

Abstract
The RNA world hypothesis proposes that nucleic acids were once responsible for both information storage and chemical catalysis, before the advent of coded protein synthesis. However, it is difficult to imagine how nucleic acid polymers first appeared, as the abiotic chemical formation of long nucleic acid polymers from mononucleotides or short oligonucleotides remains elusive, and barriers to achieving this goal are substantial. One specific obstacle to abiotic nucleic acid polymerization is strand cyclization. Chemically activated short oligonucleotides cyclize efficiently, which severely impairs polymer growth. We show that intercalation, which stabilizes and rigidifies nucleic acid duplexes, almost totally eliminates strand cyclization, allowing for chemical ligation of tetranucleotides into duplex polymers of up to 100 base pairs in length. In contrast, when these reactions are performed in the absence of intercalators, almost exclusively cyclic tetra- and octanucleotides are produced. Intercalator-free polymerization is not observed, even at tetranucleotide concentrations > 10,000-fold greater than those at which intercalators enable polymerization. We also demonstrate that intercalation-mediated polymerization is most favored if the size of the intercalator matches that of the base pair; intercalators that bind to Watson–Crick base pairs promote the polymerization of oligonucleotides that form these base pairs. Additionally, we demonstrate that intercalation-mediated polymerization is possible with an alternative, non-Watson–Crick-paired duplex that selectively binds a complementary intercalator. These results support the hypothesis that intercalators (acting as ‘molecular midwives’) could have facilitated the polymerization of the first nucleic acids and possibly helped select the first base pairs, even if only trace amounts of suitable oligomers were available.
Keywords: base pair selection, origin of life, RNA world, polymerization, molecular evolution
Over the past two decades, significant evidence has been presented in support of the RNA world hypothesis, which proposes that RNA polymers predated coded proteins in early life (1, 2). Current support for this hypothesis includes the fact that contemporary life still uses RNA as an informational polymer and in chemical catalysis (3). The ability of RNA to catalyze reactions is exemplified by natural and artificial ribozymes that promote a wide variety of chemical reactions (4) as well as the observation that the catalytic core of the ribosome is comprised of RNA (5). Despite the attractiveness of the RNA world as a hypothetical stage of early life, it remains unclear how RNA [or a predecessor of RNA (6–11)] would initially have been synthesized without the aid of protein enzyme catalysis.
Several distinct proposals have been presented for the abiotic origin of the first RNA polymers (10, 12–17). Perhaps the most notable is that of Ferris and coworkers, in which mineral surfaces are used to locally concentrate and promote the polymerization of ligation-activated mononucleotides, an approach that allows formation of single-stranded RNA strands up to ca. 50 nucleotides in length (18). Additionally, Sawai et al. demonstrated the use of 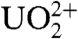 in the solution-state polymerization of activated monomers into linear and cyclic oligomers up to decanucleotides (19, 20), and Steltsov et al. have shown that the tripeptide trivaline facilitates the ligation of d(pGTT) (21). However, it is not clear how any of these mechanisms alone would have selectively produced nucleic acids containing only bases capable of forming Watson–Crick base pairs from a prebiotic chemical inventory that likely contained a complex mixture of molecules (22). Earlier work by Orgel and coworkers demonstrated that oligonucleotides (of rather restricted nucleotide sequence) can serve as templates for the polymerization of activated mononucleotides in solution, resulting in duplexes with Watson–Crick base pairs (23–25). However, it remains an open question precisely how the first polymers of RNA (or a chemically related proto-RNA) could have assembled from mononucleotides, or even short oligonucleotides, without preexisting templates.
in the solution-state polymerization of activated monomers into linear and cyclic oligomers up to decanucleotides (19, 20), and Steltsov et al. have shown that the tripeptide trivaline facilitates the ligation of d(pGTT) (21). However, it is not clear how any of these mechanisms alone would have selectively produced nucleic acids containing only bases capable of forming Watson–Crick base pairs from a prebiotic chemical inventory that likely contained a complex mixture of molecules (22). Earlier work by Orgel and coworkers demonstrated that oligonucleotides (of rather restricted nucleotide sequence) can serve as templates for the polymerization of activated mononucleotides in solution, resulting in duplexes with Watson–Crick base pairs (23–25). However, it remains an open question precisely how the first polymers of RNA (or a chemically related proto-RNA) could have assembled from mononucleotides, or even short oligonucleotides, without preexisting templates.
