

SUMMARY
Cytoplasmic dynein, the 1.2 MDa motor driving minus-end-directed motility, has been reported to move processively along microtubules, but its mechanism of motility remains poorly understood. Here, using S. cerevisiae to produce recombinant dynein with a chemically controlled dimerization switch, we show by structural and single-molecule analysis that processivity requires two dynein motor domains but not dynein’s tail domain or any associated subunits. Dynein advances most frequently in 8 nm steps, although longer as well as side and backward steps are observed. Individual motor domains show a different stepping pattern, which is best explained by the two motor domains shuffling in an alternating manner between rear and forward positions. Our results suggest that cytoplasmic dynein moves processively through the coordination of its two motor domains, but its variable step size and direction suggest a considerable diffusional component to its step, which differs from Kinesin-1 and is more akin to myosin VI.
INTRODUCTION
Cytoplasmic dynein performs nearly all minus-end-directed microtubule-based movement in most eukaryotic cells; its many functions include spindle formation and chromosome segregation and the transport of numerous cargoes including viruses, RNA, signaling molecules, and organelles (Vallee et al., 2004). The partial loss of cytoplasmic dynein function has been implicated in lissencephaly, schizophrenia, and motor neuron degeneration (Gerdes and Katsanis, 2005). Thus, dissecting the molecular mechanism of dynein motility is essential for understanding a wide array of biological processes and the molecular basis of several types of human disease.
Unlike the two other cytoskeletal motor protein families, kinesin and myosin, for which detailed structural and mechanistic models exist, relatively little is known about the mechanism of dynein motility. Moreover, whereas kinesin and myosin share many mechanistic similarities because of their common evolutionary origin (Vale and Milligan, 2000), dynein is a divergent evolutionary branch of the AAA+ family of ATPases (Neuwald et al., 1999). Thus, dynein is likely to share more mechanistic features with this group of ATP-hydrolyzing protein “unfoldases” than with the kinesin/myosin motors. Like many other AAA+ ATPases, dynein’s motor domain is composed of a ring of 6–7 AAA+ domains, which, in the case of dynein, are concatenated into a single large polypeptide (Burgess et al., 2003; Samso and Koonce, 2004). Four of the AAA+ domains can bind ATP (Figure 1A) (Gibbons et al., 1991; Kon et al., 2004; Reck-Peterson and Vale, 2004; Takahashi et al., 2004), with AAA1 being the principal site of hydrolysis (Gibbons et al., 1987) and AAA3 also contributing an essential role in dynein function (Kon et al., 2005; Reck-Peterson and Vale, 2004; Silvanovich et al., 2003). A 10–15 nm antiparallel coiled-coil stalk emerges after AAA4, and a globular microtubule binding domain is located at the tip of the coiled-coil stalk (Figure 1A) (Gee et al., 1997; Goodenough and Heuser, 1984; Koonce, 1997). NH2 terminal to the AAA domains is the “linker” domain that has been suggested to interact with the AAA ring and has been proposed to be involved in force generation (Burgess et al., 2003; Kon et al., 2005). The NH2-terminal region of the dynein heavy chain (the “tail” domain) is involved in dimerization, binding to dynein associated proteins, and interactions with cargo [reviewed in Vallee et al. (2004)].
Figure 1. Purification of Native Yeast Dynein and Observation of Single-Molecule Processivity.

(A) Schematic of the native dynein heavy-chain dimer. The NH2 terminus of the motor is labeled with GFP (green asterisk), and the COOH terminus of the motor is labeled with the HaloTag and covalently bound to tetramethylrhodamine (TMR, red asterisk).
(B) Purification of native yeast dynein expressed from its endogenous promoter. Dynein, NH2-terminally tagged with two copies of the Protein A IgG binding domain, followed by a TEV cleavage site, was bound to IgG beads and then released from the beads with TEV protease. Subsequently, dynein was bound to microtubules in the absence of ATP, microtubule-dynein complexes were collected by centrifugation, and then dynein was released from microtubules by addition of 5 mM ATP (see Supplemental Data for details). A silver-stained gel shows the full-length dynein heavy chain after the final purification step. Tubulin is also present from the last affinity step.
(C) Tagging native yeast dynein (the construct described in [A] and [B]) does not affect its function in nuclear segregation in vivo. For monitoring nuclear segregation, the number of binucleate mitotic cells was determined after growth at 16°C for 16 hr. dyn1Δ is a yeast strain bearing a deletion in the dynein heavy-chain gene. The mean and standard error of the mean are shown (n > 300).
(D) Histogramof velocities of single native dynein molecules moving along axonemal microtubules (fit with a Gaussian function; black line). The mean velocity is 85 ± 30 nm/s (SD); n = 334.
(E) Histogram of run lengths of single dynein molecules moving along axonemal microtubules. The run length is 1.9 ± 0.2 μm (mean ± standard error [SE]; n = 334; Figure S3). See Movie S1 for images of native dynein moving along axonemes.
Studies of bead movement driven by cytoplasmic dynein in vitro suggest that single dynein molecules are processive, i.e., that single-motor molecules are capable of taking multiple steps along their microtubule track without detaching (King and Schroer, 2000; Mallik et al., 2004; Wang et al., 1995). The mechanisms of processivity of Kinesin-1 and myosin V have been well studied, and it has been shown that nucleotide-driven conformational changes of their mechanical elements power the hand-over-hand stepping of their two identical motor domains (Vale and Milligan, 2000; Yildiz and Selvin, 2005). In contrast, the mechanism of processivity of dynein is much less well understood, and dynein’s distinct evolutionary origin and structural features suggest that its mechanism could differ considerably from other cytoskeletal motors. Indeed, a single-headed axonemal dynein has been shown to move processively (Sakakibara et al., 1999), suggesting a mechanism other than one involving the coordination of two motor domains. The requirement for one or two motor domains, however, has not been explored for dimeric dyneins, and current models for cytoplasmic dynein motility evoke the actions of only a single dynein motor domain (Kon et al., 2005; Mallik et al., 2004).
Studies of kinesin and myosin processivity have been aided by the ability to produce recombinant motors that can be mutated and studied at a single-molecule level. Comparable studies with cytoplasmic dynein have been challenging because of its large size, and the production of a recombinant dynein capable of processive motion has not been achieved. Here, using S. cerevisiae to produce full-length dynein and a variety of truncated and artificially dimerized dyneins, we show that two cytoplasmic dynein motor domains are needed for processivity, but that the cargo binding region and dynein-associated chains are not required for this process. We have also been able to label dynein in specific locations with fluorescent dyes or quantum dots and then track single molecules with a precision of a few nanometers to reveal dynein’s stepping behavior. In contrast to a previous report of very large steps of cytoplasmic dynein under low load (24–32 nm) (Mallik et al., 2004), we find that dynein, like Kinesin-1, moves primarily in 8 nm increments through alternating movements of its two motor domains. However, unlike Kinesin-1, we find that dynein steps are variable in their size and direction. On the basis of these findings, we provide a molecular model for how processive motion is achieved by cytoplasmic dynein.
RESULTS
Native Yeast Cytoplasmic Dynein Is a Processive Motor
For dissection of the structural basis of dynein processivity, it is essential to develop a system in which one can readily mutate or engineer the large dynein heavy-chain gene and produce biochemical quantities of protein for analysis. Previously, we have shown that S. cerevisiae affords an attractive system for manipulating the nonessential genomic copy of DYN1 by homologous recombination (Reck-Peterson and Vale, 2004). In this current study, we purified full-length cytoplasmic dynein from S. cerevisiae by using an NH2-terminal affinity tag (Figures 1A and 1B; Supplemental Data available online). We also inserted a COOH- or NH2-terminal HaloTag, a 26 kDa protein tag that enables covalent linkage of fluorescent dyes or biotin. These tags did not affect the in vivo function of yeast cytoplasmic dynein, as revealed by a nuclear segregation assay (Figure 1C). Full-length purified yeast dynein (Dyn1471 kDa; Figure 1B), which contains several associated subunits (Figure S1), bound to and released from microtubules and elicited in vitro microtubule gliding at 93 ± 27 nm/s (mean ± standard deviation [SD]; n = 302). This velocity is an order of magnitude slower than that described for cytoplasmic dyneins from other species, but exceeds the rates of dynein-dependent microtubule sliding in living S. cerevisiae (20 nm/s) (Adames and Cooper, 2000).
