement of purified microtubules along a glass coverslip and movement of carboxylated latex beads along microtubules (Vale et al., 1985b). Native microtubules in extruded squid axoplasm also move along a glass coverslip (Allen et al., 1985). The soluble translocator from axoplasm that induces bead movement might also be responsible for moving inert beads after they are microinjected into axons or cultured cells (Adams and Bray, 1983; Beckerle, 1984). The velocities of microtubule and bead movements are the same (0.4 μm/sec), but both movements are 3 to 4 fold slower than organelle movements (Vale et al., 1985b). However, their similar sensitivities to vanadate suggest that organelle, bead, and microtubule movements may be driven by the same ATPase.
We describe the purification, from squid axoplasm and optic lobes, of a translocator protein that induces movement of microtubules on glass and movement of beads along microtubules. We also have identified a homologous protein in bovine brain. The characteristics of these proteins are distinct from myosin and dynein and appear to define a novel class of force-generating molecules.
Results
In Vitro Movement Assays
A high speed supernatant from axoplasm (S2 supernatant) promoted linear, ATP-dependent movement of purified microtubules (freed of microtubule-associated proteins by 1 M NaCl extraction) at 0.4 μm/sec along a glass coverslip (Table 1). In the presence of S2 supernatant, microtubules in solution also moved relative to one another, to form a contracted aggregate of microtubules. Carboxylated latex beads treated with S2 supernatant also moved at 0.4 μm/sec along microtubules immobilized by poly-D-lysine treatment of the coverslip (Table 1). Microtubules or beads incubated with ATP in the absence of S2 supernatant did not exhibit directed movement. Furthermore, the number of organelles moving along microtubules was enhanced severalfold by S2 supernatant (Table 1; see also Vale et al., 1985b). The translocator is a protein whose activity is inhibited by vanadate (100 μM) and by a nonhydrolyzable ATP analog, adenylyl imidodiphosphate (AMP-PNP) (Vale et al., 1985b).
Table 1.
Movements Supported by Squid and Bovine Translocators
| Sample | Microtubule Movement along Glass | Microtubule Movement in Solution | Bead Movement | Organelle Movementb |
|---|---|---|---|---|
| Buffer | – | − | – | + |
| S2 axoplasmic supernatanta | 0.44 ± 0.07 μm/sec | + | 0.52 ± 0.06μm/sec | + + + to + + + + |
| Squid gel filtration | 0.35 ± 0.06 μm/sec | + | 0.34 ± 0.05 μm/sec | + + to + + + + |
| Squid hydroxyapatite | 0.37 ± 0.03 μm/sec | + | 0.37 ± 0.06 μm/sec | + + to + + + + |
| Bovine gel filtration | 0.41 ± 0.05 μm/sec | + | 0.59 ± 0.01 μm/sec | + to + + |
Microtubule, bead, and organelle movements promoted by S2 axoplasmic supernatant or purified squid or bovine translocator; movement occurred consistently in preparations for which rates are shown. Squid gel filtration peak fractions (n = 7; equivalent to lane 30 in Figure 4) and hydroxyapatite peak fractions (n = 3; equivalent to lane 40, 41 in Figure 5) were tested in motility buffer or 100 mM KCl, 50 mM Tris (pH 7.6), 5 mM MgCl2, 0.5 mM EDTA, 2 mM ATP at final protein concentrations between 10 and 150 μg/ml. Bovine gel filtration peak fractions (n = 3; equivalent to lane 30 in Figure 7) were tested in KCl/Tris buffer at protein concentrations between 20 and 50 μg/ml. Organelle movement was assessed by viewing a 20 μm × 20 μm field of microtubules for 2.5–15 min and counting the number of different organelles that made directed movements along microtubules. The following rating system was employed: −, no movement in all preparations; +, 0–3 movements/min; + +, 3–7 movements/min; + + +, 7–15 movements/min; + + + +, 15–34 movements/min. The range of organelle movement in different preparations is reported. Microtubule, bead, or organelle movement assays are described in Experimental Procedures.
Values from Vale et al. (1985b).
The velocity of organelle movement in all samples was approximately 1.64 ± 0.24 μm/sec.
Because microtubule movement on glass and in solution is a sensitive and rapid assay of movement-inducing activity, we used this assay to purify the translocator protein in axoplasmic supernatant. Samples or fractions were scored positive for microtubule movement if movement was observed either on the glass coverslip or in solution.
Cosedimentation of the Translocator with Microtubules
Squid Axoplasm
Our previous studies suggested that the translocator does not bind tightly to microtubules (Vale et al., 1985b). Indeed, no microtubule movement occurred when microtubules were treated with S2 supernatant, centrifuged, washed, and resuspended in an ATP-containing buffer. However, it has been shown that AMP-PNP induces attachment of axoplasmic organelles to microtubules dissociated from squid axoplasm (Lasek and Brady, 1984). We thought that if this attachment of organelles were due to an increase in the affinity of a soluble translocator for microtubules, it might be possible to bind the translocator to microtubules with AMP-PNP to provide a means of affinity purification.
Addition of AMP-PNP to purified microtubules and axoplasmic supernatant (in the presence of 1 mM ATP) markedly increased the amount of a polypeptide (110 kilodaltons in SDS polyacrylamide gels) that copelleted with microtubules (Figure 1). This band often appeared as a doublet, possibly indicating that there are two forms of the protein that differ slightly in charge or molecular weight. This 110 kd polypeptide could be released from the microtubules with 0.1 M KCl and 5 mM ATP. When the released material was applied to new microtubules, they translocated along the coverslip and aggregated in solution (Figure 1). Two prominent proteins with molecular weights higher than 300 kd also associated with microtubules, but the amount bound was not influenced by AMP-PNP. Proteins released from microtubules treated with S2 supernatant in the absence of AMP-PNP, which included the 300 kd proteins but not the 110 kd protein, did not induce microtubule movement. The translocator also cosedimented with microtubules in the absence of AMP-PNP if the S2 supernatant was first depleted of ATP using hexokinase and glucose (not shown).
Figure 1.

Cosedimentation of a Soluble Axoplasmic Translocator with Purified Microtubules in the Presence of AMP-PNP and Its Subsequent Release by ATP
Axoplasmic supernatant (S2, lane b) was incubated with purified microtubules (lane a) at 100 μg/ml (with or without 10 mM AMP-PNP) for 10 min at 23°C, and then the microtubules were pelleted at 37,000 × g for 30 min at 4°C. Proteins released by ATP (see details below) from AMP-PNP-treated microtubules (lane d) show a prominent band at 110 kd and also induce microtubule movement (+). Neither this band nor movement (−) is associated with material released by ATP from control (AMP-untreated) microtubules (lane c), or with the microtubule pellets of AMP-PNP-treated (lane f) and untreated samples (lane e) after the ATP release.
Conditions for ATP release. After the original centrifugation of microtubules, the supernatant was discarded and microtubules were resuspended in 75 μl MTG buffer with or without 10 mM AMP-PNP and then centrifuged as before. The microtubule pellet was washed with MTG buffer (50 μl), then resuspended in 30 μl MTG plus 5 mM ATP and 0.1 M KCl for 30 min at 23°C to release associated proteins. Microtubules were pelleted as before, the supernatants were collected (lanes c and d), and the microtubules were resuspended in 30 μl of MTG (lanes e and f). The positions of molecular weight standards are noted at left.
