

Abstract
Nucleophosmin (NPM1), one of the most abundant nucleolar proteins, is a frequent target of oncogenic mutations in acute myeloid leukaemia (AML). Mutation-induced changes at the C-terminal domain of NPM1 (Cter-NPM1) compromise its stability and cause the aberrant translocation of NPM1 to the cytosol. Hence, this protein represents a suitable candidate to investigate the relations between folding and disease. Since Cter-NPM1 folds via a compact denatured state, stabilization of the folded state of the mutated variants demands detailed structural information on both the native and denatured states. Here, we present the characterization of the complete folding pathway of Cter-NPM1 and provide molecular details for both the transition and the denatured states. The structure of the transition state was assessed by Φ-value analysis, whereas residual structure in the denatured state was mapped by evaluating the effect of mutations as modulated by conditions promoting denatured state compaction. Data reveal that folding of Cter-NPM1 proceeds via an extended nucleus and that the denatured state retains significant malleable structure at the interface between the second and third helices. Our observations constitute the essential prerequisite for structure-based drug-design studies, aimed at identifying molecules that may rescue pathological NPM1 mutants by stabilizing the native-like state.
Keywords: kinetics, mutagenesis, protein folding
Nucleophosmin (NPM1) is a ubiquitously expressed protein that belongs to the nucleoplasmin family of nuclear chaperones and is one of the most represented nucleolar proteins (1). NPM1 is a key component of several cellular processes, ranging from ribosome biogenesis, control of centrosome duplication, maintenance of genomic stability, and cell-cycle regulation also by its direct interaction with the tumor suppressors p53 and p14arf (2). A distinctive feature of NPM1 resides in its property to rapidly shuttle from the nucleus to the cytoplasm and backwards (3), a property that may require some structural malleability.
The NPM1 gene has been found overexpressed in several solid human cancers and is also a frequent target of translocations occurring in hematopoietic tumors. Mutation of the NPM1 gene is the most frequent genetic lesion in acute myeloid leukemia (AML) displaying a normal kariotype (4). Such mutations map at the C-terminal domain of the protein, and all result in its denaturation and appearance of an additional nuclear export signal. These two signatures concur and are both necessary to cause the stable and aberrant cytoplasmic localization of NPM1 mutants (1). Furthermore, NPM1 mutants enhance the translocation in the cytoplasm of several interacting partners, including the tumor suppressor p14arf and the c-Myc E3-ubiquitin ligase Fbw7-γ (5, 6). It has been proposed that this feature is involved in the mechanism for the oncogenic properties of NPM mutants (7).
Recent work (8) has shown that some pathological mutations affect the stability of proteins, yielding partial or total denaturation of the mutants even under physiological conditions. Within this perspective, the study of the folding properties of proteins whose stability is altered in specific pathologies is gaining momentum, with the possible goal of restoring the functional native structure of destabilized mutant proteins by appropriate pharmacological chaperones (9, 10). In fact, because folding is an equilibrium between native and denatured states, molecules binding preferentially to the native state can in theory restore the activity of unfolded mutants, if they do not disrupt the activity by binding to or near active sites or hinder critical recognition surfaces.
Recently, we showed that the C-terminal domain of NPM1 (Cter-NPM1) folds via a two-state mechanism, but its denatured state retains significant residual structure (11). In addition, we observed that a compaction of the denatured state results in the acceleration of the folding process, implying that the residual structure should contain mainly native-like interactions; therefore, a structural characterization of the denatured state is a prerequisite to design specific chaperone-active ligands. Because of its elusive nature, unveiling the structure of the denatured state under conditions favoring folding is a challenging task. In fact, it requires reduction of the population of the native state without the addition of denaturants. The experimental approaches used so far include reduction of intramolecular disulfide bridges (12), removal of cofactors (13), truncation of a few residues at the termini of the protein (14, 15), or conservative site-directed mutagenesis (16).
In this work we present the structural dynamics of the folding of Cter-NPM1 using kinetics and protein engineering. We obtained structural information on both the transition state for folding and the denatured state. In particular, while the structure of the transition state was assessed using standard Φ-value analysis (17), the residual structure in the denatured state was inferred by comparing the mutation-induced changes in stability as obtained in the absence and in the presence of a stabilizing salt. In fact, as detailed below, quantitative comparison of the datasets obtained under the two experimental conditions allows to estimate for each of the tested positions, a ΔΔΔGD-N value that reflects the contribution of a specific residue to the structure of the denatured state. Data reveal that folding of Cter-NPM1 proceeds via an extended nucleus. Moreover, the denatured state of Cter-NPM1 appears to retain significant residual structure at the interface between helices H2 and H3, whereas H1 is largely unfolded. Interestingly, we demonstrate a linear correlation between mD-N values and ΔΔΔGD-N values, which suggests that the structure of the denatured state is highly malleable and perturbed by mutagenesis.
Our results provide a unique, detailed structural description of the folding transition state and the denatured state of Cter-NPM1. This information paves the way for future studies focused on the design of small molecules aimed at rescuing the activity of naturally occurring pathological NPM1 mutants.