Several previous studies examined the movement of artificial beads with different numbers of bound dynein molecules and concluded via statistical methods that a single dynein can take multiple steps per encounter with its microtubule track (King and Schroer, 2000; Mallik et al., 2004; Wang et al., 1995). To observe processive movement of the dynein motor directly, we fluorescently labeled dynein and examined single molecules by using total internal reflection fluorescence (TIRF) microscopy. Instead of using GFP, which is suboptimal for single-molecule fluorescence studies because of its low quantum efficiency, blinking behavior, and photobleaching rate, we covalently linked tetramethylrhodamine (TMR) to a COOH-terminal HaloTag. Single-molecule imaging was confirmed by observation of single- or two-step photobleaching of TMR-labeled dynein (Figure S2A). Single TMR-labeled dynein molecules attached to and moved along sea urchin axonemes with a velocity of 85 ± 30 nm/s (mean ± SD; Figure 1D; Movie S1). A histogram of the run lengths (Figure 1E) revealed an exponential distribution with a decay constant of 1.9 ± 0.2 μm (defined as the mean run length, Figure S3). We also observed processive movement of dynein on porcine brain microtubules; the mean run length (1.7 ± 0.2 μm) and velocity (90 ± 32 nm/s) were comparable to those on axonemes (Figure S4; Movie S2).
Dimerization of the Dynein Heavy Chains Is Required for Processivity
We next examined whether one or two motor domains are required for processivity. To address this question, we engineered a monomeric cytoplasmic dynein motor by truncating NH2-terminal sequences that are responsible for dimerization. This monomeric dynein (named Dyn1331 kDa on the basis of its predicted molecular weight of 331 kDa) is truncated at the equivalent site to the 380 kDa truncated dynein constructs from Dictyostelium and mammalian cytoplasmic dynein (Hook et al., 2005; Koonce and Samso, 1996; Nishiura et al., 2004), although the yeast protein is shorter than these counterparts because it lacks sequence at its COOH terminus. As shown previously for the 380 kDa Dictyostelium dynein (Nishiura et al., 2004), Dyn1331 kDa is a minus-end-directed motor in a multiple-motor microtubule gliding assay (65 ± 12 nm/s; Figure 3E). However, when individual Dyn1331 kDa molecules were examined by TIRF microscopy, processive motion was not observed (not shown).
Figure 3. Defining the Minimal Processive Dynein Motor.
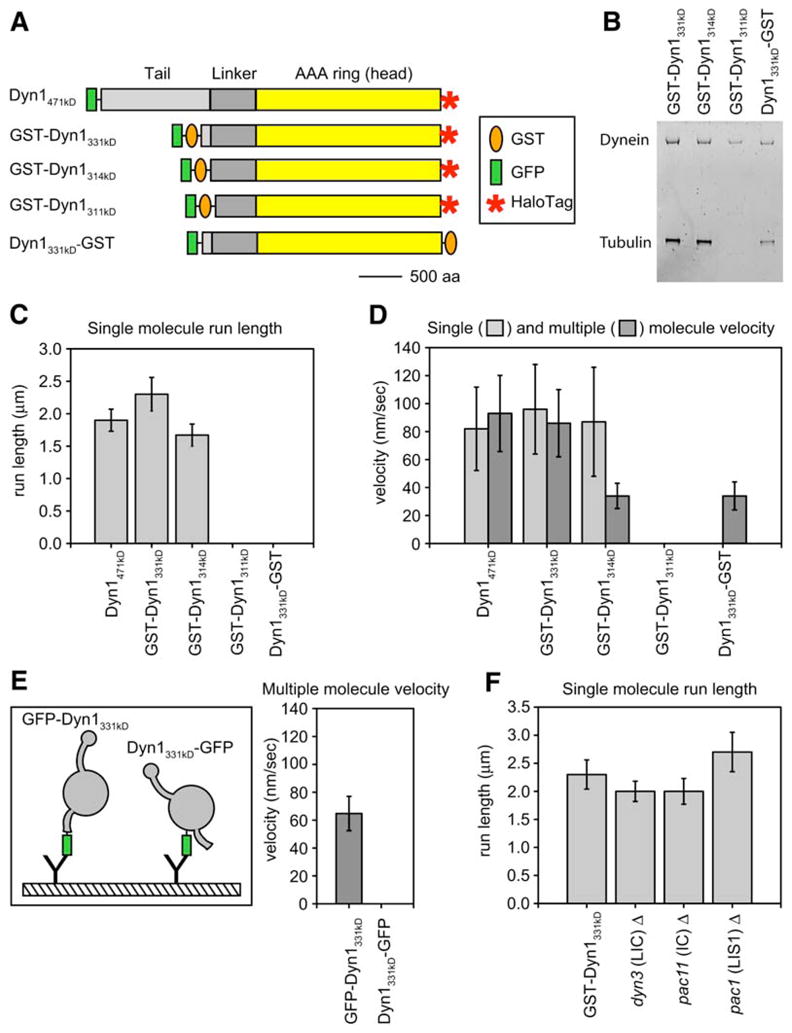
(A) Schematic showing dynein heavy-chain truncations and tags. A HaloTag at the COOH terminus allowed attachment of TMR for single-molecule assays. The orange oval represents GST, which was used to dimerize monomeric constructs either at the NH2 or the COOH terminus. GFP (green rectangle) was added to the NH2 terminus of all proteins and was used to anchor dynein molecules to glass with a GFP antibody in microtubule gliding assays. Here we functionally define the term “linker” as the ~400 amino acids, located NH2-terminal to the start of the AAA domain, that we have found necessary for dynein motility. The scale bar represents 500 aa.
(B) SYPRO-ruby (Molecular Probes)-stained protein gel of the truncated dynein proteins. The gel shows GST-Dyn1331 kDa, GST-Dyn1314 kDa, and Dyn1331 kDa-GST after IgG and microtubule affinity-purification steps, and GST-Dyn1311 kDa is shown after the first affinity-purification step because this protein releases poorly from microtubules in the presence of ATP.
(C) The mean single-molecule run length ± SE for each of the dynein constructs is shown. GST-Dyn1311 kDa and Dyn1331 kDa-GST both bound to microtubules in the single-molecule assay, but no moving molecules were observed.
(D) Mean single (light gray) and multiple motor (dark gray) velocity ± SD for each of the dynein constructs. GST-Dyn1311 kDa bound to microtubules and axonemes, but showed no motility in either assay. Dyn1331 kDa-GST bound, but was not capable of moving on, axonemes in the single-molecule assay.
(E) The tail domain is required for the dynein power stroke. Monomeric Dyn1331 kDa was anchored to glass for microtubule gliding assays via GFP attached either to its tail (NH2 terminus; GFP-Dyn1331 kDa) or end of its motor domain (COOH terminus; Dyn1331 kDa-GFP). GFP-Dyn1331 kDa moved microtubules along the glass (mean ± SD is shown), whereas Dyn1331 kDa-GFP could bind microtubules but not move them under our standard buffer conditions (including 50 mM KAcetate). Under higher ionic strength conditions (300 mM KAcetate), very slow (3 ± 1 nm/s) movement of Dyn1331 kDa-GFP was observed.
(F) GST-Dyn1331 kDa purified from yeast strains lacking the light intermediate chain (Dyn3), the intermediate chain (Pac11), or the Lis 1 protein (Pac1) shows a similar run length to a yeast strain that does not harbor these deletions (GST-Dyn1331 kDa). The bar graph shows the mean single-molecule run length ± SE for each strain background (Figure S3). The single-molecule velocities for dynein purified from each of these yeast strains was also similar to the control yeast strain (mean velocity ± SD = 103 ± 32, 108 ± 33, 98 ± 46, and 98 ± 32 nm/s for GST- Dyn1331 kDa, dyn3Δ, pac11Δ, and pac1Δ, respectively). n values for the data in this figure are between 184 and 334.