Squid Optic Lobes
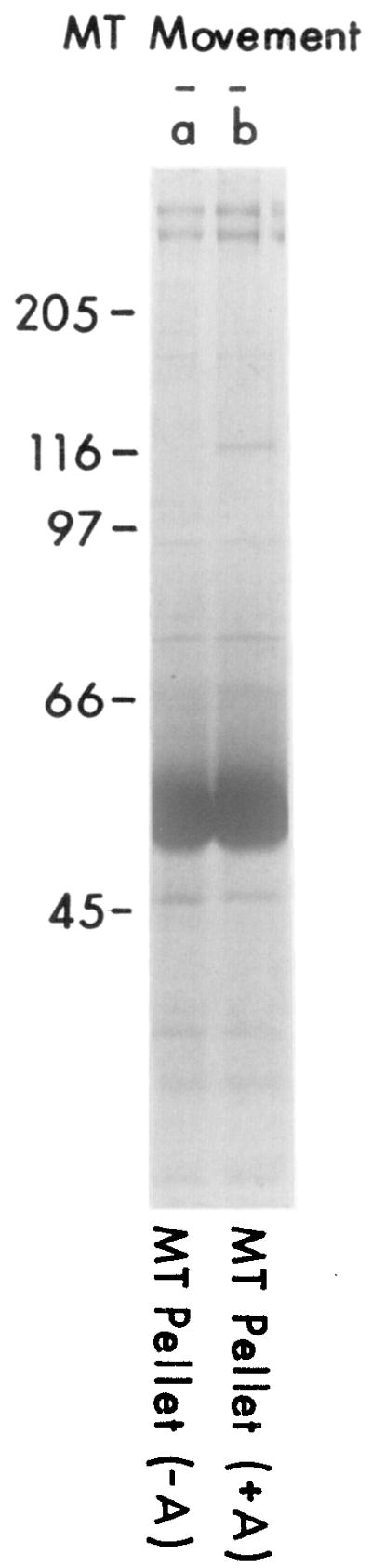
A similar microtubule cosedimentation experiment was performed using a high speed supernatant (S3) from squid optic lobes in order to obtain larger quantities of translocator. When S3 supernatant from optic lobes was combined with microtubules and 5 mM AMP-PNP, a polypeptide of 110 kd copelleted with the microtubules. This prominent band was not seen in the absence of AMP-PNP treatment (Figure 2). Minor bands corresponding to molecular weights of 65, 70, and 80 kd were also more intense after AMP-PNP treatment. Two high molecular weight proteins (>300 kd) also associated with the microtubules (Figures 2 and 3). We refer to the higher and lower molecular weight species of the 300 kd polypeptides as HMW1, and HMW2, respectively. The amount of HMW2 was somewhat variable, which may reflect its dissociation from microtubules during washing of the microtubule pellet (see below). Association of HMW1 and HMW2 with microtubules generally was unaffected by AMP-PNP, although in some experiments a small (2-fold) increase in the amount of HMW1 was found in AMP-PNP-treated samples.
Figure 2.
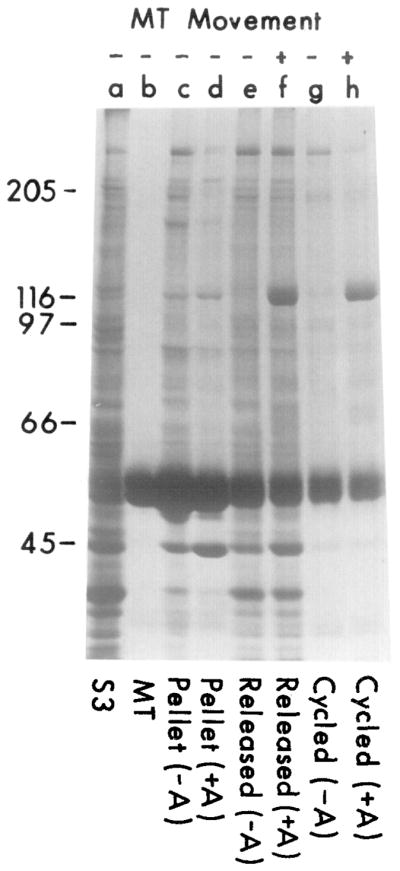
Cosedimentation of a Translocator from Squid Optic Lobes with Purified Microtubules in the Presence of AMP-PNP, and Its Subsequent Release by ATP
Soluble supernatant from squid optic lobes (S3, lane a) was incubated with 100 μg/ml of purified microtubules (lane b) in the presence or absence of 5 mM AMP-PNP as described in Experimental Procedures. Material released from microtubules by 5 mM ATP and 0.1 M KCl (45 min; 4°C) from AMP-PNP-treated samples (lane f) shows both a prominent band at 110 kd and movement-inducing activity; neither characteristic is associated with the supernatant of the untreated samples (lane e) or the final microtubule pellets resuspended in an equal volume of MTG with 2 mM ATP (lanes c and d) after ATP release.
The ATP-released translocator (lane f) was tested for rebinding to microtubules. The sample was diluted 5-fold in MTG (to reduce the concentration of ATP/KCl) and incubated with new microtubules (100 μg/ml) in the presence or absence of 5 mM AMP-PNP (15 min; 23°C). The microtubules were pelleted and washed, and proteins were released with 5 mM ATP and 0.1 M KCl for 45 min at 23°C. Material released from the AMP-PNP-treated sample contained the 110 kd polypeptide (lane h) and also induced movement (+); neither characteristic was prominent in the control (AMP-PNP-untreated) sample (lane g).
Figure 3.

ATP- and KCl-Dependent Release of the Translocator from Microtubules
S3 supernatant was incubated with microtubules and AMP-PNP as described in Experimental Procedures. Many contaminating proteins (lane a), but not the translocator, were removed by resuspending the MT pellet in 2 ml of MTG buffer containing 10 mM AMP-PNP, centrifuging the microtubules, and removing the AMP-PNP-containing supernatant. The translocator could then be obtained by washing the pellet with MTG, releasing with 1 ml of MTG plus 10 mM ATP/0.1 M KCl (30 min; 23°C), centrifuging the microtubules, and collecting the supernatant that contains movement-inducing activity (lane b). The final microtubule pellet was then resuspended in 1 ml of MTG (lane c). To define further the conditions that release translocator from microtubules, S3 supernatant (8 ml) was incubated with microtubules and AMP-PNP, and the microtubule pellet was washed as described above. Microtubules were then aliquoted into eight 100 μl samples in MTG containing the indicated amounts of ATP and KCl and incubated for 30 min at 23°C (lanes d–k). The microtubules were pelleted, and 50 μl of each supernatant was run per lane. Lanes a–c are from a different experiment than lanes d–k.
Unlike S2 supernatant from axoplasm, S3 supernatant from optic lobes did not induce microtubule movement, either because the concentration of translocator was too low or because inhibitory proteins were present. The AMP-PNP-dependent binding of the translocator to microtubules provided a method for concentrating its activity. Indeed, microtubules that were incubated with S3 optic lobe supernatant and AMP-PNP, and then pelleted, washed, and resuspended in ATP-containing buffer moved along each other and on the glass coverslip. When microtubule-associated proteins, including the 110 kd polypeptide, were released with 5 mM ATP and 0.1 M KCl and then applied to new microtubules, these microtubules moved in a manner indistinguishable from the movement produced by S2 axoplasmic supernatant (Figure 2). Proteins released from microtubules not treated previously with AMP-PNP, which included HMW1 and HMW2 but not the 110 kd polypeptide, did not induce microtubule movement.