Strand cyclization is a formidable problem facing most proposed systems for the prebiotic synthesis of nucleic acid polymers. Specifically, short ligation-activated oligonucleotides (i.e., di- to octanucleotides) undergo efficient intramolecular ligation (i.e., cyclization), limiting their potential for polymerization (26). It is a general principle in polymer chemistry that the length of polymers formed by irreversible reactions can be greatly limited by cyclization (27). When a nascent polymer becomes sufficiently long to sample conformations that allow for intramolecular bond formation, cyclization occurs, and polymer growth ceases. A polymer’s persistence length (which relates to its rigidity) largely determines the length at which a growing polymer favors cyclization over continued growth. The persistence length of single-stranded nucleic acids is only 3 to 6 nucleotides (28), whereas the persistence length of a base-paired duplex is 150 to 300 base pairs (29). To form appreciable equilibrium amounts of base-paired duplexes, oligonucleotides must be ca. five or more residues in length (30), even at temperatures near the freezing point of water and at moderately high oligonucleotide concentrations (10-4 - 10-3 M). Thus, given a pool of chemically activated di-, tri-, and tetranucleotides, one would predict that such short oligonucleotides would cyclize efficiently. This prediction is borne out by experiment, including model prebiotic reactions in which activated mononucleotides are condensed on a mineral surface, where cyclic products can have yields that are still comparable to those of linear products (12). Even activated hexanucleotides of 100% GC content, designed to assemble into polymeric structures, have been shown to form primarily cyclic products (26). Clearly, strand cyclization would have inhibited the prebiotic production of nucleic acid polymers, unless a mechanism existed to increase the persistence length of oligonucleotides during polymerization.
Here, we report that molecules that intercalate the base pairs of nucleic acid duplexes can circumvent the problem of oligonucleotide cyclization. Specifically, we demonstrate that certain intercalators [or “molecular midwives” (13)] promote the coupling of activated tetranucleotides into long duplex polymers, whereas in the absence of intercalators, only short cyclic oligonucleotides are formed. Further, we show that intercalator-mediated polymer formation is ligand and base-pair specific; size matching is required between the intercalator and the base pairs of a duplex. These data lend support to the hypothesis that the original structure of nucleic acids, including that of the base pairs, could have been templated by intercalators (13, 31).
Results
Intercalators Prevent Oligonucleotide Cyclization Via Duplex Stabilization.
We have previously demonstrated that the free energy associated with the intercalation of nucleic acid duplexes by small, planar molecules can promote the chemical ligation of oligonucleotides (31, 32). However, our previous experimental systems were limited to the coupling of two monofunctional oligonucleotides that were incapable of multiple couplings (i.e., polymerization) and, therefore, they were not subject to the problem of oligonucleotide cyclization. To determine if intercalation-mediated assembly can also circumvent the strand cyclization problem (Fig. 1A), we investigated the chemical ligation of the tetranucleotide d(pCGTA) as a model system. The sequence of this oligonucleotide is such that, when base-paired, it is capable of forming long, extensively nicked concatemeric (or tiling) Watson–Crick duplexes, with the 5′ and 3′ ends of the nick sites in close proximity for chemical ligation (Fig. 1B).
Fig. 1.