The lack of processive motion of Dyn1331 kDa suggested that a dimer of two motor domains may be needed for processivity. To examine this possibility, we produced a dynein whose monomer-dimer state could be controlled by a small molecule. Dyn1331 kDa was fused at the NH2 terminus (tail) to either the FRB or FKBP12 (referred to as FKBP) domain, which can be crosslinked by the natural product rapamycin to create a FRB-FKBP heterodimer (Ho et al., 1996). In the absence of FKBP-Dyn1331 kDa, TMR-labeled FRB-Dyn1331 kDa rarely displayed processive movement (Figure 2A, Movie S3) either with or without rapamycin. Photobleaching studies indeed confirmed that FRB-Dyn1331 kDa is monomeric under the conditions of this assay (Figure S2B). After the addition of FKBP-Dyn1331 kDa and rapamycin, TMR-labeled FRB-Dyn1331 kDa exhibited robust processive movement (Figure 2B, Movie S3). The frequency of processive movement of FKBP-Dyn1331 kDa-rapamycin-FRB-Dyn1331 kDa was ~300-fold greater than FRB-Dyn1331 kDa alone. (The very rare movements of FRB-Dyn1331 kDa [~1 event per 15 μm axoneme per 15 min of observation] could be attributable to a rare dimer or aggregate in the population.) The velocity of FKBP-rapamycin-FRB-Dyn1331 kDa (64 ± 32 nm/s) and run length (1.2 ± 0.1 μm) were slightly lower than those of full-length dynein. The slightly lower run length might be due to the release of rapamycin [the dissociation constant of the FKBP-rapamycin-FRB ternary complex to FKBP-rapamycin and FRB is 12 nM (Banaszynski et al., 2005)], which would be expected to terminate a run by dissociating the dynein dimer. These experiments showing rapamycin- based control of dynein processivity indicate that a dimer of two dynein motor domains is required for processivity and that the NH2-terminal ~200 kDa of the dynein heavy chain is not required for this activity.
Figure 2. Artificial Dimerization of Dynein Monomers Induces Processive Movement.

(A) TMR-labeled FRB-Dyn1331 kDa plus rapamycin did not move processively in the single-molecule TIRF assay. Kymographs were made by horizontally stacking line scans along the axonemal axis. Some stationary dynein molecules were seen (vertical lines), but most molecules bound transiently, and no moving molecules were observed (see diagonal lines in Figure 2B).
(B) Dimerization of TMR-labeled FRB-Dyn1331 kDa and FKBP12-Dyn1331 kDa by rapamycin produces processive runs in the single-molecule TIRF assay. Diagonal lines in the kymograph represent dynein molecules that are moving along the axoneme over time (see also Movie S3). A histogram of rapamycin-dimerized dynein run lengths shows a run length of 1.2 ± 0.1 μm (mean ± SE; n = 223, Figure S3).
A Minimal Dynein Motor that Displays Processive Motion
We next sought to make a stable dynein homodimer that exhibits processive motion and define a minimal motor that displays processive motion. To accomplish this, we placed glutathione S-transferase (GST), which forms a constitutive homodimer, at the NH2 terminus (tail) of Dyn1331 kDa (Figure 3A). TMR-labeled GST-Dyn1331 kDa exhibited robust processive movement in the single-molecule TIRF assay with a run length of 2.3 ± 0.3 μm and a velocity of 102 ± 32 nm/s (Figures 3B and 3C; Movie S4). These values are similar to those of full-length dynein and indicate that a nonnative dimerization domain can produce a fully functional motor.
To further define the minimal size of a dynein motor that enables processive movement, we truncated an additional 145 aa (GST-Dyn1314 kDa) and 171 aa (GST-Dyn1311 kDa) from the NH2-terminal motor sequence of GST-Dyn1331 kDa (Figures 3A and 3B; see Figure S6 for a detailed sequence alignment). GST-Dyn1314 kDa was processive in the single-molecule TIRF assay with a run length of 1.7 ± 0.2 μm and a velocity of 87 ± 39 nm/s (Figure 3C). However, the removal of an additional 26 amino acids produced a motor (GST-Dyn1311 kDa) that could bind to axonemes in the single-molecule TIRF assay, but was neither processive (Figure 3C) nor produced movement in a multiple-motor microtubule gliding assay (Figure 3D). In contrast to GST-Dyn1331 kDa, which showed robust microtubule-stimulated ATPase activity (kcat, 16.1 ± 0.3 ATP/dimer/s, Km MT, 0.39 ± 0.06 μM tubulin dimer; Figure S5), GST-Dyn1311 kDa exhibited 4-fold lower basal ATPase activity and did not display microtubule-stimulated ATPase activity (Figure S5). We have also found that the majority of GST-Dyn1311 kDa dynein molecules exhibit 2-step photobleaching, indicating that its impaired activity is not due to lack of dimerization (data not shown). Additional truncations beyond GST-Dyn1311 kDa also did not produce active motor proteins (data not shown). Thus, we conclude that the 26 amino acids that separate GST-Dyn1314 kDa and GST-Dyn1311 kDa are essential for motility and microtubule-stimulated ATPase activity and that the GST-Dyn1314 kDa construct represents a minimal functional dynein motor.
To explore whether dynein needs to be dimerized via its NH2 terminus or COOH terminus, we attached GST to the COOH terminus of the AAA ring (Dyn1331 kDa-GST) rather than the NH2 terminus (tail). Dyn1331 kDa-GST did not move in the single-molecule TIRF assay, but, when tethered to the glass via its NH2-terminal GFP tag, was capable of moving microtubules in a multiple-motor gliding assay and displayed microtubule-stimulated ATPase activity (kcat 4.3 ± 0.3 ATP/dimer/s; Figures 3C and 3D; Figure S5). In addition, we find that Dyn1331 kDa-GST forms a dimer by sucrose gradient centrifugation (Dyn1331 kDa-GST and GST-Dyn1331 kDa have an S value of ~15S, while monomeric Dyn1331 kDa has an S value of ~10S). Thus, COOH-terminal GST dimerization does not permit coordinated processive motion, but does not inactivate the motor.
Recent structural experiments have shown that the NH2-terminal linker domain of dynein undergoes ATP-dependent movements that have been hypothesized to produce a dynein power stroke (Burgess et al., 2003; Kon et al., 2005). To functionally test the idea that a movement of the linker domain is involved in the power stroke, we attached GFP to the COOH terminus (Dyn1331 kDa-GFP) of a dynein monomer rather than the NH2 terminus (GFP-Dyn1331 kDa). When attached to glass via the end of its motor domain, Dyn1331 kDa-GFP bound to, but did not move, microtubules in a multiple-motor gliding assay under our standard buffer conditions (containing 50 mM KAcetate), but moved very slowly (3 ± 1 nm/s) at higher ionic strength (300 mM KAcetate). In contrast, GFP-Dyn1331 kDa tethered via its linker domain was capable of movement (Figure 3E). Thus, when the anchor point for motility is moved from the linker to the COOH terminus of the dynein ring, motility is nearly abolished. These results suggest that the dynein linker domain, and not the elongate microtubule binding stalk or an element of the AAA ring, is the primary transmitter of mechanical motion (see Discussion).
We next wished to determine whether dynein-associated proteins are required for processive movement. A number of associated proteins are essential for yeast cytoplasmic dynein’s in vivo function, including the intermediate chain (Pac11), the light intermediate chain (Dyn3), light chain (Dyn2), the dynactin complex (Nip100, Jnm1, Arp1, Arp11, Cap1, and Cap2), and the lissencephaly 1/Lis1 (Pac1) and nudel (Ndl) proteins. Because the truncated dynein dimer GST-Dyn1331 kDa is overexpressed behind the galactose promoter, any chains that might copurify are unlikely to be present stoichiometrically. Nevertheless, we purified GST-Dyn1331 kDa from tagged yeast strains and performed immunoblots to probe for the presence of the dynein-associated subunits. We found no evidence for copurification of the p150glued subunit (Nip100) of the dynactin complex with GST-Dyn1331 kDa (Figure S1). The dynein light intermediate chain and the dynein intermediate chain were present after the first step of purification, but were not detectable after the second step of purification (microtubule affinity), whereas the Lis 1 protein was detectable after both purification steps (Figure S1). Because we are working at the single-molecule level in our TIRF assays, it remained possible that we were observing movement of rare dynein molecules with associated dynein light intermediate or intermediate chains that were present at levels below the detection of immunoblot. Thus, to ask whether the Lis 1 protein [which recruits Nudel (Li et al., 2005)] or the dynein light intermediate or intermediate [which recruits the dynactin complex and the dynein light chain (Karki and Holzbaur, 1995; Lo et al., 2001)] chains are required for processivity, we purified GST-Dyn1331 kDa from yeast strains with gene deletions in these associated subunits. GST-Dyn1331 kDa remained processive with a similar run length and velocity when purified from yeast strains lacking Lis 1 or the light intermediate or intermediate chains (Figure 3F). Thus, we conclude that dynein processivity does not require any of the known dynein-associated subunits in the yeast genome.