Results
To characterize quantitatively the folding pathway of Cter-NPM1 we carried out extensive Φ value analysis (17). In addition, to obtain structural information on the denatured state we extended the analysis of all mutants also in the presence of 0.5 M sodium chloride, which promotes denatured state compaction. Thirty-two conservative mutants insisting on 25 positions were synthetized (Table 1 and Tables S1 and S2). Five of these (K248A, A248G, M251A, F268A, and F276A) expressed too poorly to be included in the analysis; the remaining 27 were subjected to both equilibrium and kinetic folding experiments. Overall Φ values could be calculated for 19 positions (Table 1 and Tables S1 and S2).
Table 1.
Kinetic and thermodynamic parameters for the folding of Cter-NPM1 site-directed mutants, at pH 7.0 and 283 K.
| mD-N* (kcal mol-1 M-1) | ΔΔGD-N* (kcal mol-1) | Φ * | ΔΔGD-N† (kcal mol-1) | Φ † | mD-N‡ (kcal mol-1 M-1) | Φ ‡ | ΔΔΔGD-N (kcal mol-1) | |
| Low ionic strength | High ionic strength (0.5 M NaCl) | |||||||
| V244A | 1.02 ± 0.06 | 0.98 ± 0.16 | 0.16 ± 0.09 | 0.70 ± 0.08 | 0.23 ± 0.12 | 0.80 ± 0.06 | −0.44 ± 0.19 | 0.32 ± 0.20 |
| E245A | 0.91 ± 0.03 | 0.03 ± 0.14 | - § | −0.23 ± 0.03 | - § | 0.72 ± 0.04 | - § | −0.06 ± 0.23 |
| A245G | 0.92 ± 0.04 | 0.56 ± 0.11 | 0.31 ± 0.10 | 0.32 ± 0.03 | 0.54 ± 0.16 | 0.75 ± 0.04 | 0.16 ± 0.13 | −0.15 ± 0.18 |
| D246A | 0.94 ± 0.03 | 0.19 ± 0.17 | - § | −0.28 ± 0.03 | - § | 0.77 ± 0.17 | - § | 0.42 ± 0.19 |
| A246G | 0.97 ± 0.04 | 0.31 ± 0.16 | - ¶ | 0.40 ± 0.15 | - ¶ | 0.78 ± 0.11 | - ¶ | 0.23 ± 0.18 |
| I247V | 1.00 ± 0.04 | 0.50 ± 0.14 | 0.14 ± 0.12 | 1.03 ± 0.11 | 0.07 ± 0.06 | 0.70 ± 0.14 | 1.49 ± 1.78 | 0.44 ± 0.16 |
| K248A ∥ | - | - | - | - | - | - | - | - |
| A248G ∥ | - | - | - | - | - | - | - | - |
| A249G | 0.89 ± 0.07 | 1.42 ± 0.16 | 0.29 ± 0.07 | 1.59 ± 0.18 | 0.26 ± 0.06 | 0.66 ± 0.09 | 0.05 ± 0.03 | −0.11 ± 0.17 |
| K250A | 0.85 ± 0.04 | −0.60 ± 0.27 | - § | −0.51 ± 0.07 | - § | 0.80 ± 0.02 | - § | 0.23 ± 0.28 |
| A250G | 0.85 ± 0.08 | 1.11 ± 0.13 | 0.45 ± 0.08 | 0.85 ± 0.06 | 0.67 ± 0.10 | 0.75 ± 0.05 | 0.05 ± 0.05 | −0.01 ± 0.27 |
| M251A ∥ | - | - | - | - | - | - | - | - |
| A253G | 0.99 ± 0.07 | 1.30 ± 0.16 | 0.26 ± 0.08 | 1.75 ± 0.20 | 0.19 ± 0.06 | 0.62 ± 0.07 | 0.25 ± 0.05 | 0.09 ± 0.18 |
| I255V | 0.81 ± 0.04 | 0.77 ± 0.19 | 0.38 ± 0.13 | 1.18 ± 0.14 | 0.25 ± 0.07 | 0.64 ± 0.13 | 0.62 ± 0.34 | 0.45 ± 0.22 |
| L261A | 0.95 ± 0.24 | 1.95 ± 0.31 | −0.04 ± 0.07 | 2.06 ± 0.25 | −0.04 ± 0.07 | 0.94 ± 0.05 | −0.22 ± 0.05 | 0.89 ± 0.31 |
| A266G | 1.02 ± 0.05 | 0.49 ± 0.17 | 0.58 ± 0.29 | 0.40 ± 0.05 | 0.71 ± 0.27 | 0.83 ± 0.07 | 1.65 ± 1.09 | 0.68 ± 0.20 |
| F268A ∥ | - | - | - | - | - | - | - | - |
| I269V | 1.00 ± 0.03 | 0.23 ± 0.16 | - ¶ | 0.13 ± 0.02 | - ¶ | 0.86 ± 0.11 | - ¶ | 0.82 ± 0.20 |
| N270A | 0.84 ± 0.04 | −0.43 ± 0.24 | - § | −0.72 ± 0.07 | - § | 0.73 ± 0.02 | - § | 0.00 ± 0.25 |
| A270G | 0.93 ± 0.04 | 1.07 ± 0.16 | 0.84 ± 0.14 | 0.87 ± 0.16 | 1.03 ± 0.21 | 0.74 ± 0.04 | 0.52 ± 0.14 | −0.27 ± 0.18 |
| V272A | 1.22 ± 0.08 | 1.23 ± 0.15 | 0.40 ± 0.09 | 1.35 ± 0.16 | 0.36 ± 0.09 | 0.86 ± 0.05 | 0.36 ± 0.08 | 0.55 ± 0.18 |
| K273A | 0.92 ± 0.04 | 0.78 ± 0.16 | - § | 0.41 ± 0.06 | - § | 0.77 ± 0.05 | - § | 0.02 ± 0.18 |
| A273G | 0.93 ± 0.08 | 0.74 ± 0.16 | 0.92 ± 0.08 | 1.39 ± 0.16 | 0.49 ± 0.09 | 0.78 ± 0.04 | 0.70 ± 0.30 | −0.08 ± 0.18 |
| F276A ∥ | - | - | - | - | - | - | - | - |
| A283G | 0.88 ± 0.04 | 0.84 ± 0.17 | 0.56 ± 0.14 | 1.11 ± 0.14 | 0.42 ± 0.09 | 0.86 ± 0.07 | −0.17 ± 0.28 | 1.20 ± 0.20 |
| I284V | 1.12 ± 0.06 | 0.98 ± 0.15 | 0.13 ± 0.10 | 1.56 ± 0.21 | 0.08 ± 0.06 | 0.74 ± 0.06 | 0.86 ± 0.26 | 0.61 ± 0.17 |