The released 110 kd polypeptide, if diluted to lower the concentration of ATP and KCl, could rebind to microtubules in the presence of AMP-PNP, and when rereleased, it still induced microtubule movement (Figure 2). This cycling of the translocator protein with microtubules also removed many of the minor protein bands. The 110 kd polypeptide did not pellet to a significant extent with microtubules when cycled in the absence of AMP-PNP, and the subsequently released supernatant induced little or no microtubule movement. The 65/70 kd doublet and 80 kd polypeptides also cycled selectively with microtubules in the presence of AMP-PNP.
HMW2 and a variety of other proteins were released from microtubules by dilution in the absence of ATP or KCl, but the 110 kd polypeptide and HMW1 remained attached (Figure 3, lane a). Hence, the purity of the 110 kd polypeptide could be increased without cycling by resuspending microtubules in buffer containing AMP-PNP to release HMW2 and other proteins that dissociate upon dilution (Figure 3). When the 110 kd polypeptide was subsequently released with KCl or ATP, it comprised between 15% and 45% of the released protein, as determined by densitometry. The protein sample at this stage is referred to as MT-purified translocator. The major contaminants include the HMW proteins as well as tubulin, which was present in variable amounts because of some cold instability of the microtubules. Assuming 100% association of the translocator with microtubules in the presence of AMP-PNP, the concentration of translocator in the S3 supernatant is estimated to be approximately 5–10 μg/ml.
The 110 kd polypeptide dissociated from microtubules in either 0.1–0.3 M KCl or 1–10 mM ATP (Figure 3, lanes d–k). ATP was a more effective releasing agent than KCl, and combinations of ATP and KCl did not produce a greater effect than ATP alone. KCl (0.3 M) also released certain proteins that remained bound when only ATP was added. Between 40% and 90% of the 110 kd polypeptide was released by a single 30 min incubation at 23°C with 5 mM ATP and 0.1 M KCl, and the remainder was generally released by subsequent extraction under the same conditions. The HMW1 protein was also released from microtubules by ATP, as were the 65/70 kd doublet and 80 kd polypeptides (Figure 3, lanes d–k).
Chromatographic Purification of the Translocator from Squid
Gel Filtration Chromatography
Supernatant containing the 110 kd and 65/70 kd doublet polypeptides released from microtubules promoted microtubule movement. To determine whether it is these polypeptides that induce movement, the MT-purified translocator was further purified by chromatography. The microtubule translocator eluted from a Bio-Gel A5m gel filtration column (Figure 4) as a single peak that corresponded precisely to the elution peak of the 110 kd polypeptide, as determined by SDS gel electrophoresis. The 110 kd polypeptide, which appeared as two bands on SDS polyacrylamide gels, eluted between the HMW1 and HMW2 proteins with a Kav of 0.45, approximately the elution peak of the globular protein thyroglobulin (molecular weight of 667 kd). Since the 110 kd polypeptide peak was clearly separated from the two HMW protein peaks, it is unlikely that the 110 kd polypeptide is in a complex with either of these proteins. On the other hand, the 65/70 kd doublet, which also demonstrated AMP-PNP-dependent binding to microtubules (see Figure 2), comigrated with the 110 kd peak, indicating that these polypeptides are tightly associated.
Figure 4.

Gel Filtration Chromatography of Squid Translocator
(A) Microtubule-purified translocator was applied to a Bio-Gel A5m column, and the microtubule-movement-inducing and ATPase activities of the eluant fractions were determined. (B) The polypeptide compositions of fractions 21–45 were analyzed by SDS polyacrylamide gel electrophoresis (60 μl loaded per lane); fraction 30 contains approximately 65 μg/ml of protein. (C) A densitometer scan is shown of a lane from the fraction (30) with the greatest movement-inducing activity from another column. Chromatographic conditions: A 0.75 ml sample of microtubule-purified translocator (which moved microtubules at a 1:20 dilution) was applied to a 1 × 47 cm column equilibrated in 100 mM KCl, 50 mM Tris (pH 7.6), 5 mM MgCI2, 0.5 mM EDTA, and 1 mM ATP. The column was run at 4°C at 5 cm/hr; 0.75 ml fractions were collected. Excluded (Vo) and included (VT) volume fractions are noted at top. Microtubule movement was measured by serial dilution as described in Experimental Procedures. The tubulin doublet at 55 kd that streaks across the gel is not typical, but is an artifact of this particular gel.
Densitometry scans of Coomassie-stained gels of the column fractions indicated a stoichiometric ratio of the 110 to 65/70 kd doublet polypeptides which ranged from 1.5:1 to 2.2:1 (measurements from five columns). The 65 and 70 kd polypeptides were present in approximately equal amounts. An 80 kd polypeptide also comigrated with the 110 kd polypeptide, but the molar ratio of 110 kd to 80 kd polypeptide (4:1 to 10:1) was less consistent between different preparations. Silver or nickel staining of 7.5% polyacrylamide gels of the column fractions did not reveal additional polypeptides that consistently copurified with the 110 kd polypeptide (not shown).
The major peak of ATPase activity eluting from the gel filtration column corresponded to the HMW1 protein, while relatively little activity eluted with fractions that induced microtubule movement and contained the 110 kd polypeptide (Figure 4). The ATPase activities in the fractions corresponding to the peak elutions of HMW1 and the 110 kd polypeptides were, respectively, 0.13 and 0.01 μmol/min/mg (in 100 mM KCl, 50 mM Tris, 5 mM MgCl2, 0.5 mM EDTA, pH 7.6). In the 110 kd peak fractions, the ATPase was activated 1 to 2 fold by 0.25% Triton X-100 or 0.8 M KCl, but it was not significantly inhibited by 2.5 mM N-ethyl maleimide (NEM). The ATPase activity of the HMW1 peak, on the other hand, was inhibited 80%–90% by 2.5 mM NEM when incubated 15 min at 23°C followed by 20 mM dithiothreitol (DTT). However, microtubule movement (as assayed by serial dilution) was not inhibited by treating the 110 kd peak fraction with 5 mM NEM for 30 min at 23°C followed by 20 mM DTT inactivation. Because microtubule movement occurred under conditions in which the HMW1 ATPase was inhibited, this ATPase presumably is not involved in powering microtubule movement.
Translocator purified by gel filtration chromatography induced microtubule movement at total protein concentrations as low as 5 μg/ml and at ATP concentrations of 10–20 μM (at protein concentrations of 50 μg/ml). Microtubule movement also occurred at GTP concentrations of 0.5 mM; but CTP, TTP, and UTP did not support movement at concentrations of 5 mM. Microtubule movement induced by purified squid translocator was abolished by 100 μM vanadate and was only slightly inhibited by 25 μM vanadate. A similar dependence on vanadate concentration was observed with microtubule, bead, and organelle movements induced by S2 axoplasmic supernatant (Vale et al., 1985b).