Stepping Behavior of Quantum-Dot-Labeled Dynein
To learn more about the mechanism of cytoplasmic dynein processivity, we tracked individual fluorescently labeled GST-Dyn1331 kDa molecules with high spatial precision to observe their stepping behavior (Yildiz and Selvin, 2005). In this method, light emitted from a single fluorescent molecule is collected with a sensitive CCD camera, and the point-spread function of the image is fit with a 2D Gaussian distribution; the center of the Gaussian distribution can be determined with a precision of a few nanometers and tracked over time. To collect as many photons as possible for precise tracking and to minimize photobleaching, we labeled GST-Dyn1331 kDa with a bright quantum dot (Qdot), attaching it via a streptavidin-biotin link to the HaloTag under conditions that prevent aggregation (see Supplemental Data).
We first placed the Qdot at the NH2-terminal tail to observe the presumed center-of-mass movements of the motor. To best observe stepping behavior, we decreased the motor speed by reducing the ATP concentration to 4 μM. With the high photostability of the Qdot, GST-Dyn1331 kDa movement could be tracked for several minutes, and step-wise movement was evident in the traces (Figure 4A, Figure S7). The displacement fluctuations of the Qdot when dynein paused in between steps varied, but in favorable cases was ~2 nm. These fluctuations were comparable to that observed for Qdot-GST-Dyn1331 kDa bound in rigor to axonemes (2.5 nm) and exceeded that of a Qdot fixed onto a glass surface (1.7 nm; reflecting the noise inherent in our measurements). Several general features of dynein movement were apparent from high-precision spatial tracking. First, the majority of dynein molecules showed clear step-wise movement, and typically >50 steps can be discerned in the movement of a single dynein. Second, although the motion was primarily unidirectional, every dynein molecule analyzed (n > 80) exhibited backward stepping, which is rarely displayed by Kinesin-1 or myosin V under low load (Yildiz et al., 2003, 2004). Usually, only one or two consecutive backward steps were observed, although one record showed a 70 nm backward run comprising eight successive steps (top trace in Figure S7). A third obvious feature of the traces is a considerable variation in step size, with small (~8 nm) and large (12–24 nm) steps appearing in the same records. It is unlikely that the larger steps are due to successive quick steps taken by the motor, because the average dwell time (~2 s) greatly exceeds the temporal resolution of our measurements (70 ms).
Figure 4. Stepping Behavior of Tail- or Head-Labeled GST-Dyn1331 kDa.

(A)GST-Dyn1331 kDa labeled with a Qdot at its NH2 terminus (center of mass of the dimer) takes discrete steps along axonemal microtubule tracks. The raw data are shown with black circles and lines connecting the data points, and steps detected by a step finding program (see Experimental Procedures) are shown in red (step size values are noted in red). Data was acquired at 4 μM ATP every 70 ms. Traces from four different dynein molecules are shown and more representative examples can be found in Figure S7.
(B) A single Qdot positioned at the COOH terminus of one motor domain in the GST- Dyn1331 kDa dimer shows larger steps than NH2-terminally labeled GST-Dyn1331 kDa. Data were acquired at 10 μM ATP every 100 ms. Traces from four different dynein molecules are shown, and more representative examples can be found in Figure S8.
To ensure objective and unbiased step-size analysis, we applied a step-finding algorithm to our stepping traces (Kerssemakers et al., 2006). This program assumes that steps are hidden in normal-distributed noise, but makes no assumptions of step size or duration. A histogram of the step sizes of tail-labeled dynein revealed a major peak at ~8 nm, which is similar to the distance between adjacent tubulin dimers in a microtubule protofilament (8.3 nm; Figure 5A). However, there was a trailing shoulder of larger steps (Figure 5A). A population of backward steps (8–16 nm) was also observed, which constituted 20% of the total steps measured.
Figure 5. Histograms of Dynein Step Sizes and Dwell Times.

(A) A histogram of tail-labeled Qdot-GST-Dyn1331 kDa step sizes reveals a major peak at ~8 nm with a considerable tail of longer steps. Included in the histogram are 1342 steps from 27 moving dynein molecules. Backward steps make up 20% of the total. A histogram of dwell times between steps is fit with a single exponential function, and the decay constant reveals a stepping rate (k) of 0.13 ± 0.01 per second per μM ATP.
(B) A histogram of head-labeled GST-Dyn1331 kDa-Qdot step sizes reveals a major peak at ~16–18 nm with a considerable tail of longer steps. Included in the histogram are 1690 steps from 29 moving dynein molecules. Backward steps make up 13% of the total steps. The dwell-time histogram is fit with a convolution of two exponential functions with equal decay constants (the fit indicates a dynein stepping rate (k) of 0.14 ± 0.01 per second per μMATP).
We next tracked the stepping behavior of a single dynein motor domain by labeling the COOH terminus of GST-Dyn1331 kDa with a Qdot, which places the label adjacent to the AAA6 domain of the motor ring (termed head-labeled dynein). In our labeling conditions, only one head per dimer was labeled with a Qdot (see Supplemental Data). Labeling dynein with a Qdot on its head domain did not affect its function, as indicated by normal single-molecule velocity at 1 mM ATP (87 ± 44 nm/s, mean ± SD, n = 129). Stepwise displacement of head-labeled GST-Dyn1331 kDa was obvious in virtually all records of dynein motility (Figure 4B, Figure S8). Similar to the tail-labeled dynein, a variety of step sizes as well as backward steps were observed (Figure 4B, Figure S8). A heterodimer of Dyn1331 kDa labeled with TMR on a single head domain (with the FRB-rapamycin-FKBP dimerization system) moving on either axonemes or microtubules showed similar stepping to a dynein motor labeled with a Qdot (Figures S10A and S10B), indicating that neither the Qdot nor axonemes perturb the basic behavior of the motor.
The histogram of step sizes for a Qdot-labeled head domain in GST-Dyn1331 kDa revealed a major peak at ~16 nm with a broad shoulder of larger steps, as well as a broad distribution of backward steps (13% of total; Figure 5B). A similar histogram of step sizes was observed for head-labeled native dynein (Figure S9). Thus, the step size of a single head domain in a dimer is approximately 2-fold greater than the step size of the presumed center of mass.
Dwell-Time Analysis of the Dynein Stepping
The approximately 2-fold-greater step size exhibited by individual dynein heads compared with the presumed center- of-mass step size can be explained by a model in which one head remains bound to a tubulin subunit while the partner head detaches, moves past its stationary partner head, and then binds to a new tubulin binding site toward the microtubule minus end, as has been suggested for myosin V and Kinesin-1 (Yildiz et al., 2003, 2004). In this model, the center of mass moves during each ATPase cycle, whereas an individual head moves only once per two ATPase cycles (moving past a stationary forward head in one ATPase cycle, but remaining stationary itself in the next cycle). Corroborating evidence for this model came from the findings that the dwell-time distribution for the center-of-mass steps at low ATP concentrations could be fit to a single exponential (consistent with a single ATP binding event driving a step), whereas the dwell-time distribution for head movements were best fit by a convolution of two exponentials (consistent with two ATP binding events driving a step) (Yildiz et al., 2003, 2004).
The histogram of dwell times of tail-labeled GST-Dyn1331 kDa (derived from the automated step-detection program) fits very well to a single-exponential distribution (Figure 5A). In contrast, a dwell-time histogram of the single head-labeled dynein (GST-Dyn1331 kDa or native Dyn1) was best fit by a convolution of two exponentials (Figure 5B, Figure S9). Although alternative mechanisms are possible (see Discussion; Figure S11), the distinct dwell-time distributions of center-of-mass- and head-labeled dyneins is best explained by an alternating front-rear stepping pattern for the two motor domains in the dynein dimer.