Illustration of the strand cyclization problem and intercalation-promoted assembly. (A) Schematic illustration of the strand cyclization problem (Top Route) and the circumvention of this problem by intercalation (Bottom Route). In the absence of intercalators, chemically activated oligonucleotides cyclize; in the presence of intercalators (shown as Gray Rectangles), they polymerize. (B) The nicked duplex resulting from intercalator-mediated assembly of d(pCGTA), used in the experimental studies presented in the text, is also illustrated. Triangles indicate nicks that can be sealed by condensation after chemical activation of the terminal phosphate group.
To test the ability of intercalators to promote oligonucleotide polymerization and prevent cyclization, d(pCGTA) was activated for ligation with N-cyanoimidazole in the presence and absence of ethidium, a well-known intercalator of Watson–Crick duplexes. The early products of the ligation reaction (cyclic tetranucleotide, linear octanucleotide, and cyclic octanucleotide) were monitored by HPLC. Example chromatograms and kinetic traces derived from analysis of these reactions are shown in Fig. 2. In the absence of ethidium, the major product is cyclic tetranucleotide (Fig. 2B), consistent with the very short persistence length of single-stranded oligonucleotides. About one-fourth as much cyclic octanucleotide and only trace linear octanucleotide were detected. The lack of appreciable linear octanucleotide formation indicates that the rate of cyclization is faster than the rate of formation for the linear octanucleotide. In contrast, in the presence of ethidium, linear octanucleotide formation is greatly favored over tetranucleotide cyclization (Fig. 2C). The rate of octanucleotide formation in the presence of ethidium is slower than the rate of tetranucleotide cyclization in the absence of ethidium. Thus, ethidium-mediated assembly of d(pCGTA) inhibits formation of the cyclic tetranucleotide, in addition to promoting linear octanucleotide formation (Fig. 1B).
Fig. 2.

HPLC analysis of early reaction products. (A) HPLC chromatographs of d(pCGTA) (200 μM) activated with N-cyanoimidazole (25 mM) and incubated at 4 °C for 24 h. The Upper Trace is from a reaction carried out in the absence of ethidium, and the Lower Trace is from a reaction carried out in the presence of ethidium (600 μM). Product labels are L4, linear tetranucleotide (starting material); C4, cyclic tetranucleotide; L8, linear octanucleotide; and C8, cyclic octanucleotide. Products were identified using pure standards and phosphatase shift experiments (B) Kinetic analysis of the reaction described in A in the absence of ethidium. (C) Kinetic analysis of the reaction described in A in the presence of ethidium (600 μM). Kinetic analyses were as described in the Materials and Methods.
Ethidium also suppresses strand cyclization in the absence of duplex formation. For example, the initial rate of cyclization of the tetranucleotide d(pCCTA), which cannot tile, is depressed twofold when ethidium is present (Fig. S1). In contrast, the diminution of this rate in the case of d(pCGTA), which does tile, is nearly 2,000-fold, indicating that assembly of oligonucleotides into Watson–Crick-paired duplexes is the dominant mechanism for suppression of cyclization. As we discuss in the follow section, the ability of intercalators to selectively protect base-pairing oligonucleotides from cyclization (and promote their polymerization) could have helped select the first informational polymers of life.
Intercalators Promote Polymer Formation.
The oligonucleotide d (pCGTA) does not ligate appreciably in the absence of an intercalator (Fig. 3). In contrast, when ethidium is added to the same reactions, polymers of up to 100 nucleotides in length are observed (i.e., 24 linear couplings) (Fig. 3). This analysis also illustrates that the ratio of linear to cyclic products increases in the presence of ethidium for all polymer lengths (Fig. 3). For example, relatively low amounts of linear octa- and dodecanucleotide are detected compared to cyclic octa- and dodecanucleotide when ethidium is absent (Fig. 3, Lane 1), whereas approximately equal amounts of linear and cyclic octa- and dodecanucleotide products are observed when ethidium is present at a stoichiometry of one ethidium per tetranucleotide (Fig. 3, Lane 2). For higher ethidium to tetranucleotide stoichiometries, the relative amounts of cyclic products are far lower than linear products of the same length (Fig. 3, Lanes 3–5). An exploration of the range of tetranucleotide concentrations over which ethidium promotes polymerization revealed polymerization at oligonucleotide concentrations as low as 5 μM, whereas, in the absence of ethidium, polymerization is not observed, even at a tetranucleotide concentration of 60 mM (Fig. S2). Thus, ethidium increases the concentration range over which d(pCGTA) can be polymerized by a factor of at least 104.