| L287A | 1.58 ± 0.52 | 2.31 ± 0.18 | 0.06 ± 0.05 | 2.09 ± 0.30 | 0.06 ± 0.06 | 0.66 ± 0.15 | 0.24 ± 0.20 | −0.30 ± 0.54 |
| W288F | 1.15 ± 0.25 | 2.20 ± 0.26 | 0.15 ± 0.10 | 2.95 ± 0.28 | 0.11 ± 0.08 | 0.83 ± 0.05 | −0.39 ± 0.11 | 1.03 ± 0.28 |
| W290F | 0.91 ± 0.10 | 2.24 ± 0.14 | 0.28 ± 0.04 | 1.69 ± 0.10 | 0.38 ± 0.05 | 0.85 ± 0.09 | 0.27 ± 0.28 | 0.84 ± 0.40 |
| K292A | 0.94 ± 0.04 | 0.67 ± 0.16 | - § | 1.66 ± 0.07 | - § | 0.72 ± 0.07 | - § | 0.20 ± 0.19 |
| A292G | 0.94 ± 0.09 | 0.97 ± 0.17 | 0.36 ± 0.13 | 1.07 ± 0.05 | 0.33 ± 0.10 | 0.66 ± 0.08 | 0.23 ± 0.14 | −0.17 ± 0.23 |
| L294A | 0.67 ± 0.14 | 2.15 ± 0.23 | 0.26 ± 0.06 | 2.20 ± 0.03 | 0.25 ± 0.06 | 0.95 ± 0.07 | −0.13 ± 0.07 | 1.67 ± 0.24 |
The errors reported are the standard errors. Repetition of individual experiments and calculation of standard deviations on wild-type Cter-NPM1 and some selected mutants revealed that obtained standard deviations were in the same order of magnitude of the standard errors.
*Calculated from chevron plot analysis. Data recorded in the absence of NaCl.
†Calculated from thermal melt equilibrium denaturations in the absence of NaCl.
‡Calculated from chevron plot analysis. Data recorded in the presence of 0.5 M NaCl.
§Ala-Gly scanning mutants. A solvent exposed residue in the helix was mutated into Ala and Gly, and a Φ value was calculated by comparing the folding kinetics of the Gly variant against its Ala counterpart.
¶ΔΔGD-N was too low to calculate a reliable Φ value.
∥Mutants expressed poorly and could not be characterized.
Unfolding and refolding kinetics were satisfactorily fitted to single-exponential time courses, under all conditions. Semilogarithmic plots of the observed unfolding and refolding rate constants versus denaturant concentration (chevron plots) for wild-type Cter-NPM1 and selected mutants are shown in Fig. 1A. Fitted parameters are reported in Table 1 and Fig. S1.
Fig. 1.

Chevron plots of wt and representative mutants of Cter-NPM1. A and B refer to data recorded in the absence and in the presence of 0.5 M NaCl, respectively. Data refer to wt Cter-NPM1 (Closed Circles), V244A (Squares), A266G (Diamonds), and I255V (Triangles). Lines are the best fit to a two-state folding model (51).
Because of its small size (about 6500 Da) (18), Cter-NPM1 displays a low mD-N value (mD-N = 0.9 ± 0.1 Kcal mol-1 M-1; as obtained from equilibrium urea-induced denaturation) (11) that jeopardizes an accurate determination of the equilibrium unfolding free energy by equilibrium urea-induced denaturation experiments. Hence, all mutants were investigated by thermal denaturation unfolding (Fig. S2 and Tables S1 and S2). Following Fersht (19), reliable ΔΔGD-N values induced by mutations were calculated as follows:
| [1] |
The kinetic and equilibrium ΔΔGD-N values were both used to calculate Φ values (Table 1); these two sets of Φ values were found to be essentially the same within experimental error, confirming that, in the absence of stabilizing salt, the protein folds following a simple two-state model. In all cases Φ values were fractional (Table 1) and their distribution along the structure revealed the presence of a diffuse nucleus, comprising elements of each of the three helices (Fig. 2). Overall, the structural distribution and the magnitude of Φ values both suggest that Cter-NPM1 folds via a nucleation-condensation like mechanism (20–24), whereby folding proceeds via concurrent formation of secondary and tertiary structures, and the transition state should resemble a distorted version of the native state (Fig. S3).
Fig. 2.