Dynein Can Step Laterally as well as Forward
Past work has shown that dynein-coated beads make more lateral movements than kinesin-coated beads (Wang et al., 1995). With single-molecule fluorescence tracking, we could now explore this behavior with high spatial precision and without bead attachment. With head-labeled GST-Dyn1331 kDa-Qdot, we observed clear “jumps” perpendicular to the microtubule axis of dynein, which most likely represent a displacement to a neighboring protofilament (Figures 6C and 6D). One concern might be that such off-axis displacements are a result of the relatively large Qdot. However, kinesin labeled with a Qdot did not show substantial off-axis displacement (Figure S10C), and GST-Dyn1331 kDa labeled with TMR on a single-motor domain also showed off-axis stepping similar to Qdot-labeled dynein (Figure S10A). Qdot-labeled dynein moving on microtubules also displayed off-axis steps, indicating that such movements are not limited to dynein moving on axonemes (Figure S10B). Careful analysis of the off-axis steps of GST-Dyn1331 kDa-Qdot revealed that the off-axis steps usually occurred simultaneously with an on-axis step (Figures 6C and 6D). This result indicates that as the dynein motor domain takes a forward step, it has the reach or flexibility to occasionally land on an adjacent protofilament, or, in the case of an axoneme, possibly even a neighboring microtubule. However, off-axis stepping was less common than on-axis stepping. Of 1690 forward steps scored from 29 dynein molecules (all molecules showing some off-axis stepping), 298 off-axis steps were observed (ranging in size from 4–40 nm), representing 18% of the steps. In conclusion, unlike Kinesin-1, we find that cytoplasmic dynein has the capability to reach laterally to an adjacent protofilament as it takes a forward step.
Figure 6. Dynein Can Step to Neighboring Protofilaments.

(A) Diagram of an axoneme and the direction of on-axis (red arrow) and off-axis (blue arrow) movement.
(B–D) Three examples of on-axis (parallel to the microtubule long axis) and off-axis (perpendicular to the microtubule long axis) stepping by head-labeled GST-Dyn1331 kDa-Qdot. (B) shows an example of no off-axis stepping, (C) shows the most typical trace of a few off-axis steps, and (D) shows a rare trace of many large off-axis displacements. On-axis stepping position is represented by the black circles (raw data), and the red line shows a fit with the step-finding program, whereas off-axis stepping position is represented by blue circles (raw data) and lines. Dashed orange lines illustrate that on- and off-axis steps occur simultaneously. The panels on the right depict off-axis (x axis) versus on-axis (y axis) movements. The direction of movement is toward the top of the page. Each step is represented by a black circle, and the steps are connected with a black line. See Figure S10 for additional traces and discussion of off-axis displacement.
DISCUSSION
The combination of single-molecule assays and protein engineering have contributed significantly to our understanding of kinesin and myosin motility. Dynein’s large size has hindered comparable efforts, although pioneering studies have led to the production of a functional dynein motor domain in Dicytostelium (Nishiura et al., 2004) and insect cells (Hook et al., 2005). Here, we have used S. cerevisiae to produce the first functional recombinant dimeric dynein (both full-length as well as truncated, artificially dimerized motors) and have developed the first single-molecule fluorescence assays to observe processive motion of dynein directly. The combination of these approaches has allowed us to dissect the structural basis of dynein processivity and to develop strategies for linking fluorophores to specific sites on dynein to enable nanometer-precision stepping analysis of cytoplasmic dynein in the absence of load. The results derived from these structure- function and single-molecule fluorescence motility studies allow us to propose the first model for how dimeric, processive dynein moves along its microtubule track.
Defining a Minimal Motor Domain for Movement and Processivity
Previous work has established that a monomeric 380 kDa Dictyostelium (331 kDa for S. cerevisiae) dynein construct is a functional motor in a multiple-motor motility assay (Nishiura et al., 2004). Our work has further defined the minimal motor domain of dynein. Because S. cerevisiae dynein lacks the COOH-terminal 285 aa (32 kDa) found in Dictyostelium and other dyneins, this region does not appear to be essential for movement. Moreover, we show that NH2-terminal truncation to aa 1390 (a 314 kDa motor, equivalent to 365 kDa for the Dictyostelium dynein) does not substantially perturb motility. However, removing an additional 26 amino acids (Dyn1311 kDa) causes a complete loss of motility, thus establishing Dyn1314 kDa as reasonably close to the minimal size motor for dynein motility. Dyn1314 kDa lacks a number of highly conserved sequence motifs (Figure S6), which must serve some role but do not appear to be essential for movement. Interestingly, the NH2 terminus of Dyn1314 kDa is still 394 aa (46 kDa) from the beginning of the first AAA domain. A likely interpretation is that this ~400 aa sequence forms the “linker” domain, which has been proposed to wrap around the AAA ring and undergo an ATP-driven power stroke (Burgess et al., 2003; Kon et al., 2005). Truncation of the 26 aa that are missing in Dyn1311 kDa may interfere with the ability of the linker domain to either interact with the AAA ring or undergo a conformational change. Also consistent with the linker/AAA ring powering a dynein power stroke (Burgess et al., 2003), we show that dynein anchored to the glass NH2-terminal to the linker enables microtubule gliding, whereas anchoring the same construct to the glass via the COOH terminus of the AAA ring drastically impairs dynein’s ability to transmit movement to the microtubule.
This work also has established that two motor domains are required for cytoplasmic dynein processivity. Unlike inner-arm axonemal dynein bound to beads, which was reported to show processive movement as a monomer (Sakakibara et al., 1999), we find that cytoplasmic dynein monomers are incapable of processive movement. However, dimerization of monomeric constructs can restore processivity, and the minimal size construct that enables monomer-based movement (Dyn1314 kDa) also is sufficient for processive motion when artificially dimerized. Interestingly, the native dynein dimerization domain is not required for processivity and can be substituted by two structurally distinct mechanisms of dimerization, either GST homodimerization or chemically induced heterodimerization using FKBP-rapamycin-FRB, both of which enable processive motion that is very similar to that generated by native dynein. In contrast, normal processivity by Kinesin-1 requires the precise positioning of a coiled coil immediately after its mechanical element (the neck linker); substitution of this coiled coil with FKBP-rapamycin- FRB does not enable Kinesin-1 to move processively (M. Tomishige and R.D.V., unpublished data). We also show that processivity requires placement of the dimerization domain NH2-terminal to the linker domain and not COOH terminal to the AAA ring. This result indicates that the linker domains must be connected to enable head-head communication for processive motion.
We show that dynein-associated proteins (dynein light, light intermediate, and intermediate chains and Lis 1, nudel, and dynactin) are not required for processive movement, thereby simplifying investigations of the basic mechanism of dynein processivity. However, it is possible that the associated proteins could enhance processivity, as has been reported for brain dynein in the presence of the dynactin complex in vitro (King and Schroer, 2000).
Stepping Behavior of Cytoplasmic Dynein
Mallik et al. (2004) have reported that single mammalian dynein molecules primarily take large steps (24–32 nm) at low load, but advance in 8 nm steps when multiple dyneins interact with a microtubule and contribute to movement (Mallik et al., 2005). Another study has reported an 8 nm step size of cytoplasmic dynein carrying peroxisomes in living cells (Kural et al., 2005). However, the numbers of dynein molecules involved in this movement could not be established or controlled in this in vivo study, and it was difficult to establish whether plus-end-directed steps were caused by a kinesin stepping forward or a dynein stepping backward. While this paper was under review, Toba et al. (2006) reported a regular 8 nm step size of mammalian cytoplasmic dynein carrying beads in an optical trap or with an attached Qdot. Thus, cytoplasmic dynein stepping behavior remains controversial.
Here, using covalently attached Qdots at defined locations in the molecule, we have been able to collect more stepping data than in previous studies. The majority of dynein molecules show clear stepping behavior, and within an individual run, many steps (>50) can be observed enabling robust statistical analysis (n > 3500 steps for the dynein’s reported in Figures 4 and 5 and Figures S7, S8, and S9). The results of our analysis differ from those of Mallik et al. (2004) and Toba et al. (2006) as we observe primarily 8 nm steps, as well as a broad shoulder of larger step sizes. Moreover, Toba et al. randomly labeled dynein with Qdots with a nonspecific crosslinker, in which case head labeling should be similar to tail labeling on the basis of their relative masses. Therefore, our results with site-specific labeling of dynein would predict a much greater frequency of 16 nm and >16 nm steps than was reported by Toba et al. Further work will be required to resolve these differences in reported dynein stepping behavior, with particular attention paid to possible species variation in dynein, protein preparation, and assay/labeling conditions.