Fig. 3.

Polyacrylamide gel electrophoresis analysis of reaction products of activated d(pCGTA) in the presence of varying amounts of ethidium. Ethidium concentrations are given above Lanes 1–5. With no intercalator present (Lane marked 0 μM), only small amounts of cyclic octanucleotide (C8) and cyclic dodecanucleotide (C12) are produced, and almost no linear octanucleotide (L8) or linear dodecanucleotide (L12) is produced. The presence of ethidium in the reaction dramatically promotes linear polymerization, to ca. 100mer products (Lanes marked 100 to 600 μM). The tetranucleotide starting material and cyclic tetranucleotide products observed in HPLC analyses did not stain efficiently; band intensities for ≥8 mers are ca. linear with total nucleic acid (Fig. S5). Additional reaction details are provided in Materials and Methods. The gel Lane marked M contains marker bands generated by the enzymatic ligation of a tiling decanucleotide.
Ethidium-Promoted Ligation Requires Watson–Crick Base Pairing.
To demonstrate that the ethidium-promoted ligation of oligonucleotides requires Watson–Crick base pairing (and that nonspecific, i. e., hydrophobic or electrostatic, interactions are insufficient), we examined the ligation of two other oligonucleotides, d(pCCTA) and d(pGGTA). Individually, neither tetranucleotide can form a fully Watson–Crick base-paired tiled duplex, whereas a 1∶1 mixture of the two strands can do so. Neither tetranucleotide alone ligates appreciably when activated, irrespective of the presence of ethidium (Fig. 4). By contrast, ligation occurs when both activated tetranucleotides are in solution with ethidium (Fig. 4), demonstrating the importance of Watson–Crick base pairing in ethidium-mediated ligation. Furthermore, d(TTTT), a tetranucleotide that can neither form a Watson–Crick base-paired tiled duplex nor polymerize on its own (due to lack of a terminal phosphate), does not interfere with the intercalation-mediated polymerization of d(pCGTA), even when it comprises > 99% of the total oligonucleotide present (Fig. S3). Thus, ethidium selectively promotes the polymerization of Watson–Crick base-pairing oligonucleotides, even in the presence of a substantial excess of non-base-pairing oligonucleotides.
Fig. 4.

Condensation reactions demonstrating that ethidium-mediated oligonucleotide polymerization requires Watson–Crick base pairs. Reactions loaded in the Six Right Lanes contained various compositions of d(pCCTA) and d(pGGTA), as indicted above lanes, with each being 200 μM total tetranucleotide and 600 μM ethidium, when present. The efficient ligation demonstrated in Lane 7 was observed only when both complementary oligonucleotides and ethidium were present. Additional reaction details are provided in Materials and Methods.
Intercalators Can Promote the Polymerization of Duplexes Containing Non-Watson–Crick Base Pairs.
Intercalators can drive the assembly of a variety of base-pairing structures. Previously, we have demonstrated that a ligand for pyrimidine triplexes promotes the ligation of two Hoogsteen-paired strands in a triplex (31). With this principle in mind, we sought to achieve ligand-dependent polymerization of nucleic acids by using a system comprised of ligand-dependent non-Watson–Crick base pairs. The recent discovery that the azacyanine aza3 (Fig. 5) promotes the formation of duplex nucleic acids with A·A base pairs (33–38) suggested to us that homo-dA oligonucleotides might be suitable for a ligation system promoted by these molecules. Indeed, we found that aza3 promotes the assembly and ligation of d(pA6). In contrast, ethidium, which promotes the ligation of oligonucleotides that form Watson–Crick base pairs, does not promote the ligation of d(pA6) (Fig. 5). Similarly, aza3, which has low affinity for Watson–Crick duplexes (39), does not promote the ligation of d(pCGTA) (Fig. 5).