Φ value analysis on Cter-NPM1. Φ values are highlighted on the structure and clustered in three groups: 0 < Φ < 0.3 kcal mol-1 (Red); 0.3 < Φ < 0.5 kcal mol-1 (Magenta); Φ > 0.5 kcal mol-1 (Blue). A broad diffused folding nucleus, consistent with a nucleation-condensation type folding mechanism, may be identified. The two panels (A and B) refer to two different orientations of the molecule.
In a recent study, we characterized the folding of Cter-NPM1 under different experimental conditions (11). Analysis of the kinetic m values according to Sanchez and Kiefhaber (25) revealed that the denatured state retains some residual native-like compact structure in the presence of stabilizing salt (0.5 M NaCl), while it corresponds to an extended random coil at destabilizing conditions (such as low pH); at physiological ionic strength and pH conditions a somewhat intermediate behavior was observed. Characterizing the structural features of a denatured state is a particularly difficult task. In this case we resorted to compare the effect of mutagenesis at low ionic strength with that obtained in the presence of a stabilizing salt (in this case NaCl). The rationale of such an approach is as follows.
While the Φ values analysis allows characterization of the structure of folding transition states, it is extremely difficult to unveil interaction patterns in denatured states by protein engineering. In fact, by reference to a square thermodynamic cycle (19), changes in stability upon mutation may be calculated as follows:
| [2] |
where D′ and N′ represent the denatured and native states of the mutant protein, respectively. Because residues are generally taken not to interact in completely random unfolded conformations, the observed 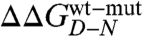 typically equals the ΔΔGN-N′: i.e., in the absence of residual structure in the denatured state, the observed change in stability reflects the perturbation in the free energy of the native state induced by the mutation. In this work, we focused on the analysis of the residual structural features of the denatured state of Cter-NPM1, as induced by high salt concentrations (11). Hence, considering a cubic thermodynamic scheme (Fig. 3A), and comparing the changes in the stability induced by a mutation as obtained in the absence and in the presence of 0.5 M NaCl, the coupling free energy
typically equals the ΔΔGN-N′: i.e., in the absence of residual structure in the denatured state, the observed change in stability reflects the perturbation in the free energy of the native state induced by the mutation. In this work, we focused on the analysis of the residual structural features of the denatured state of Cter-NPM1, as induced by high salt concentrations (11). Hence, considering a cubic thermodynamic scheme (Fig. 3A), and comparing the changes in the stability induced by a mutation as obtained in the absence and in the presence of 0.5 M NaCl, the coupling free energy 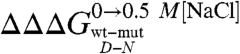 can be calculated by the following equation:
can be calculated by the following equation:
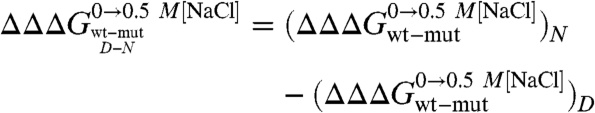 |
[3] |
Fig. 3.

Denatured state structure of Cter-NPM1 as obtained from protein engineering. (A) Double perturbation cycle. The cube depicts the different states of the native (N) and denatured (D) conformations populated as a result of a double perturbation (i.e., mutagenesis and addition of stabilizing salt). (B and C) Coupling free energies mapped on the structure shown in two orientations (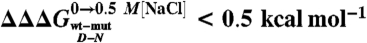 (Black);
(Black); 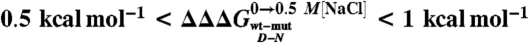 (Green);
(Green); 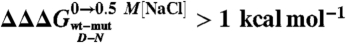 (Blue).
(Blue).
This equation, formally similar to the analysis of double-mutant cycles (19, 26), implies that the measured coupling free energy for each mutant in the absence and presence of 0.5 M NaCl is equal to the difference in coupling energy between the native and the denatured states. If we assume that the free energy of the native state in high salt solutions is unlikely to be affected by conservative mutations mostly involving buried hydrophobic side-chains (27), 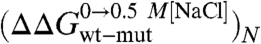 may be taken to be zero. Since we have shown that there is a compaction of the denatured state upon addition of salt, we consider that
may be taken to be zero. Since we have shown that there is a compaction of the denatured state upon addition of salt, we consider that 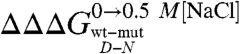 is a measure of the contribution of a specific residue to the structure of the denatured state alone. It should be noted, however, that while Φ values represent direct structural indexes,
is a measure of the contribution of a specific residue to the structure of the denatured state alone. It should be noted, however, that while Φ values represent direct structural indexes, 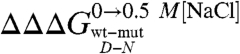 simply reflects the contribution of each mutated side-chain to the stabilization of the structure nucleus of the denatured state when comparing data in the absence and in the presence of a stabilizing salt. Furthermore, a
simply reflects the contribution of each mutated side-chain to the stabilization of the structure nucleus of the denatured state when comparing data in the absence and in the presence of a stabilizing salt. Furthermore, a  does not necessarily imply that the probed residue is unstructured in the denatured state, but that there is no change in coupling free energy when considering the denatured state upon addition of salt. Hence, while analysis of
does not necessarily imply that the probed residue is unstructured in the denatured state, but that there is no change in coupling free energy when considering the denatured state upon addition of salt. Hence, while analysis of 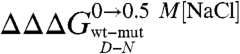 can in theory provide structural information about compact denatured states, the calculated values should be interpreted with caution.
can in theory provide structural information about compact denatured states, the calculated values should be interpreted with caution.