Hydroxyapatite Chromatography
Fractions containing the 110 kd polypeptide from the Bio-Gel A5m column were pooled and applied to an hydroxyapatite column. The fraction that induced the most microtubule movement corresponded with the peak of the 110 kd polypeptide, which eluted at a phosphate concentration of approximately 0.18–0.2 M (Figure 5). The HMW proteins eluted earlier than the 110 kd polypeptide, and before the fractions that promoted microtubule movement. The 65/70 kd doublet and 80 kd polypeptides coeluted with the 110 kd polypeptide. Densitometry again revealed a stoichiometric ratio of the 110 kd polypeptides to the 65/70 kd doublet polypeptide that ranged from 1.5:1 to 2:1 (not shown). The extent of purification of the translocator after hydroxyapatite chromatography is at least 1000- fold, assuming a concentration of the translocator in S3 supernatant of 10 μg/ml (see above).
Figure 5.

Hydroxyapatite Chromatography of Squid Translocator Purified by Gel Filtration
Samples corresponding to fractions 28–32 from the gel filtration column shown in Figure 4 were pooled and applied to a 1 × 6 cm hydroxyapatite column equilibrated with gel filtration buffer at pH 7.4. Proteins were eluted with a 0–0.3 M phosphate gradient, and 0.5 ml fractions were collected. Microtubule-movement-inducing activity, assayed by serial dilution, correlated with the presence of 110 and 65 kd polypeptides in eluant fractions. Fraction 40 contains approximately 35 μg/ml of protein.
MT-purified translocator could be further purified by DEAE-Sephadex chromatography. Movement-inducing activity as well as the 110 kd and 65/70 kd doublet polypeptides bound to this column and were eluted together by 0.2 M KCl (not shown), also indicating that these polypeptides are involved in microtubule movement.
Purification of a Translocator from Bovine Brain
A protein homologous to the squid optic lobe translocator may be present in the vertebrate nervous system; therefore, we attempted to purify it by its affinity for microtubules in the presence of AMP-PNP. When S3 supernatant from bovine brain (which itself did not induce movement of microtubules) was incubated with squid microtubules, several proteins, including two of high molecular weight that might correspond to MAP1 and MAP2 (Vallee and Bloom, 1984), copelleted with the microtubules (Figure 6). If AMP-PNP was also added, another major polypeptide, of 120 kd, copelleted with the microtubules. This polypeptide attached with approximately equal affinity to squid or bovine microtubules in the presence of AMP-PNP (not shown). This and a minor polypeptide of approximately 62 kd were the only ones that demonstrated AMP-PNP-dependent binding to microtubules. However, microtubules associated with this protein did not move on glass or in solution (Figure 6). The 120 kd polypeptide could be released from the microtubules with 1–10 mM ATP, and when added to new microtubules, the released proteins still did not induce microtubule movement on glass or in solution.
Figure 6.
Cosedimentation of a 120 Kilodalton Bovine Brain Polypeptide with Squid Microtubules Depends upon AMP-PNP
Bovine brain S3 supernatant was incubated with squid optic lobe microtubules (100 μg/ml) in the absence (lane a) or presence (lane b) of 5 mM AMP-PNP for 15 min at 23°C. Microtubules were pelleted, washed once with PEM-taxol-GTP buffer, resuspended in 1/40 of the original volume with the same buffer containing 5 mM ATP and 0.1 M KCl, and then assayed for movement-inducing activity.
When the MT-purified and ATP-released 120 kd polypeptide from bovine brain was applied to a Bio-Gel A5m column, the 120 kd polypeptide eluted in the same position as the 110 kd polypeptide from squid (Figure 7). The 120 kd polypeptide clearly separated from the high molecular weight polypeptides, but coeluted with the 62 kd polypeptide. The 120 kd and 62 kd polypeptides typically appeared as single bands rather than doublets. Densitometry indicated that the stoichiometric ratio of 120 kd polypeptide to 62 kd polypeptide was approximately 2:1 (measurements from three columns).
Figure 7.

Gel Filtration Chromatography of the Microtubule-Purified Bovine Translocator
(A) Microtubule-purified bovine proteins (0.75 ml; purified as described in Experimental Procedures) were run on a Bio-Gel A5m column like that described in Figure 4, yielding the profile of ATPase and movement-inducing activities shown. (B) The polypeptide compositions of fractions 21–37 were analyzed by SDS polyacrylamide gel electrophoresis (40 μl per lane). (C) A densitometer scan of the Coomassie stained gel is shown of the lane corresponding to the fraction with greatest movement-inducing activity (30, which contains approximately 40–50 μg/ml of protein). Void (Vo) and excluded (VT) volumes are indicated.
Unlike the material applied to the gel filtration column, fractions eluting from the column which contained the 120 kd polypeptide induced movement of squid and bovine microtubules in solution and along glass (Figure 7). Microtubule movement was detected at protein concentrations as low as 5 μg/ml. Fractions from the column, however, exhibited very little ATPase activity. Microtubule movement induced by fractions containing the bovine 120 kd polypeptide was inhibited by 100 μM vanadate but not by 5 mM NEM.
Movements Induced by Column-Purified Squid and Bovine Translocators
The S2 supernatant from axoplasm induces movements of microtubules on glass, microtubules in solution, latex beads on microtubules, and organelles on microtubules (Vale et al., 1985b; Table 1). The squid and bovine translocators were purified by microtubule affinity and column chromatography and assayed for their ability to move microtubules in solution and along glass as described above. The velocity of microtubule movement on glass was 0.3–0.5 μm/sec, whether induced by purified squid or bovine translocators or axoplasmic S2 supernatant (Table 1). Column-purified squid and bovine translocator also induced movement of carboxylated latex beads along microtubules at 0.3–0.5 μm/sec (Table 1). Preliminary studies suggest that bead movement along microtubules is unidirectional in the presence of purified translocator. The copurification of the factors inducing bead and microtubule movements suggests that they depend on activities of the same protein.
The squid translocator purified by gel filtration, or by gel filtration and hydroxyapatite chromatography, increased the number of organelles moving along purified microtubules severalfold compared to the number that moved in an ATP-containing buffer alone. Some purified protein preparations enhanced organelle movement to the same extent or to a greater extent than the S2 axoplasmic supernatant, although there was more variability in the amount of organelle movement between different preparations of purified squid translocator than between preparations of S2 axoplasmic supernatant. The purified bovine translocator, on the other hand, did not consistently increase the frequency of movement of organelles isolated from squid axoplasm. Organelle movement induced by S2 supernatant or purified translocator appears not to depend on non-specific association of the translocator with the organelle surface, because trypsinized organelles, lipid vesicles, and vesicles, which are isolated with microtubules during microtubule purification, do not exhibit directed movement, even in the presence of active translocator (see also Vale et al., 1985b).
The velocity of organelle movement on purified microtubules was approximately 1.6 μm/sec in buffer alone, in S2 axoplasmic supernatant, or in the presence of column-purified 110 kd squid translocator (Table 1); this velocity is the same as that of organelles in dissociated axoplasm (Vale et al., 1985a, 1985b). Organelle movements in the presence of purified squid translocator or S2 supernatant were qualitatively indistinguishable.