Fig. 5.

Base pair recognition by a given intercalator is a necessary, but not sufficient, condition for achieving intercalation-mediated ligation. (A) PAGE analysis of products from the condensation of d(pCGTA) (200 μM) in the absence and the presence of various small molecules (600 μM when present). Relative to the ligand-free reaction (Lane 1), ethidium (Lane 2) promotes the polymerization of d(pCGTA), whereas proflavine (Lane 3), another Watson–Crick intercalator, does not. Neither coralyne (Lane 4) nor aza3 (Lane 5), which binds homo-A duplexes, promotes d(pCGTA) ligation. (B) PAGE analysis of products formed by the condensation of d(pA6) (500 μM) in the absence and the presence of various small molecules (750 μM when present, lanes as in A). Relative to the ligand-free reaction (Lane 1), neither Watson–Crick intercalator promotes the ligation of d(pA6) (Lanes 2 and 3). In contrast, aza3 (Lane 5) promotes the ligation of d(pA6). Although coralyne also binds homo-A duplexes, it promotes the ligation of d(pA6) only weakly (Lane 4). The N1–N7 base pair shown for homo-adenine duplexes has been shown to be consistent with molecular modeling and chemical probing studies (33). Additional reaction details are provided in Materials and Methods. The gel Lane marked M in panel A contains marker bands generated by ligation of a tiling decanucleotide, the gel Lane marked M in panel B contains DNA oligonucleotides of length 10, 32, and 110 nt.
Although coralyne binds duplexes with A·A base pairs with an even greater affinity than aza3 (34), coralyne promotes d(pA6) ligation only weakly. Similarly, proflavine, which binds Watson–Crick DNA with an affinity comparable to that of ethidium, does not promote ligation (Fig. 5). Taken together, these data illustrate that molecular recognition between an intercalator and base pair may be necessary for the intercalation-mediated stabilization of a duplex structure, but it is clearly not sufficient for the promotion of intercalation-mediated polymerization.
Discussion
It is widely assumed that the abiotic synthesis and template-directed replication of nucleic acid polymers (or their predecessors) was an early and critical step in the origin of life (40–43). Strand cyclization is an inherent and general obstacle to the growth of polymers from bifunctional mono- and oligomers, and it, therefore, would have thwarted the abiotic synthesis of early phosphodiester-linked RNA or RNA-like polymers, unless a mechanism existed to organize mono- and short oligonucleotides prior to the bond formation step of polymerization. Previous chemical polymerization studies required moderate to high concentrations of di- and tetranucleotide substrates (10-3 - 10-1 M) (44, 45), or the use of some monofunctional substrates to prevent strand cyclization, albeit at the expense of chain termination (41). Here, we have shown that intercalator binding can circumvent strand cyclization and enable polymerization, even at low (i.e., 10-6 M) substrate concentrations. Additionally, intercalators can drive the polymerization of base-pairing oligonucleotides, even when they are a trace component (< 1%) in a high-concentration pool of a non-base-pairing, non-polymerizable oligonucleotide. While we do not propose that the intercalators used in the present study are prebiotic, it is possible that the reactions that gave rise to the first nucleobases produced other heterocycles that might have functioned as molecular midwives for the base-pair selection, assembly, and polymerization of the earliest proto-RNA polymers.