Analysis of the dwell times between steps can provide information on the number of ATPs hydrolyzed per step. Our dwell-time histogram for the tail-labeled dynein at low ATP concentrations is fit by a single exponential, which suggests that steps are triggered by the binding of a single ATP molecule. A similar finding was reported for brain cytoplasmic dynein stepping in an optical trap (Mallik et al., 2004; Toba et al., 2006). Our measured ATPase rates (kcat of ~16 Pi/s/dimer) and stepping measurements (~8 nm steps with a 0.8 probability of forward stepping) also are consistent with a single ATP hydrolysis per step producing our measured velocities of movement (~100 nm/s) assuming all motors in our preparation are active. These results are intriguing, since dynein contains four ATP binding sites and mutagenesis studies have shown that ATP binding and hydrolysis at two sites (AAA1 and AAA3) are essential for dynein function in vivo (Reck-Peterson and Vale, 2004; Silvanovich et al., 2003) and in vitro (Kon et al., 2005). It is still unclear how the single- exponential distribution of dwell times can be reconciled with the mutagenesis studies, although one possible explanation is that more than one ATP binds per step but with considerably different association rates.
Our dynein stepping data also reveal interesting similarities and differences with other cytoskeletal motors. Like kinesin, dynein can take 8 nm steps along a microtubule track. However, kinesin motion at low load is very regular, exhibiting little variation in step size and very few backward steps. In contrast, cytoplasmic dynein exhibits a broad step-size distribution and frequently makes backward and sideways steps. This behavior is most reminiscent of myosin VI, which also shows a broad step-size distribution as well as occasional backward steps at low load [e.g., (Rock et al., 2005)]. The variation in myosin VI step sizes is thought to be due to flexibility in its “neck” domain, which allows Brownian excursions to several potential actin binding sites (Rock et al., 2005). A similar model may explain the variation in dynein step sizes, as discussed below.
Models for Cytoplasmic Dynein Motility
By selectively placing fluorescent labels in the dynein molecule, we have discovered that the dynein tail (center of mass of the dimer) takes steps that are ~2 fold smaller than the head. In addition, the dwell-time distribution of tail-labeled dynein is fit by a single exponential, whereas the dwell-time distribution of head-labeled dynein is best fit by a convolution of two exponentials. Taken together, these data are consistent with two possible models for dynein stepping (Figure 7, Figure S11). In the first model, the two dynein heads alternate taking 16 nm steps, whereas the centroid position of the molecule moves by 8 nm for each step (Figure 7). We term this motion “alternating shuffling” rather than “hand-over-hand” movement, since the large dimensions of the AAA ring make it unlikely that the heads are completely separated into nonoverlapping forward and rear positions as is true of Kinesin-1 and myosin V (Figure 7; see discussion below). In the second model (“asymmetric inchworm”) (Hua et al., 2002), a single head always takes the leading position, extending forward to a new binding site while the rear head remains stationary, and then the rear head catches up in the next cycle (Figure S11). We disfavor the asymmetric inchworm model (Figure 7C, Figure S11B), however, because it requires a very large separation of the dynein heads in order for the tail to take >8 nm steps. For example, to achieve a 24 nm center-of-mass step (seen in our traces) with an asymmetric inchworm mechanism, the centers of the two dynein heads would have to separate by 48 nm, thus requiring the linkers to extend by 36 nm between the rings and the central dimerization domain (Figure S11B). Although separation of the two dynein heads may be possible (discussed below), it seems unlikely that the linker regions in GST-Dyn1331 kDa could accommodate such a large head-head separation (Burgess et al., 2003; Samso and Koonce, 2004). However, a final proof of the alternating shuffling mechanism will require labeling of each head with different fluorophores, as has been done for Myosin V (Churchman et al., 2005; Warshaw et al., 2005). We have attempted this experiment with dynein, but have thus far not been able to label a single molecule with two different Qdots (see Supplemental Data).
Figure 7. Model for Processive Movement of Dimeric Cytoplasmic Dynein.
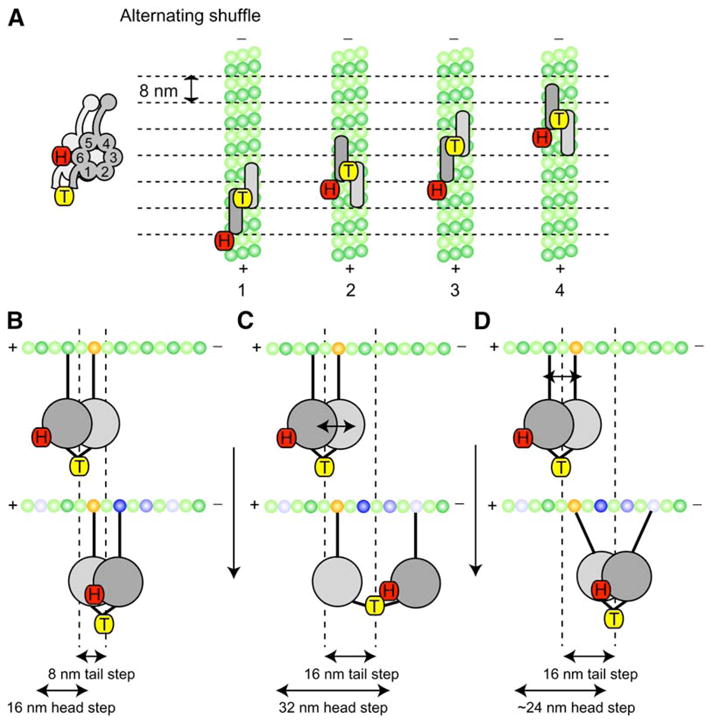
A stepping model for cytoplasmic dynein that takes into account the step-size and dwell-time data for head- and tail-labeled dynein. The head (COOH terminus)- labeled dynein (H) is represented by the red tag, and the center- of-mass-labeled dynein (T) is represented by the yellow tags (the AAA ring is shown schematically from this top view as a rectangle with rounded edges; the AAA rings and tubulin dimers are shown approximately to scale).
(A) In this Alternating Shuffle model, the two dynein motor domains alternate between forward and rear positions; the tail-labeled dynein takes predominantly successive 8 nm steps (the distance between tubulin dimers). In contrast, a single dynein motor domain takes a 16 nm step and then remains stationary (a 0 nm step) as its partner ring advances. Structural considerations suggest that the rings may align parallel to one another and partially overlap (see Discussion). Although dynein is shown here moving along a single protofilament, we have shown that dynein can also move on neighboring protofilaments (see Figure 6).
(B–D) Another view of the Alternating Shuffle model illustrating how dynein could take 8 nm as well as 16 nm steps from either a greater separation of the motor rings (C) or angular movements of the microtubule binding stalks (D) (a combination of these mechanisms is also possible). In (B)–(D), possible tubulin binding sites (both forward and rearward) are indicated by the blue-shaded tubulin subunits, with the darkest blue representing the most frequently observed step size of 8 nm.
A stepping model also must take into account the dimensions of the dynein molecule and the step sizes. The dimensions of the dynein ring (13–15 nm in diameter) and stalk (10–15 nm in length) are large compared to the spacing of tubulin subunits (8.3 nm) (Burgess et al., 2003; Samso and Koonce, 2004). Whereas dimeric kinesin needs to extend via its neck linkers to span the distance between tubulin subunits, the large dynein dimer may need to adopt a compact form to constrain itself to taking 8 nm steps. A compact dynein dimer could be achieved by overlapping the dynein rings, which are only 4 nm in width (Burgess et al., 2003; Samso and Koonce, 2004), parallel to the microtubule axis (Figure 7B). Consistent with this idea, electron microscopy of axonemal dyneins suggests that the motor rings may indeed stack (Lupetti et al., 2005; Nicastro et al., 2005), analogous to other AAA+ proteins [e.g., (Fletcher et al., 2003)]. A compact dynein dimer is further supported by the fact that the artificial dimerization domains used in this study are likely to be located within a few nanometers of the ring (Kon et al., 2004). The close proximity of the rings also raises the possibility that ring-ring communication could occur, as observed for other AAA proteins (Cashikar et al., 2002; DeLaBarre and Brunger, 2003).