Discussion
Purification of a Protein Involved in Microtubule-Based Movements
A translocator protein that induces movement of microtubules on a glass coverslip and in solution has been partially purified from squid axoplasm, squid optic lobes, and bovine brain. The principal step in purification involves cosedimentation of the translocator with microtubules in the presence of an ATP analog, adenylyl imidodiphosphate. Microtubule affinity has also been employed in the purification of other proteins such as MAP1, tau, and dynein (Vallee and Bloom, 1984; Cleveland et al., 1977; Nasr and Satir, 1985).
Microtubule movement occurs only in the presence of a major polypeptide with a molecular weight of approximately 110–120 kd and with less abundant polypeptides with molecular weights of approximately 65, 70, and 80 kd. Binding of these polypeptides to microtubules is enhanced by AMP-PNP and is diminished by ATP, conditions that correlate with induction of microtubule movement. Moreover, the peak activity of the microtubule translocator coincides with the peak fractions of the 110 kd squid polypeptide in gel filtration, hydroxyapatite, and DEAE- Sephadex chromatography. We believe that the 110 kd squid and the 120 kd bovine polypeptides, although slightly different in molecular weight, are homologous proteins, because they share the ability to induce movement of microtubules and carboxylated latex beads, they bind to microtubules in the presence of AMP-PNP, they coelute in gel filtration columns, and they have similar sensitivities to NEM and vanadate.
The two high molecular weight microtubule-associated proteins are unlikely to be involved directly in microtubule movement. Neither protein coelutes from gel filtration and hydroxyapatite columns with the proteins that induce microtubule movement. Although HMW1 is released from microtubules by ATP, its binding to them is typically not enhanced by AMP-PNP. Furthermore, the ATPase activity of HMW1 is inhibited by NEM, while microtubule movement is not.
Several polypeptides from squid axons or optic lobes with molecular weights of 110, 80, 70, and 65 kd are consistently associated with microtubule movement. The 65/70 kd doublet polypeptides coelute with the 110 kd polypeptide in gel filtration and hydroxyapatite columns with a constant ratio of Coomassie blue staining intensity. A similar ratio of 120 kd to 62 kd polypeptides is observed with the bovine translocator. An 80 kd polypeptide also copurifies with the squid translocator, but typically not with the bovine translocator. This polypeptide, which was present in variable and generally less than stoichiometric amounts, could represent breakdown of the 110 kd polypeptide. In gel filtration columns, both the squid and bovine translocators elute with an apparent molecular weight of 600 kd. This result is consistent with the idea that both the bovine and squid translocators are complexes of the 110–120 kd and 62–70 kd polypeptides, but additional experiments are needed to establish the precise quaternary structure of this complex.
The AMP-PNP-dependent binding of the translocator to microtubules and its release by ATP indicates that it has a nucleotide binding site. The vanadate sensitivity of the translocator-induced microtubule movement, as well as the fact that ATP is a more effective substrate than GTP by an order of magnitude, suggests the presence of an ATPase. However, the fractions that induce the most movement have very little associated solution ATPase activity (ATP turnover rate of 0.1 sec−1) compared to the ATPase activity of dynein (1.5–30 sec−1; Bell et al., 1982). To obtain maximal ATPase hydrolysis of the translocator, a ternary complex consisting of translocator; microtubule; and bead, organelle, or glass surface may be necessary, though conditions for producing such complexes in high concentrations have not yet been found.
Kinesin: A Novel Class of Force-Generating Molecules
Dynein is the only protein known to bind to microtubules and perform work (Gibbons, 1981). Axonemal and cytoplasmic dyneins have been isolated from a variety of sources and have a number of common features that distinguish them as dyneins. By several structural and enzymatic criteria, however, the translocators isolated from squid and bovine neural tissue are not dyneins.
Dyneins are characterized by an ATPase subunit of high molecular weight (>300 kd) whereas the translocators described here contain no polypeptide with a molecular weight higher than 120 kd. Axonemal dyneins of sea urchin (Bell et al., 1979; Gibbons and Fronk, 1979), Tetrahymena (Porter and Johnson, 1983), and Chlamydomonas (Piperno and Luck, 1979) are quite large (18S–30S) and contain two or three high molecular weight ATPase subunits as well as several intermediate and low molecular weight subunits. The molecular weights of these dynein complexes have been estimated by scanning transmission electron microscopy to be between 1 million and 2 million daltons (Witman et al., 1983; Johnson and Wall, 1983). Lower molecular weight forms (10S–14S) have also been isolated, particularly by dissociating larger dynein complexes at low ionic strength, but they still contain a single high molecular weight ATPase (Gibbons and Fronk, 1979; Porter and Johnson, 1983; Pfister and Witman, 1984).
Cytoplasmic forms of dynein have been isolated (Pratt, 1980; Scholey et al., 1984; Asai and Wilson, 1985; Hisanga and Sakai, 1983; Hollenbeck et al., 1984). These proteins also contain a high molecular weight ATPase subunit that interacts with microtubules, but they have not yet been shown capable of generating force. Since the microtubule translocators isolated from neural tissue contain no 300 kd subunits, even in the presence of protease inhibitors, they appear to be structurally distinct from cytoplasmic and axonemal dyneins. The translocator also is distinct from high MW microtubule-associated proteins that have been characterized (Vallee and Bloom, 1984).
The translocator also differs from dynein with respect to its enzymatic properties. While AMP-PNP decreases the affinity of dynein for microtubules (Mitchell and Warner, 1981) and of myosin for actin (Greene and Eisenberg, 1980), it dramatically increases the affinity of the translocator for microtubules, even in the presence of equimolar concentrations of ATP. While NEM inhibits force generation by dynein (Mitchell and Warner, 1981; Cosson and Gibbons, 1978), as well as dynein’s ability to bind to microtubules (Shimizu and Kimura, 1974), microtubule movement induced by squid or bovine translocators is largely unaffected by NEM. We conclude that the squid and bovine translocators represent a novel class of motility proteins that are structurally as well as enzymatically distinct from dynein, and we propose to call these translocators kinesin (from the Greek kinein, to move).
Possible Biological Roles for Kinesin
Kinesin appears to have a site for binding to a structure to be moved and another site capable of cyclic on and off interactions with microtubules to produce mechanical displacement (Vale et al., 1985b). If attached to an organelle or a bead, kinesin could act as a motor for organelle or bead translocation. The attachment to the organelle may be mediated by a protein on the organelle surface, since trypsinization of organelles blocks their movement (Vale et al., 1985b) whereas attachment to the bead would be through absorption to the anionic surface. Only those molecules in the correct orientation relative to the microtubule would generate forces resulting in unidirectional translocation, analogous to the manner in which myosin-coated beads move unidirectionally on Nitella actin cables (Sheetz and Spudich, 1983).
Although our results are consistent with the notion that kinesin powers organelle translocation along microtubules, the evidence is circumstantial. Column-purified kinesin, like axoplasmic supernatant, can increase the number of organelles moving along purified microtubules. Kinesin also induces latex beads to move along microtubules, although at a lower velocity than the organelles. Furthermore, the motor powering organelle transport appears to form a stable attachment between vesicles and native axoplasmic microtubules in the presence of AMP-PNP (Lasek and Brady, 1984), and kinesin also forms a stable attachment to microtubules in the presence of AMP-PNP. Finally, organelles (Vale et al., 1985a) as well as kinesin can form rigor-like attachments with microtubules in the absence of ATP. On the basis of these findings, it seems likely that kinesin binds to organelles and moves them along microtubules, although there is no direct proof of an interaction of kinesin with organelles.