Here, we have presented results from experiments involving the ligation of DNA oligonucleotides that provide an experimentally practical model system for exploring intercalation-promoted polymerization. Nevertheless, these results are also relevant to the nonenzymatic polymerization of RNA that has a duplex persistence length somewhat greater than that of DNA (29), but a single-stranded persistence length that still renders RNA oligonucleotides susceptible to cyclization (12). Indeed, activated r(pCGUA) exhibits enhanced ligation in the presence of ethidium, albeit to a much lesser extent than observed for d(pCGTA) (Fig. S6). The diminished performance of RNA in this system might result from the activation chemistry and intercalators tested thus far being less compatible with RNA oligonucleotides or the intercalated A-form helix. As noted above, not all intercalators tested were found to facilitate DNA ligation, even though some possess binding affinities comparable to those that do facilitate ligation. Current efforts in our laboratory include the search for an intercalator that more efficiently promotes RNA ligation.
Consistent with our previous work, the present ligation system exhibits sensitivity to the particular intercalator used to promote assembly, even among intercalators that exhibit similar affinity for the duplex to be ligated. In particular, proflavine did not promote ligation in our Watson–Crick system, despite the fact that proflavine and ethidium have similar association constants for Watson–Crick duplex DNA. Interestingly, in a previous study of a ligation system using a different activation chemistry, we found that proflavine, but not ethidium, promotes oligonucleotide ligation (32). Thus, the efficiency of an intercalator for promoting a given ligation reaction appears not to be fully expressed by its association constant for a nucleic acid assembly. In retrospect, this observation is not surprising; covalent bond formation requires an energetically accessible reactant complex with appropriate bond geometries, and individual intercalators make structure-specific helical contacts that could modify the conformational landscape of the polymer backbone in an intercalator-specific manner. This feature could prove useful for selecting specific backbone linkages in intercalation-mediated reactions.
We have used tetranucleotides as a model system to illustrate the nonenzymatic production of polymers from short oligonucleotides. With regard to a more complete model for the origin of RNA-like polymers, it is certainly desirable to demonstrate polymerization with even shorter oligonucleotides and, ultimately, mononucleotides. Thus far, our attempts to use intercalators to drive the polymerization of mononucleotides and dinucleotides in the absence of a preexisting template strand have proven unsuccessful. In the case of dinucleotides, we surmise that the nearest neighbor exclusion principle (46–48), which states that intercalators can bind at most between every other base pair, could potentially inhibit the coupling of dinucleotides, as dinucleotides are known to crystallize in the presence of intercalators with ligands bound both within and outside each minihelix (49), an arrangement that could block backbone coupling between dinucleotides. To find a possible means by which to circumvent this problem, we have initiated studies of intercalation of nucleic acids with alternative backbone linkages (50, 51). Given the wide variety of RNA-like polymers with nucleoside elements other than ribofuranosides that also form duplexes (11, 52, 53), it is possible that the backbone of the first informational polymers may not have utilized ribofuranosides. If this was the case, the original pre-RNA backbone might have allowed intercalation between every dinucleotide step. With regard to mononucleotide polymerization, it appears that the poor stacking of the pyrimidine nucleotides precludes assembly of mononucleotides by intercalating molecular midwives. The poor stacking of pyrimidine bases has prompted our laboratory, and several others (54–56), to reconsider an early proposal by Crick that the first RNA-like polymers might have used purine-purine base pairs (57). Our demonstration in the present work that intercalators can promote the polymerization of homo-adenine oligonucleotides represents the first step towards the realization of intercalation-mediated mononucleotide polymerization in a homo-purine system.
Recently, Szostak and coworkers demonstrated the nonenzymatic template-directed synthesis of RNA inside model protocells (58). These model protocells exhibited high permeability for small, minimally charged species. Therefore, before the advent of protein enzymes, molecules similar to the intercalator midwives discussed in the present work, which contain only a single positive charge, could have permeated protocells along with nucleotides, thus assisting in the assembly of encapsulated nucleic acid polymers.
Finally, it was recently proposed that the tendency for Watson–Crick DNA duplexes to form liquid crystals could reflect the process by which prebiotic nucleic acids were organized (and partitioned) prior to polymerization (59). The experimental results presented here demonstrate that intercalators can promote the assembly of oligonucleotides of comparable length at concentrations on the order of 100,000-fold lower than those required for liquid crystal formation (59).