However, an “extended” conformation of the dynein dimer also is likely to exist, as evidenced by the relatively common occurrence of >8 nm and off-axis steps. Two mechanisms (which are not mutually exclusive) might allow dynein to extend to more distant subunits and take larger steps. First, the connection between the AAA ring and the dimerization domain might be flexible, allowing the rings to separate and the lead head to reach more distant tubulin subunits (Figure 7C). Second, angular motions of the elongate (10–15 nm) stalk [observed in EM studies (Burgess et al., 2003; Samso and Koonce, 2004)] could enable the microtubule binding domain to reach forward or laterally (Figure 7D). Stalk movements, however, do not preserve a precise 2:1 coupling of head:tail step sizes, whereas ring-ring separations do (Figure 7C versus Figure 7D). Thus, simultaneous dual-color imaging of fluorophores on the head and tail could be used to distinguish between these models.
Still lacking in our model is a structural understanding of how ATP-driven protein conformational changes cause one dynein head to advance past its partner. In Kinesin-1 and myosin V, conformational changes in the lead head-neck linker and lever arm domains, respectively, pull the rear head forward (Vale and Milligan, 2000). The dynein linker domain may serve a similar role in dynein motility. The single-molecule tools described here could be further developed to obtain information on the conformations of the linker domains in the leading and trailing heads in order to derive a more detailed structural model for dynein processivity.
EXPERIMENTAL PROCEDURES
Protein Constructs
The yeast strains used in this study are listed in Table S1. Nuclear segregation assays were performed as described (Reck-Peterson and Vale, 2004). All tagging and truncation of the dynein heavy chain was performed by homologous recombination into the genomic copy of DYN1 in haploid yeast cells. For purification and analysis, a ZZ tag (two copies of the IgG binding domain of protein A), a TEV protease cleavage site, a short linker sequence (DYDIPTTENLYFQG) that enhances the efficiency of TEV protease cleavage, GFP (used to tether dynein to glass in microtubule gliding assays), and a 3XHA tag (used to detect dynein by immunoblot) were added to the 5′ end of most dynein constructs (VY206 contained GFP at its 3′ end, and VY207 and VY209 did not contain GFP). All truncated dynein proteins were expressed behind the galactose promoter, which was placed just upstream of the purification tag, whereas full-length dynein was expressed behind its endogenous promoter. Deletion (with NAT or URA) or tagging (with 3X Flag or 13X Myc) of the dynein-associated chains (DYN3, PAC11, PAC1, and NIP100) was performed with standard techniques (Longtine et al., 1998). Further details of construct design, protein purification, and fluorescent labeling are described in the Supplemental Data.
Motility Assays
Microtubule gliding assays were performed by attaching dynein via its GFP tag to glass surfaces coated with GFP antibodies, analogous to gliding assays reported for kinesin motors (Case et al., 1997). Details are described in the Supplemental Data.
For single-molecule TIRF microscopy, Cy5-labeled sea urchin axonemes were added directly to the coverslip of the motility chamber (to which they adhere tightly), and the chamber was washed to remove free dynein. Dynein motors were then added to the motility chamber, where they adhered in a rigor state to axonemes. After a ~2 min incubation, the chamber was washed again and motility buffer [30 mM HEPES (pH 7.2), 50 mM KAcetate, 2 mM MgAcetate, 1 mM EGTA, 10% glycerol, 1 mM DTT, 1 mM Mg-ATP, + an oxygen scavenger system (Yildiz et al., 2003)] was added. Single dynein molecules moving along Cy5-labeled axonemes were visualized with a custom-built total internal reflection microscope using objective-style TIRF and an Argon laser with 514 nm illumination at 3 mW. Images were acquired (every 2 s for 8 min) with a cooled, intensified CCD camera (Mega10-S30Z, Stanford Photonics). See Supplemental Data for details on run length and velocity analysis. Every construct was tested for processivity from at least two independent protein preparations.
High spatial-precision measurements of fluorescently labeled dynein were performed with an objective-type TIRF microscope as described previously (FIONA method) (Yildiz et al., 2003). HaloTagged yeast dynein was first labeled with TMR (Promega) or dPEG-biotin (Promega). Biotin-labeled dynein molecules were then labeled with 655 Qdot Streptavidin (Quantum dots Inc) either on the NH2 or the COOH terminus while bound in rigor to axonemes, as described above, to prevent possible aggregation on Qdots. The sample was excited by a 488 nm Argon Ion laser line (for Qdots) or a 532 nm NdYAG laser (for TMR) and imaged through a 1.45 N.A. oil immersion objective (Zeiss). The signal was detected with a Cascade 512B (Photometrics), back-thinned electron multiplier CCD camera. Stepping experiments were performed in motility buffer lacking KAcetate (which increases the run length) and supplemented with an ATP regenerating system (1% pyruvate kinase and 10 mM phosphenolpyruvate). A 2D Gaussian- fit procedure and data analysis were performed as described previously (Yildiz et al., 2003). Steps were determined by using a step-finding program developed by Kerssemakers et al. (2006). We then visually screened the data and included steps in the histogram that could be visually separated and contained no more than one data point between two dwell plateaus (two or more data points between obvious plateaus could represent either noise or steps that were too fast to be temporally resolved at our image acquisition rate).
Supplementary Material
Acknowledgments
The authors wish to thank Nicole Mahoney for early contributions, Nico Stuurman and Adam Douglass for advice on microscopy and image analysis, members of the Vale lab and Andres Leschziner for stimulating discussions, and Roberto Albanese for media preparation. We thank Jacob Kerssemakers and Marileen Dogterom for generously providing their step-finding program ahead of publication. This work has been supported by the National Institutes of Health (P01-AR42895 [R.D.V.] and F32-GM67403-02 [S.R.-P.]), the Jane Coffin Childs Foundation (A.G. and A.P.C.), the German Research Foundation (GE 1609/1 [A.G.]), the Agouron Institute (A.P.C.), and the Howard Hughes Medical Institute.
Footnotes
Supplemental Data include four movies, Supplemental Experimental Procedures, one table, and eleven figures and are available with this article online at: http://www.cell.com/cgi/content/full/126/2/335/DC1/.