A complete equilibrium and kinetic characterization of all the mutants in the absence and in the presence of 0.5 M NaCl allowed calculating folding parameters and the resulting 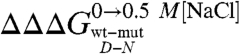 values, all listed in Table 1. As depicted in a color-coded representation in Fig. 3 B and C, a structural distribution of
values, all listed in Table 1. As depicted in a color-coded representation in Fig. 3 B and C, a structural distribution of 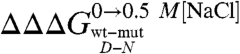 clearly shows that mutations displaying the highest values (> 0.5 Kcal mol-1) all map onto the helices H2 and H3 and their interface, whereas helix H1 includes only residues with smaller
clearly shows that mutations displaying the highest values (> 0.5 Kcal mol-1) all map onto the helices H2 and H3 and their interface, whereas helix H1 includes only residues with smaller 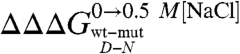 values (< 0.5 Kcal mol-1). These data clearly suggest that the region comprised between helices H2 and H3 is likely to partially retain its native conformation in the denatured state.
values (< 0.5 Kcal mol-1). These data clearly suggest that the region comprised between helices H2 and H3 is likely to partially retain its native conformation in the denatured state.
There is a growing consensus that denatured states of proteins do not behave as fully unfolded chains (15, 16, 28–31). However, little is known about the mechanism whereby their residual structure may be affected by denaturants or mutagenesis. Is the denatured state under physiological conditions in equilibrium with the fully unfolded conformation or does it unfold via a gradual melting of structure? Does the denatured state unfold via a first- or a second-order type transition? Taking advantage of the folding and unfolding parameters obtained for the site-directed mutants of Cter-NPM1, we addressed this issue by analyzing the dependence of the observed mD-N values on 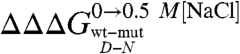 . In fact, while according to the analysis outlined above
. In fact, while according to the analysis outlined above 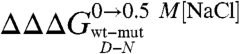 values reflect the effect of any given mutation on the free energy of the denatured state, the mD-N value would indicate its overall degree of compaction. Interestingly, data in Fig. 4 show a linear dependence of mD-N on
values reflect the effect of any given mutation on the free energy of the denatured state, the mD-N value would indicate its overall degree of compaction. Interestingly, data in Fig. 4 show a linear dependence of mD-N on 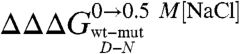 (R = 0.73), with greater values of
(R = 0.73), with greater values of 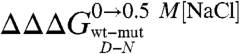 (i.e., stronger destabilization of D) corresponding to greater values of mD-N(i.e., more unfolded D). Furthermore, it may be noted that such dependence is mainly due to an effect on the mF value, the calculated mU values being essentially independent of
(i.e., stronger destabilization of D) corresponding to greater values of mD-N(i.e., more unfolded D). Furthermore, it may be noted that such dependence is mainly due to an effect on the mF value, the calculated mU values being essentially independent of 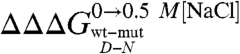 (R = 0.17). These data strongly suggest that the denatured state responds to destabilizing mutations by altering its structure and indicate that the variable two-state scenario (32) would represent the best model to describe the folding and unfolding behavior of Cter-NPM1 and its mutants.
(R = 0.17). These data strongly suggest that the denatured state responds to destabilizing mutations by altering its structure and indicate that the variable two-state scenario (32) would represent the best model to describe the folding and unfolding behavior of Cter-NPM1 and its mutants.
Fig. 4.

Correlation between mF (Open Circles), mU (Diamonds), and mD-N (Closed Circles) values (recorded in the presence of 0.5 M NaCl) versus 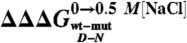 values. Lines are the best fit to a linear function (mF, R = 0.84; mU, R = 0.17; mD-N, R = 0.73). As described in the text, the observed correlation of both mF and mD-N suggests that the denatured state retains a malleable structure, which is perturbed by mutagenesis.
values. Lines are the best fit to a linear function (mF, R = 0.84; mU, R = 0.17; mD-N, R = 0.73). As described in the text, the observed correlation of both mF and mD-N suggests that the denatured state retains a malleable structure, which is perturbed by mutagenesis.
Discussion
Nucleophosmin is a key nucleolar chaperone that was discovered to be mutated in several haematological malignancies. In general, mutations compromise the physiological cellular traffic of the protein: Indeed, more than 30% of adult patients affected by AML show aberrant nucleophosmin localization in the cell cytoplasm (33). This led to the recent inclusion of AML with mutated NPM1 into the World Health Organization classification of myeloid neoplasms as a new provisional entity (34). All NPM1 mutations, in spite of remarkable heterogeneity (about 50 mutations identified so far), result in a shift of the reading frame and cause changes at the C terminus of the protein that are responsible for its partial or total denaturation (1). This observation suggests that the design of small molecules capable of stabilizing NPM1 native fold compromised in pathologic mutants might have a therapeutic value, possibly enhancing relocation of the mutants in their native compartment. Recently, we have shown that Cter-NPM1 retains some residual structure in the denatured state in the presence of salt (11). Because folding relies on the equilibrium between the native and denatured states, mapping the residual structure in the denatured state is a key step in any attempt to design structure-based drugs (8, 9, 35).