If kinesin serves as a motor for organelle transport, a particularly intriguing problem is how bidirectional movement of organelles on single microtubules is generated (Schnapp et al., 1985). Myosin (Sheetz and Spudich, 1983) and dynein (Gibbons, 1981) generate force in only one direction with respect to their filamentous substrates. Furthermore, movement of organelles, beads, and microtubules along purified microtubules in the presence of axoplasmic S2 supernatant (Vale et al., 1985b) or kinesin is primarily unidirectional, as is the movement of negatively charged beads injected into crab axons (Adams and Bray, 1983). Are there two different translocators that move organelles in opposite directions, or is kinesin modified in the axon to induce movement in both directions? Current data do not allow us to distinguish between these two alternatives.
In addition to organelle transport, there are other microtubule-based forms of intracellular motility for which the molecular motors are unknown. The most widely studied of such phenomena is the sliding of the microtubule in the spindle apparatus during mitosis. The ability of kinesin to move microtubules in solution suggests that it may have two microtubule-binding domains, at least one of which is capable of force generation. In a solution of microtubules, these forces would cause microtubule aggregation and contraction, as observed here and by Weisenberg and Ciani (1984). The same kinesin-induced microtubule–microtubule (or even microtubule–organelle) movements in an organized array of microtubules could generate microtubule sliding similar to that which occurs during mitosis. Similarly, kinesin could generate force between microtubules and the plasma or organelle membranes, which could account for some forms of cellular motility.
Experimental Procedures
Materials
Squid, Loligo pealeii, were obtained from the Marine Resources Department at the Marine Biological Laboratory at Woods Hole, Massachusetts. Giant axons were dissected from the squid as previously described (Vale et al., 1985a) and were frozen in liquid nitrogen. Freshly removed squid optic lobes were also frozen in liquid nitrogen. Fresh bovine brain was obtained from a slaughter house, placed on ice, and used within 1 hr of slaughtering. Carboxylated latex beads (0.15 μm diameter; 2.5% solid/solution) were obtained from Polyscience Inc. (Warrington, Pennsylvania) and glass coverslips (type 0) from Clay Adams (Parisippany, New Jersey). Taxol was a gift from Dr. Matthew Suffness at the National Cancer Institute. Bid-Gel A5m and hydroxyapatite were obtained from Bio-Rad Inc. (Richmond, New York). All other reagents were purchased from Sigma Chemical Co. (St. Louis, Missouri).
Preparation of Axoplasmic Supernatant and Organelles
S2 axoplasmic supernatant and organelles were prepared as previously described (Vale et al., 1985b) with the following modification in the sucrose gradient. A low speed (40,000 × g/min) supernatant (about 150 μl) from a homogenate of seven to ten axoplasms was incubated with 20 μM taxol and 1 mM GTP, and applied to a sucrose gradient in a 5 × 41 mm Ultra-Clear centrifuge tube (Beckman Inst., Palo Alto, California) consisting of 7.5% (125 μl), 15% (125 μl), and 35% (200 μl) sucrose layers. Sucrose solutions were made in motility buffer (175 mM potassium asparate, 65 mM taurine, 85 mM betaine, 25 glycine, 20 mM Hepes [pH 7.2], 6.4 mM MgCl2, and 5 mM EGTA) containing 20 μM taxol and 1 mM GTP (this buffer is referred to as MTG buffer) and 2 mM ATP. The sucrose gradient was centrifuged at 135,000 × g for 70 min at 4°C in a SW50.1 rotor. The organelle band at the 15%–35% sucrose interface as well as 75–100 μl of the S2 supernatant above the gradient were collected.
Assays for Microtubule, Bead, and Organelle Movement
These assays (Vale et al., 1985b), as well as a complete account of the procedure for visualizing objects by video-enhanced differential interference contrast microscopy (B. Schnapp, submitted), will be described elsewhere. To measure microtubule movement, a 1 μl suspension of microtubules essentially free of microtubule-associated proteins (0.5–1 mg/ml solution; see below for preparation) was combined with 3–4 μl of a test sample on a glass coverslip. In some instances, a microtubule pellet was directly assayed for movement by placing an aliquot of the microtubules directly on a coverslip in an ATP-containing buffer. Microtubule movement on glass or microtubule movement relative to other microtubules in solution was visualized with the video microscope. A sample was scored positive for microtubule movement if movement was observed on glass or in solution. The presence or absence of movement in different parts of a preparation was so consistent that only a minute or two of observation was required to evaluate a sample. To determine the amount of movement-inducing activity of a sample, the maximum dilution that still supported microtubule movement was determined.
For determining movement of carboxylated latex beads, glass coverslips were treated overnight with a 1 mg/ml solution of poly-D-lysine, which prevented microtubules from moving along the glass; bead movements on microtubules could then be examined without the complication of simultaneous microtubule movement. A test sample (10 μl) was combined with beads (4 μl of a 200-fold dilution of a 2.5% solid stock solution in sample buffer) for 5 min at 4°C. Then 3–4 μl of this mixture was combined with 1 μl of microtubules on a poly-D-lysine-coated coverslip, and movement of beads along the microtubules was evaluated.
Organelle movement was assayed by combining 1 μl of microtubules with 3 μl of organelles (in motility buffer and 2 mM ATP) and 3 μl of test sample. The purified protein sample in motility buffer plus 2 mM ATP or in 100 mM KCl, 50 mM Tris (pH 7.6), 5 mM MgCl2, 0.5 mM EDTA, 2 mM ATP yielded equivalent results with regard to organelle movement. High concentrations of phosphate, however, inhibited organelle movement, so fractions from the hydroxyapatite column were equilibrated with motility buffer using a Centricon filter (molecular weight cutoff of 30 kd; Amicon Corp., Massachusetts) precoated with bovine serum albumin. A video recording of a 20 × 20 μm field of microtubules was made over 2–5 min, and the number of different organelles moving per minute was determined. Velocities of microtubule, bead, and organelle movements were determined over distances of 3–10 μm.
ATPase Assay
ATPase activity was measured according to the procedure of Clarke and Spudich (1974). Protein samples (22.5 μl) in 0.1 M KCl, 50 mM Tris (pH 7.6), 5 mM MgCl2, 0.5 mM EDTA were combined with 2.5 μl of 1–10 mM ATP containing 32P-ATP (approximately 150,000 cpm) in 1.5 ml microfuge tubes at 23°C for 5 to 40 min.
Microtubule Affinity Purification of Squid Kinesin
Squid optic lobes (40 gm wet weight; either freshly dissected or previously frozen in liquid N2) were homogenized in 1½–2 vol of motility buffer containing 1 mM ATP, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml leupeptin, 10 μg/ml N-a-p-tosyl-L-arginine methyl ketone (TAME), 100 μg/ml soybean trypsin inhibitor, and 0.5 mM DTT with 40 strokes in a Dounce homogenizer. Homogenization and all subsequent steps were performed at 4°C unless specified. The homogenate was centrifuged for 30 min at 40,000 × g, and the supernatant was collected and recentrifuged at 150,000 × g for 60 min. The supernatant from this centrifugation (S2) was incubated with 20 μM taxol and 1 mM GTP for 25 min at 23°C to polymerize tubulin into microtubules (see Preparation of Microtubules).