References
- Adames NR, Cooper JA. Microtubule interactions with the cell cortex causing nuclear movements in Saccharomyces cerevisiae. J Cell Biol. 2000;149:863–874. doi: 10.1083/jcb.149.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Liu CW, Wandless TJ. Characterization of the FKBP. rapamycin FRB ternary complex. J Am Chem Soc. 2005;127:4715–4721. doi: 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- Burgess SA, Walker ML, Sakakibara H, Knight PJ, Oiwa K. Dynein structure and power stroke. Nature. 2003;421:715–718. doi: 10.1038/nature01377. [DOI] [PubMed] [Google Scholar]
- Case RB, Pierce DW, Hom-Booher N, Hart CL, Vale RD. The directional preference of kinesin motors is specified by an element outside of the motor catalytic domain. Cell. 1997;90:959–966. doi: 10.1016/s0092-8674(00)80360-8. [DOI] [PubMed] [Google Scholar]
- Cashikar AG, Schirmer EC, Hattendorf DA, Glover JR, Ramakrishnan MS, Ware DM, Lindquist SL. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol Cell. 2002;9:751–760. doi: 10.1016/s1097-2765(02)00499-9. [DOI] [PubMed] [Google Scholar]
- Churchman LS, Okten Z, Rock RS, Dawson JF, Spudich JA. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc Natl Acad Sci USA. 2005;102:1419–1423. doi: 10.1073/pnas.0409487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–863. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- Fletcher RJ, Bishop BE, Leon RP, Sclafani RA, Ogata CM, Chen XS. The structure and function of MCM from archaeal M. Thermoautotrophicum. Nat Struct Biol. 2003;10:160–167. doi: 10.1038/nsb893. [DOI] [PubMed] [Google Scholar]
- Gee MA, Heuser JE, Vallee RB. An extended microtubule-binding structure within the dynein motor domain. Nature. 1997;390:636–639. doi: 10.1038/37663. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Katsanis N. Microtubule transport defects in neurological and ciliary disease. Cell Mol Life Sci. 2005;62:1556–1570. doi: 10.1007/s00018-005-5007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons IR, Gibbons BH, Mocz G, Asai DJ. Multiple nucleotide-binding sites in the sequence of dynein beta heavy chain. Nature. 1991;352:640–643. doi: 10.1038/352640a0. [DOI] [PubMed] [Google Scholar]
- Gibbons IR, Lee-Eiford A, Mocz G, Phillipson CA, Tang WJ, Gibbons BH. Photosensitized cleavage of dynein heavy chains. Cleavage at the “V1 site” by irradiation at 365 nm in the presence of ATP and vanadate. J Biol Chem. 1987;262:2780–2786. [PubMed] [Google Scholar]
- Goodenough U, Heuser J. Structural comparison of purified dynein proteins with in situ dynein arms. J Mol Biol. 1984;180:1083–1118. doi: 10.1016/0022-2836(84)90272-9. [DOI] [PubMed] [Google Scholar]
- Ho SN, Biggar SR, Spencer DM, Schreiber SL, Crabtree GR. Dimeric ligands define a role for transcriptional activation domains in reinitiation. Nature. 1996;382:822–826. doi: 10.1038/382822a0. [DOI] [PubMed] [Google Scholar]
- Hook P, Mikami A, Shafer B, Chait BT, Rosenfeld SS, Vallee RB. Long range allosteric control of cytoplasmic dynein ATPase activity by the stalk and C-terminal domains. J Biol Chem. 2005;280:33045–33054. doi: 10.1074/jbc.M504693200. [DOI] [PubMed] [Google Scholar]
- Hua W, Chung J, Gelles J. Distinguishing inchworm and hand-over-hand processive kinesin movement by neck rotation measurements. Science. 2002;295:844–848. doi: 10.1126/science.1063089. [DOI] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem. 1995;270:28806–28811. doi: 10.1074/jbc.270.48.28806. [DOI] [PubMed] [Google Scholar]
- Kerssemakers JWJ, Munteanu EL, Laan L, Noetzel TL, Janson ME, Dogterom M. Assembly dynamics of microtubules at molecular resolution. Nature. 2006 doi: 10.1038/nature04928. in press. Published online June 25, 2006. [DOI] [PubMed] [Google Scholar]
- King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2:20–24. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- Kon T, Mogami T, Ohkura R, Nishiura M, Sutoh K. ATP hydrolysis cycle-dependent tail motions in cytoplasmic dynein. Nat Struct Mol Biol. 2005;12:513–519. doi: 10.1038/nsmb930. [DOI] [PubMed] [Google Scholar]
- Kon T, Nishiura M, Ohkura R, Toyoshima YY, Sutoh K. Distinct functions of nucleotide-binding/hydrolysis sites in the four AAA modules of cytoplasmic dynein. Biochemistry. 2004;43:11266–11274. doi: 10.1021/bi048985a. [DOI] [PubMed] [Google Scholar]
- Koonce MP. Identification of a microtubule-binding domain in a cytoplasmic dynein heavy chain. J Biol Chem. 1997;272:19714–19718. doi: 10.1074/jbc.272.32.19714. [DOI] [PubMed] [Google Scholar]
- Koonce MP, Samso M. Overexpression of cytoplasmic dynein’s globular head causes a collapse of the interphase microtubule network in Dictyostelium. Mol Biol Cell. 1996;7:935–948. doi: 10.1091/mbc.7.6.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: A tug-of-war or coordinated movement? Science. 2005;308:1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- Li J, Lee WL, Cooper JA. NudEL targets dynein to microtubule ends through LIS1. Nat Cell Biol. 2005;7:686–690. doi: 10.1038/ncb1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KW, Naisbitt S, Fan JS, Sheng M, Zhang M. The 8-kDa dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J Biol Chem. 2001;276:14059–14066. doi: 10.1074/jbc.M010320200. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lupetti P, Lanzavecchia S, Mercati D, Cantele F, Dallai R, Mencarelli C. Three-dimensional reconstruction of axonemal outer dynein arms in situ by electron tomography. Cell Motil Cytoskeleton. 2005;62:69–83. doi: 10.1002/cm.20084. [DOI] [PubMed] [Google Scholar]
- Mallik R, Carter BC, Lex SA, King SJ, Gross SP. Cytoplasmic dynein functions as a gear in response to load. Nature. 2004;427:649–652. doi: 10.1038/nature02293. [DOI] [PubMed] [Google Scholar]
- Mallik R, Petrov D, Lex SA, King SJ, Gross SP. Building complexity: An in vitro study of cytoplasmic dynein with in vivo implications. Curr Biol. 2005;15:2075–2085. doi: 10.1016/j.cub.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- Nicastro D, McIntosh JR, Baumeister W. 3D structure of eukaryotic flagella in a quiescent state revealed by cryo-electron tomography. Proc Natl Acad Sci USA. 2005;102:15889–15894. doi: 10.1073/pnas.0508274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiura M, Kon T, Shiroguchi K, Ohkura R, Shima T, Toyoshima YY, Sutoh K. A single-headed recombinant fragment of Dictyostelium cytoplasmic dynein can drive the robust sliding of microtubules. J Biol Chem. 2004;279:22799–22802. doi: 10.1074/jbc.M313362200. [DOI] [PubMed] [Google Scholar]
- Reck-Peterson SL, Vale RD. Molecular dissection of the roles of nucleotide binding and hydrolysis in dynein’s AAA domains in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2004;101:1491–1495. doi: 10.1073/pnas.2637011100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rock RS, Ramamurthy B, Dunn AR, Beccafico S, Rami BR, Morris C, Spink BJ, Franzini-Armstrong C, Spudich JA, Sweeney HL. A flexible domain is essential for the large step size and processivity of myosin VI. Mol Cell. 2005;17:603–609. doi: 10.1016/j.molcel.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Sakakibara H, Kojima H, Sakai Y, Katayama E, Oiwa K. Inner-arm dynein c of Chlamydomonas flagella is a single-headed processive motor. Nature. 1999;400:586–590. doi: 10.1038/23066. [DOI] [PubMed] [Google Scholar]
- Samso M, Koonce MP. 25 Angstrom resolution structure of a cytoplasmic dynein motor reveals a seven-member planar ring. J Mol Biol. 2004;340:1059–1072. doi: 10.1016/j.jmb.2004.05.063. [DOI] [PubMed] [Google Scholar]
- Silvanovich A, Li MG, Serr M, Mische S, Hays TS. The third P-loop domain in cytoplasmic dynein heavy chain is essential for dynein motor function and ATP-sensitive microtubule binding. Mol Biol Cell. 2003;14:1355–1365. doi: 10.1091/mbc.E02-10-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Edamatsu M, Toyoshima YY. Multiple ATP-hydrolyzing sites that potentially function in cytoplasmic dynein. Proc Natl Acad Sci USA. 2004;101:12865–12869. doi: 10.1073/pnas.0403429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toba S, Watanabe TM, Yamaguchi-Okimoto L, Toyoshima YY, Higuchi H, Watanabe Y, Hayashi M, Yagi T, Kamiya R, Hachiya NS, et al. Overlapping hand-over-hand mechanism of single molecular motility of cytoplasmic dynein. Proc Natl Acad Sci USA. 2006;103:5741–5745. doi: 10.1073/pnas.0508511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Milligan RA. The way things move: Looking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Williams JC, Varma D, Barnhart LE. Dynein: An ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58:189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- Wang Z, Khan S, Sheetz MP. Single cytoplasmic dynein molecule movements: Characterization and comparison with kinesin. Biophys J. 1995;69:2011–2023. doi: 10.1016/S0006-3495(95)80071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshaw DM, Kennedy GG, Work SS, Krementsova EB, Beck S, Trybus KM. Differential labeling of myosin V heads with quantum dots allows direct visualization of hand-over-hand processivity. Biophys J. 2005;88:L30–L32. doi: 10.1529/biophysj.105.061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: Single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Selvin PR. Fluorescence imaging with one nanometer accuracy: application to molecular motors. Acc Chem Res. 2005;38:574–582. doi: 10.1021/ar040136s. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Tomishige M, Vale RD, Selvin PR. Kinesin walks hand-over-hand. Science. 2004;303:676–678. doi: 10.1126/science.1093753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.