Materials and Methods
Oligonucleotides were purchased from Oligos Etc. or IDT and HPLC purified before use. Ethidium bromide (Fisher Scientific), proflavine hemisulfate (Sigma), and coralyne chloride (Sigma) were used as received. aza3 was synthesized as previously reported (60). N-cyanoimidazaole was purchased from Toronto Research Chemicals.
Watson–Crick-Paired Chemical Ligations.
Ligation reactions were 200 μM in oligonucleotide strand (unless otherwise stated), 10 mM triethylammonium MES (pH 6), and 5 mM MnCl2. Ligation reactions in the presence of various divalent ions demonstrated that MnCl2 resulted in the highest yield of condensation products (Fig. S4). For reactions containing intercalators, specific molecules and concentrations used are provided in figure legends. After 15 min. equilibration at 4 °C, N-cyanoimidazole (from an H2O stock) was added to 250 mM. Ligation reactions were incubated for 72 h at 4 ºC, unless otherwise stated. The reaction products were then ethanol precipitated and resuspended in PAGE loading buffer. In reactions containing < 200 μM oligonucleotide, linear polyacrylamide was used as a carrier in precipitations. Reaction products were separated by using denaturing (7 M urea) PAGE (19% acrylamide:1% N, N′-methylenebisacrylamide) in TBE running buffer. The gels were then stained with SYBR Gold (Invitrogen) for band visualization.
A•A Paired Chemical Ligations.
Ligation reactions contained 500 μM d(pA6), 10 mM triethylammonium MES (pH 6), and 5 mM MnCl2. Reactions with intercalators contained 750 μM intercalator. Ligation reactions were incubated for 24 h at 4 ºC. Reactions products were separated by using denaturing (7 M urea) PAGE (19% acrylamide:1% N, N′-methylenebisacrylamide) with TBE running buffer. Gels were then stained with SYBR Gold for band visualization.
HPLC and Kinetic Analysis of Early Reaction Products.
Reactions contained 200 μM d(pCGTA), 5 mM MnCl2, 10 mM triethylammonium MES (pH 6), 600 μM ethidium (when present), and 25 mM N-cyanoimidazole. The reactions were incubated at 4 °C. At each time point, a 10 μL aliquot was removed and diluted in 90 μL 22 mM EDTA to quench the reaction. The aliquot was then immediately chromatographed (Agilent 1100, 4.6 mm × 250 mm Phenomenex Luna C18, ambient temperature). Gradient: Solvent A = 100 mM triethylammonium acetate, pH 7. Solvent B = Acetonitrile. 0–12 min, 7.5% B. 12–20 min, 7.5–20% B. 20–25 min, 20–70% B. 25–30 min, 70% B.
Supplementary Material
Acknowledgments.
This work was supported by the National Aeronautics and Space Administration (NNX08A014G) and National Science Foundation (CHE-0739189).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/cgi/content/full/0914172107/DCSupplemental.