In this work we have characterized the folding transition state structure of Cter-NPM1 and deciphered the overall structural features of its denatured state nucleus through kinetics and extensive protein engineering. In particular, we performed a conventional Φ value analysis on about 30 conservative mutants. This protein displays a folding rate constant that is only marginally slower than what predicted from its calculated contact order (36), i.e., contact order of 10.5% corresponding to a folding rate constant of circa 104 s-1 vs. the experimentally determined value of 4 × 103 s-1. Data revealed the presence of a diffused nucleus for folding, as mirrored by the presence of fractional Φ values in essentially all probed positions. Furthermore, these values were broadly distributed along the Cter-NPM1 structure, suggesting that this small protein domain folds via a nucleation-condensation mechanism (20, 21, 24). This observation is somewhat surprising, because this type of mechanism is generally invoked for proteins that display little or no residual structure in their denatured states (23, 37). Indeed, this behavior, in analogy to what recently observed for the homeodomain-like proteins, has been associated to a hybrid between the nucleation-condensation and diffusion-collision mechanisms (23, 38, 39).
In an effort to unveil the structural features of the denatured state, we subjected all mutants to equilibrium and kinetic folding experiments under conditions promoting denatured state structure, i.e., in the presence of a stabilizing salt (0.5 M NaCl). The rationale for this approach, represented in Fig. 3A and discussed in Results, relies on the emerging view that, because the denatured state ensemble can contain energetically significant interactions, mutations can exert on the denatured state effects that are comparable to those seen for the native or the transition states, (40) and references therein.
The results of the Φ value analysis in the presence of salt are shown in Table 1. Interestingly, while all the Φ values calculated in the absence of salt were fractional, five of the Φ values obtained in its presence appear to be nonconventional, i.e., negative. This finding may be explained following Cho and Raleigh (40), who argued that when a mutation changes the free energy level of all the states (denatured, native, and transition), the resulting Φ value may be negative because of a change in the free energy of the denatured state. Detailed analysis of the effect of mutations on the free energy of the denatured state has been previously reported (41–43).
As described in Results, a comparison between the datasets obtained in the absence and in the presence of salt allows an estimate of the contribution of a given mutation to the stabilization of the denatured state residual structure. Notably for Cter-NPM1, the residues with the highest values of 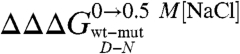 (> 0.5 Kcal mol-1) all map on helices H2 and H3 and at the surface in between, contrary to residues in H1 (
(> 0.5 Kcal mol-1) all map on helices H2 and H3 and at the surface in between, contrary to residues in H1 (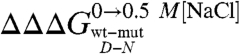 < 0.5 Kcal mol-1). These results indicate that H2 and H3 retain some residual structure in the denatured state of Cter-NPM1. Furthermore, the linear dependence of mD-N on
< 0.5 Kcal mol-1). These results indicate that H2 and H3 retain some residual structure in the denatured state of Cter-NPM1. Furthermore, the linear dependence of mD-N on 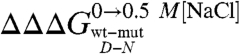 indicates that such a residual structure is malleable, at variance with discrete folding intermediates (44, 45) and similarly to what was recently observed in the Engrailed homeodomain (16), where the denatured state under physiological conditions unfolds in a gradual fashion upon destabilization. According to our data on Cter-NPM1, helix H1 would represent the structural portion of the protein to be targeted by chemicals in an effort to favor the native structure, and thereby our findings add previously undescribed information for the selection of drugs that counteract leukemic mutations. So far, in fact, the region comprising helix H2 has been mainly envisaged for ligand binding, as it includes important functional residues (e.g. K263 and K267) (46) and the Cys 275, which is responsible for the binding of the natural antitumoral compound Avrainvillamide (47).
indicates that such a residual structure is malleable, at variance with discrete folding intermediates (44, 45) and similarly to what was recently observed in the Engrailed homeodomain (16), where the denatured state under physiological conditions unfolds in a gradual fashion upon destabilization. According to our data on Cter-NPM1, helix H1 would represent the structural portion of the protein to be targeted by chemicals in an effort to favor the native structure, and thereby our findings add previously undescribed information for the selection of drugs that counteract leukemic mutations. So far, in fact, the region comprising helix H2 has been mainly envisaged for ligand binding, as it includes important functional residues (e.g. K263 and K267) (46) and the Cys 275, which is responsible for the binding of the natural antitumoral compound Avrainvillamide (47).
In the context of the fight against AML, this work paves the way to an investigation of libraries of pharmaceuticals that, examined by appropriate revealing assays (48), may restore the native fold of leukemic variants of NPM1 by stabilizing helix H1 and ultimately restore the physiological subcellular localization of nucleophosmin.
Materials and Methods
Mutagenesis, Expression, and Purification.
All mutants were realized by PCR using a QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and using the Cter-NPM1 cDNA cloned into a pET28a vector (Novagen, San Diego, CA) as template. The construct codes for the C-terminal domain of NPM1 (ΔN241) fused to a His6-tag. The presence of the His6-tag had a negligible effect on the folding and unfolding kinetics of Cter-NPM1 (Fig. S1). The expression system was Escherichia coli strain BL21(DE3) and proteins were purified using Ni2+ chelating affinity chromatography, followed by ion-exchange and size exclusion chromatography. SDS–PAGE and mass spectrometry were used to confirm the protein purity and mass. All reagents were purchased from Sigma-Aldrich Corp. (St. Louis, MO) and were of analytical grade.
Circular Dichroism (CD).