The mixture was then layered over a 15%/50% (2–4 ml of each) sucrose gradient made in MTG buffer, and the gradient was centrifuged at 100,000 × g for 60 min in a SW27 rotor. The supernatant (S3, approximately 40 ml) was collected, taking care to avoid the first interface. S3 supernatant then was incubated with microtubules (100 μg/ml) in the presence of 5 mM AMP-PNP for 15 min at 23°C, and microtubules were sedimented at 40,000 × g for 30 min. The following conditions proved best for releasing the translocator in a relatively pure form, although different conditions were used in some experiments, as described in the figure legends. The microtubule pellet was resuspended in 1–2 ml of MTG containing 10 mM AMP-PNP for 15 min at 4°C to release proteins that dissociate from microtubules by dilution. Microtubules were pelleted at 37,000 × g for 30 min, and the pellet was washed once with MTG to remove AMP-PNP. The pellet was then resuspended in 1 ml of MTG containing either 10 mM ATP or 5 mM ATP and 0.1 M KCl for 30–40 min at 23°C. Microtubules were centrifuged as before, and the supernatant containing proteins that induced microtubule movement was removed (typically containing 1–4 mg/ml total protein) and, if not used that day, was stored in liquid nitrogen without significant loss of activity. The amount of the 110 kd polypeptide in the microtubule pellet and supernatant was assayed by polyacrylamide gel electrophoresis, and when necessary, the pellet was extracted again with 1 ml of MTG containing 10 mM ATP to retrieve additional translocator protein.
Microtubule Affinity Purification of Bovine Translocator
White matter (70 gm) was obtained from brains of freshly killed cows and homogenized at 4°C in a weight:volume ratio of 1:1 of 50 mM Pipes, 50 mM Hepes, 2 mM MgCl2, 1 mM EDTA, 0.5 mM DTT, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml TAME, and 0.5 mM ATP (pH 7.0) with five bursts of 5 sec in a Polytron homogenizer. The homogenate was centrifuged at 25,000 × g for 30 min at 4°C, and the supernatant was collected and recentrifuged at 150,000 × g for 60 min at 4°C. GTP (1 mM) and taxol (10 μM) were added to the supernatant for 30 min at 25°C, and the polymerized microtubules were pelleted at 37,000 × g for 30 rain at 20°C. The supernatant was removed and the microtubule pellet was further processed as described below. The supernatant (comparable to squid S3) was then incubated with squid or bovine microtubules (100 μg/ml) and AMP-PNP (5 mM) for 15 min at 23°C, and the microtubules were pelleted at 37,000 × g for 20 min at 20°C. The supernatant was discarded, and the pellet was resuspended in 5 ml of homogenization buffer containing 5 mM AMP-PNP without ATP for 15 min at 23°C. The microtubules were pelleted as described above, and the pellet was washed with 2 ml of homogenization buffer without AMP-PNP or ATP. The bovine translocator was released by incubation in 1–3 ml of homogenization buffer containing 10 mM ATP for 30–40 min at 23°C. Microtubules were pelleted as before, and the supernatant containing released bovine translocator was collected (1–2 mg/ml). Additional translocator was obtained in some instances by further extraction with 0.1 M KCl/10 mM ATP.
Preparation of Microtubules
Taxol-polymerized microtubules essentially free of microtubule-associated proteins were prepared from squid optic lobes either as previously described (Vale et al., 1985b) or in conjunction with the preparation of squid translocator. In the latter instance, S2 supernatant from squid optic lobes (see Microtubule Affinity Purification of Squid Kinesin) was incubated with 20 μM taxol and 1 mM GTP for 25 min at 23°C to polymerize soluble tubulin into microtubules. The mixture was layered over a 15%/50% (2–4 ml of each) sucrose gradient made in MTG buffer and centrifuged for 60 min at 100,000 × g in a SW27 rotor. The microtubule pellet was then washed once with 2 ml of 100 mM Pipes (pH 6.6), 5 mM EGTA, 1 mM MgSO4 (PEM) containing 20 μM taxol and 1 mM GTP and resuspended in 4 ml of the same buffer. To extract microtubule-associated proteins, 2 ml of PEM-taxol-GTP containing 3 M NaCl was added to the microtubules, and the sample incubated at 23°C for 30 min. Microtubules were centrifuged at 37,000 × g for 30 min at 10°C, and the pellet was washed once with PEM-taxol-GTP. Microtubules were then resuspended in 2 ml of PEM-taxol-GTP at a concentration of 2–8 mg/ml and stored in liquid N2. Microtubules obtained during the purification of translocator from bovine brain (see above) were further processed by the protocol of Regula et al. (1981). Bovine microtubules were also prepared according to Vallee (1982).
Polyacrylamide Gel Electrophoresis
Electrophoresis was performed in polyacrylamide gels (7.5%) under denaturing and reducing conditions according to the method of Laemmli (1970) and stained with Coomassie blue. Densitometry was performed using a Zenith Soft Laser Scanning densitometer equipped with a helium-neon laser.
Acknowledgments
We are grateful to Eric Steuer for his outstanding technical assistance. We thank Dr. Bruce Schnapp for his assistance and the use of the video microscope facility, and we also appreciate the efforts of Mary Wisgirda and Amy Sewell in collection of squid optic lobes and axons. This work was supported in part by grants from Hoechst-Roussell Pharmaceutical and National Institutes of Health GM33351 (to M. P. S.). M. P. S. is an Established Investigator of the American Heart Association.