CD spectra of Cter-NPM1 wt and mutants were recorded between 260 and 190 nm at a concentration of 10 μM using a Jasco spectropolarimeter (Jasco, Inc., Easton, MD) and a 1-mm pathlength quartz cuvette (Hellma, Plainview, NY). Thermal denaturation was followed at 222 nm with heating from 277 to 363 K at a rate of 1 K min-1(Fig. S2). Data were fitted to a standard 2-state denaturation (19). An equation that takes into account the pre- and post-transition baselines was used to fit the observed unfolding transition (49). The buffers used were 50 mM sodium phosphate pH 7.0, 1 mM DTT either with or without 0.5 M sodium chloride.
Stopped-Flow Measurements.
Single mixing kinetic folding experiments were carried out on a Π∗ stopped-flow instrument (Applied Photophysics, Leatherhead, UK); the excitation wavelength was 280 nm and the fluorescence emission was measured using a 320-nm cut-off glass filter. In all experiments, performed at 283 K, refolding and unfolding were initiated by an 11-fold dilution of the denatured or the native protein with the appropriate buffer. Final protein concentrations were typically 1 μM. The kinetics observed were always independent of protein concentration, as expected for a monomolecular reaction without effects due to transient aggregation (50).
Data Analysis.
Analysis of observed time courses was performed by nonlinear least-squares fitting of single exponential time courses using the fitting procedures provided in the Applied Photophysics software. The chevron plots were fitted by numerical analysis based on a two-state model following the equation:
| [4] |
where kF and kU represent the folding and unfolding rate constants, respectively. The logarithm of each microscopic rate constant was assumed to vary linearly with denaturant concentration (51). Φ values were calculated from folding rate constants using standard equations (17).
Supplementary Material
Acknowledgments.
We express our thanks to Professor Brunangelo Falini (Perugia, Italy) for introducing us to nucleophosmin and for his precious collaboration. This work was supported by a grant from the Associazione Italiana Ricerca sul Cancro to M.B.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0910516107/DCSupplemental.
References
- 1.Falini B, et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: Molecular basis and clinical implications. Leukemia. 2009;23:1731–1743. doi: 10.1038/leu.2009.124. [DOI] [PubMed] [Google Scholar]
- 2.Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493–505. doi: 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- 3.Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56:379–390. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 4.Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood. 2007;109:874–885. doi: 10.1182/blood-2006-07-012252. [DOI] [PubMed] [Google Scholar]
- 5.Bolli N, et al. A dose-dependent tug of war involving the NPM1 leukaemic mutant, nucleophosmin, and ARF. Leukemia. 2009;23:501–509. doi: 10.1038/leu.2008.326. [DOI] [PubMed] [Google Scholar]
- 6.Bonetti P, et al. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. J Cell Biol. 2008;182:19–26. doi: 10.1083/jcb.200711040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Fiore PP. Playing both sides: Nucleophosmin between tumor suppression and oncogenesis. J Cell Biol. 2008;182:7–9. doi: 10.1083/jcb.200806069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joerger AC, Fersht AR. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene. 2007;26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- 9.Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- 10.Pey AL, et al. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J Clin Invest. 2008;118:2858–2867. doi: 10.1172/JCI34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaloni F, Gianni S, Federici L, Falini B, Brunori M. Folding mechanism of the C-terminal domain of nucleophosmin: Residual structure in the denatured state and its pathophysiological significance. FASEB J. 2009;23:2360–2365. doi: 10.1096/fj.08-128306. [DOI] [PubMed] [Google Scholar]
- 12.Schwalbe H, et al. Structural and dynamical properties of a denatured protein. Heteronuclear 3D NMR experiments and theoretical simulations of lysozyme in 8 M urea. Biochemistry. 1997;36:8977–8991. doi: 10.1021/bi970049q. [DOI] [PubMed] [Google Scholar]
- 13.Bai Y, Chung J, Dyson HJ, Wright PE. Structural and dynamic characterization of an unfolded state of poplar apo-plastocyanin formed under nondenaturing conditions. Protein Sci. 2001;10:1056–1066. doi: 10.1110/ps.00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neira JL, et al. Towards the complete structural characterization of a protein folding pathway: The structures of the denatured, transition and native states for the association/folding of two complementary fragments of cleaved chymotrypsin inhibitor 2. Direct evidence for a nucleation-condensation mechanism. Fold Des. 1996;1:189–208. doi: 10.1016/s1359-0278(96)00031-4. [DOI] [PubMed] [Google Scholar]
- 15.Shortle D, Meeker AK. Residual structure in large fragments of staphylococcal nuclease: Effects of amino acid substitutions. Biochemistry. 1989;28:936–944. doi: 10.1021/bi00429a003. [DOI] [PubMed] [Google Scholar]
- 16.Religa TL, Markson JS, Mayor U, Freund SM, Fersht AR. Solution structure of a protein denatured state and folding intermediate. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 17.Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 18.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fersht AR. Structure and mechanism in protein science. New York: Freeman; 1999. [Google Scholar]