References
- Adams RJ, Bray D. Rapid transport of foreign particles microinjected into crab axons. Nature. 1983;303:718–720. doi: 10.1038/303718a0. [DOI] [PubMed] [Google Scholar]
- Allen RD, Metuzals J, Tasaki I, Brady ST, Gilbert SP. Fast axonal transport in squid giant axon. Science. 1982;218:1127–1128. doi: 10.1126/science.6183744. [DOI] [PubMed] [Google Scholar]
- Allen RD, Weiss DG, Hayden JH, Brown DT, Fujiwake H, Simpson M. Gliding movement of and bidirectional organelle transport along single native microtubules from squid axoplasm: evidence for an active role of microtubules in cytoplasmic transport. J Cell Biol. 1985;100:1736–1752. doi: 10.1083/jcb.100.5.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai DJ, Wilson L. A latent activity dynein-like cytoplasmic magnesium adenosine triphosphatase. J Biol Chem. 1985;260:699–702. [PubMed] [Google Scholar]
- Beckerle MC. Microinjected fluorescent polystyrene beads exhibit saltatory motion in tissue culture cells. J Cell Biol. 1984;98:2126–2132. doi: 10.1083/jcb.98.6.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CW, Fronk E, Gibbons IR. Polypeptide subunits of dynein 1 from sea urchin sperm flagella. J Supramol Struct. 1979;11:311–317. doi: 10.1002/jss.400110305. [DOI] [PubMed] [Google Scholar]
- Bell CW, Fraser CL, Sale WS, Tang WY, Gibbons IR. Preparation and purification of dynein. Meth Enzymol. 1982;85:450–474. doi: 10.1016/0076-6879(82)85045-3. [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–1131. doi: 10.1126/science.6183745. [DOI] [PubMed] [Google Scholar]
- Clarke M, Spudich JA. Biochemical and structural studies of actomyosin-like proteins from non-muscle cells. J Mol Biol. 1974;86:209–222. doi: 10.1016/0022-2836(74)90013-8. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- Cosson MP, Gibbons IR. Properties of sea urchin sperm flagella in which the bending waves have been preserved by pretreatment with mono- and bi-functional maleimide derivatives. J Cell Biol. 1978;79:286a. [Google Scholar]
- Forman DS, Brown KJ, Promersberg MW, Adelman MR. Nucleotide specificity for reactivation of organelle movement in permeabilized axons. Cell Motil. 1984;4:121–128. doi: 10.1002/cm.970040205. [DOI] [PubMed] [Google Scholar]
- Gibbons IR. Cilia and flagella of eukaryotes. J Cell Biol. 1981;91:107–124. doi: 10.1083/jcb.91.3.107s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons IR, Fronk E. A latent adenosine triphosphatase form of dynein 1 from sea urchin sperm flagella. J Biol Chem. 1979;254:187–196. [PubMed] [Google Scholar]
- Greene LE, Eisenberg E. Dissociation of the actin sub-fragment 1 complex by adenyl-5′imidodiphosphate, ADP and PPi. J Biol Chem. 1980;225:543–548. [PubMed] [Google Scholar]
- Hayden JH, Allen RD, Goldman RD. Cytoplasmic transport in keratocytes: direct visualization of particle translocation along microtubules. Cell Motil. 1983;3:1–19. doi: 10.1002/cm.970030102. [DOI] [PubMed] [Google Scholar]
- Hisanga S, Sakai H. Cytoplasmic dynein of the saa urchin egg. II Purification, characterization and interactions with microtubules and Ca-calmodulin. J Biochem. 1983;93:87–98. doi: 10.1093/oxfordjournals.jbchem.a134182. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Suprynowicz F, Cande WZ. Cytoplasmic dynein-like ATPase cross-links microtubules in an ATP-sensitive manner. J Cell Biol. 1984;99:1251–1258. doi: 10.1083/jcb.99.4.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Wall JS. Structure and molecular weight of dynein ATPase. J Cell Biol. 1983;96:669–678. doi: 10.1083/jcb.96.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U. Cleavage of structural proteins during the assembly of the bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lasek RJ, Brady ST. Adenylyl imidodiphosphate (AMP-PNP), a non-hydrolyzable analogue of ATP produces a stable inter-mediate in the motility cycle of fast axonal transport. Biol Bull. 1984;167:503. [Google Scholar]
- Mitchell DR, Warner FD. Binding of dynein 21 S ATPase to microtubules. Effects of ionic conditions and substrate analogs. J Biol Chem. 1981;256:12535–12544. [PubMed] [Google Scholar]
- Murphy DB, Hiebsch RR, Wallis KT. Identity and origin of the ATPase activity associated with neuronal microtubules. I The ATPase activity is associated with membrane vesicles. J Cell Biol. 1983;96:1298–1305. doi: 10.1083/jcb.96.5.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr T, Satir P. Alloaffinity chromatography: a general approach to affinity purification of dynein and dynein-like molecules. Anal Biochem. 1985 doi: 10.1016/0003-2697(85)90058-2. in press. [DOI] [PubMed] [Google Scholar]
- Pfister KK, Witman GB. Subfractionation of Chlamydomonas 18 S dynein into two unique subunits containing ATPase activity. J Biol Chem. 1984;259:12072–12080. [PubMed] [Google Scholar]
- Piperno G, Luck DJL. Axonemal adenosine triphosphatases from flagella of Chlamydomonas reinhardtii. J Biol Chem. 1979;254:3084–3090. [PubMed] [Google Scholar]
- Porter ME, Johnson KA. Characterization of the ATP-sensitive binding of Tetrahymena 30 S dynein to bovine brain microtubules. J Biol Chem. 1983;258:6575–6581. [PubMed] [Google Scholar]
- Pratt MM. The identification of a dynein ATPase in unfertilized sea urchin eggs. Dev Biol. 1980;74:364–378. doi: 10.1016/0012-1606(80)90438-8. [DOI] [PubMed] [Google Scholar]
- Regula CS, Pfeiffer JR, Berlin RD. Microtubule assembly and disassembly at alkaline pH. J Cell Biol. 1981;89:45–53. doi: 10.1083/jcb.89.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliwa M. Mechanisms of intracellular organelle transport. In: Shaw JW, editor. Cell Muscle Motility. Vol. 5. New York: Plenum Publishing Co; 1984. pp. 1–81. [DOI] [PubMed] [Google Scholar]
- Schnapp BJ, Sheetz MP, Vale RD, Reese TS. Filamentous actin is not a component of transport filaments isolated from squid axoplasm. J Cell Biol. 1984;99:351a. [Google Scholar]
- Schnapp BJ, Vale RD, Sheetz MP, Reese TS. The structure of cytoplasmic filaments involved in organelle transport in the squid giant axon. Cell. 1985;40:455–462. [Google Scholar]
- Scholey JM, Neighbors B, McIntosh JR, Salmon ED. Isolation of microtubules and a dynein-like MgATPase from un-fertilized sea urchin eggs. J Biol Chem. 1984;259:6516–6525. [PubMed] [Google Scholar]
- Sheetz MP, Spudich JA. Movement of myosin-coated structures on actin cables. Cell Motil. 1983;3:484–485. doi: 10.1002/cm.970030515. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kimura I. Effects of N-ethylmaleimide on dynein adenosine-triphosphatase activity and its recombining ability with outer fibers. J Biochem. 1974;76:1001–1008. [PubMed] [Google Scholar]
- Smith RS. The short term accumulation of axonally transported organelles in the region of localized lesions of single myelinated axons. J Neurocytol. 1980;9:39–65. doi: 10.1007/BF01205226. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Ishikawa H. The movement of membranous organelles in axons. Electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol. 1980;84:513–530. doi: 10.1083/jcb.84.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP. Movement of organelles along filaments dissociated from axoplasm of the squid giant axon. Cell. 1985a;40:449–454. doi: 10.1016/0092-8674(85)90159-x. [DOI] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP. Organelle, bead and microtubule translocations promoted by soluble factors from the squid giant axon. Cell. 1985b;40:559–569. doi: 10.1016/0092-8674(85)90204-1. [DOI] [PubMed] [Google Scholar]
- Vallee RB. A taxol-dependent procedure for the isolation of microtubules and microtubule-associated proteins (MAPs) J Cell Biol. 1982;92:435–442. doi: 10.1083/jcb.92.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, Bloom GS. High molecular weight microtubule-associated proteins. In: Satir PH, editor. Modern Cell Biology. New York: Alan R. Liss; 1984. pp. 21–75. [Google Scholar]
- Weisenberg RC, Ciani C. ATP-induced gelation-contraction of microtubules assembled in vitro. J Cell Biol. 1984;99:1527–1533. doi: 10.1083/jcb.99.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witman GB, Johnson KA, Pfister KK, Wall JS. Fine structure and molecular weight of the outer arm dyneins of Chlamydomonas. J Submicrosc Cytol. 1983;15:193–197. [Google Scholar]