- 20.Abkevich VI, Gutin AM, Shakhnovich EI. Specific nucleus as the transition state for protein folding: Evidence from the lattice model. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 21.Fersht AR. Optimization of rates of protein folding: The nucleation-condensation mechanism and its implications. Proc Natl Acad Sci USA. 1995;92:10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gianni S, et al. A PDZ domain recapitulates a unifying mechanism for protein folding. Proc Natl Acad Sci USA. 2007;104:128–133. doi: 10.1073/pnas.0602770104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gianni S, et al. Unifying features in protein-folding mechanisms. Proc Natl Acad Sci USA. 2003;100:13286–13291. doi: 10.1073/pnas.1835776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analyzed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J Mol Biol. 1995;254:260–88. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez IE, Kiefhaber T. Hammond behavior versus ground state effects in protein folding: Evidence for narrow free energy barriers and residual structure in unfolded states. J Mol Biol. 2003;327:867–84. doi: 10.1016/s0022-2836(03)00171-2. [DOI] [PubMed] [Google Scholar]
- 26.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold Des. 1996;1:R121–R126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 27.Timasheff SN. The control of protein stability and association by weak interactions with water: How do solvents affect these processes? Annu Rev Biophys Biomol Struct. 1993;22:67–97. doi: 10.1146/annurev.bb.22.060193.000435. [DOI] [PubMed] [Google Scholar]
- 28.McCarney ER, Kohn JE, Plaxco KW. Is there or isn't there? The case for (and against) residual structure in chemically denatured proteins. Crit Rev Biochem Mol Biol. 2005;40:181–189. doi: 10.1080/10409230591008143. [DOI] [PubMed] [Google Scholar]
- 29.Merchant KA, Best RB, Louis JM, Gopich IV, Eaton WA. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc Natl Acad Sci USA. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mok KH, et al. A pre-existing hydrophobic collapse in the unfolded state of an ultrafast folding protein. Nature. 2007;447:106–109. doi: 10.1038/nature05728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Gunsteren WF, Bürgi R, Peter C, Daura X. The key to solving the protein-folding problem lies in an accurate description of the denatured state. Angew Chem Int Ed Engl. 2001;40:351–355. doi: 10.1002/1521-3773(20010119)40:2<351::AID-ANIE351>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 32.Ferreon AC, Bolen DW. Thermodynamics of denaturant-induced unfolding of a protein that exhibits variable two-state denaturation. Biochemistry. 2004;43:13357–13369. doi: 10.1021/bi048666j. [DOI] [PubMed] [Google Scholar]
- 33.Falini B, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 34.Falini B, Martelli MP. Anaplastic large cell lymphoma: changes in the World Health Organization classification and perspectives for targeted therapy. Haematologica. 2009;94:897–900. doi: 10.3324/haematol.2009.008250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedler A, et al. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc Natl Acad Sci USA. 2002;99:937–942. doi: 10.1073/pnas.241629998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 37.Daggett V, Fersht AR. Is there a unifying mechanism for protein folding? Trends Biochem Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 38.Karplus M, Weaver DL. Protein folding dynamics: the diffusion-collision model and experimental data. Protein Sci. 1994;3:650–668. doi: 10.1002/pro.5560030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White GW, et al. Simulation and experiment conspire to reveal cryptic intermediates and a slide from the nucleation-condensation to framework mechanism of folding. J Mol Biol. 2005;350:757–775. doi: 10.1016/j.jmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Cho JH, Raleigh DP. Denatured state effects and the origin of nonclassical phi values in protein folding. J Am Chem Soc. 2006;128:16492–16493. doi: 10.1021/ja0669878. [DOI] [PubMed] [Google Scholar]
- 41.Cho JH, Raleigh DP. Experimental characterization of the denatured state ensemble of proteins. Methods Mol Biol. 2009;490:339–351. doi: 10.1007/978-1-59745-367-7_14. [DOI] [PubMed] [Google Scholar]
- 42.Fersht AR, Sato S. Phi-value analysis and the nature of protein-folding transition states. Proc Natl Acad Sci USA. 2004;101:7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott KA, Alonso DO, Sato S, Fersht AR, Daggett V. Conformational entropy of alanine versus glycine in protein denatured states. Proc Natl Acad Sci USA. 2007;104:2661–2666. doi: 10.1073/pnas.0611182104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dalby PA, Oliveberg M, Fersht AR. Folding intermediates of wild-type and mutants of barnase. I. Use of phi-value analysis and m-values to probe the cooperative nature of the folding pre-equilibrium. J Mol Biol. 1998;276:625–646. doi: 10.1006/jmbi.1997.1546. [DOI] [PubMed] [Google Scholar]
- 45.Uversky VN, Ptitsyn OB. All-or-none solvent-induced transitions between native, molten globule and unfolded states in globular proteins. Fold Des. 1996;1:117–122. doi: 10.1016/S1359-0278(96)00020-X. [DOI] [PubMed] [Google Scholar]
- 46.Grummitt CG, Townsley FM, Johnson CM, Warren AJ, Bycroft M. Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J Biol Chem. 2008;283:23326–23332. doi: 10.1074/jbc.M801706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wulff JE, Siegrist R, Myers AG. The natural product avrainvillamide binds to the oncoprotein nucleophosmin. J Am Chem Soc. 2007;129:14444–51. doi: 10.1021/ja075327f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kau TR, Way JC, Silver PA. Nuclear transport and cancer: from mechanism to intervention. Nat Rev Cancer. 2004;4:106–117. doi: 10.1038/nrc1274. [DOI] [PubMed] [Google Scholar]
- 49.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 50.Silow M, Oliveberg M. Transient aggregates in protein folding are easily mistaken for folding intermediates. Proc Natl Acad Sci USA. 1997;94:6084–6. doi: 10.1073/pnas.94.12.6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.