References
- 1.Joyce GF. RNA evolution and the origins of life. Nature. 1989;338:217–224. doi: 10.1038/338217a0. [DOI] [PubMed] [Google Scholar]
- 2.Gesteland RF, Cech TR, Atkins JF. The RNA World. 3rd Ed. Cold Spring harbor, NY: Cold Spring Harbor Laboratory Press; 2006. p. 768. [Google Scholar]
- 3.Fedor MJ, Williamson JR. The catalytic diversity of RNAs. Nat Rev Mol Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 4.Ellington AD, Chen X, Robertson M, Syrett A. Evolutionary origins and directed evolution of RNA. Int J Biochem Cell Biol. 2009;41:254–265. doi: 10.1016/j.biocel.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 5.Cech TR. Structural biology—the ribosome is a ribozyme. Science. 2000;289:878–879. doi: 10.1126/science.289.5481.878. [DOI] [PubMed] [Google Scholar]
- 6.Heuberger BD, Switzer C. A pre-RNA candidate revisited: both enantiomers of flexible nucleoside triphosphates are DNA polymerase substrates. J Am Chem Soc. 2008;130:412–413. doi: 10.1021/ja0770680. [DOI] [PubMed] [Google Scholar]
- 7.Mittapalli GK, et al. Mapping the landscape of potentially primordial informational oligomers: oligodipeptides tagged with 2,4-disubstituted 5-aminopyrimidines as recognition elements. Angew Chem Int Edit. 2007;46:2478–2484. doi: 10.1002/anie.200603209. [DOI] [PubMed] [Google Scholar]
- 8.Bean HD, Anet FAL, Gould IR, Hud NV. Glyoxylate as a backbone linkage for a prebiotic ancestor of RNA. Origins Life Evol B. 2006;36:39–63. doi: 10.1007/s11084-005-2082-4. [DOI] [PubMed] [Google Scholar]
- 9.Benner SA, Ricardo A, Carrigan MA. Is there a common chemical model for life in the universe? Curr Opin Chem Biol. 2004;8:672–689. doi: 10.1016/j.cbpa.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Benner SA. Understanding nucleic acids using synthetic chemistry. Acc Chem Res. 2004;37:784–797. doi: 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]
- 11.Eschenmoser A. Chemical etiology of nucleic acid structure. Science. 1999;284:2118–2124. doi: 10.1126/science.284.5423.2118. [DOI] [PubMed] [Google Scholar]
- 12.Ertem G, Ferris JP. Template-directed synthesis using the heterogeneous templates produced by montmorillonite catalysis. A possible bridge between the prebiotic and RNA worlds. J Am Chem Soc. 1997;119:7197–7201. doi: 10.1021/ja970422h. [DOI] [PubMed] [Google Scholar]
- 13.Hud NV, Anet FAL. Intercalation-mediated synthesis and replication: A new approach to the origin of life. J Theor Biol. 2000;205:543–562. doi: 10.1006/jtbi.2000.2084. [DOI] [PubMed] [Google Scholar]
- 14.Leitzel JC, Lynn DG. Template-directed ligation: from DNA towards more versatile templates. Chem Rec. 2001;1:53–62. doi: 10.1002/1528-0691(2001)1:1<53::AID-TCR8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Costanzo G, Saladino R, Crestini C, Ciciriello F, Di Mauro E. Nucleoside phosphorylation by phosphate minerals. J Biol Chem. 2007;282:16729–16735. doi: 10.1074/jbc.M611346200. [DOI] [PubMed] [Google Scholar]
- 16.Anastasi C, et al. RNA: Prebiotic product, or biotic invention? Chem Biodivers. 2007;4:721–739. doi: 10.1002/cbdv.200790060. [DOI] [PubMed] [Google Scholar]
- 17.Rajamani S, et al. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Origins Life Evol B. 2008;38:57–74. doi: 10.1007/s11084-007-9113-2. [DOI] [PubMed] [Google Scholar]
- 18.Huang W, Ferris JP. One-step, regioselective synthesis of up to 50-mers of RNA oligomers by montmorillonite catalysis. J Am Chem Soc. 2006;128:8914–8919. doi: 10.1021/ja061782k. [DOI] [PubMed] [Google Scholar]
- 19.Sawai H, Totsuka S, Yamamoto K, Ozaki H. Non-enzymatic, template-directed ligation of 2′-5′ oligoribonucleotides. Joining of a template and a ligator strand. Nucleic Acids Res. 1998;26:2995–3000. doi: 10.1093/nar/26.12.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawai H, Kuroda K, Hojo T. Uranyl-ion as a highly effective catalyst for internucleotide bond formation. Bull Chem Soc Jpn. 1989;62:2018–2023. [Google Scholar]
- 21.Streltsov SA, et al. Trivaline ‘catalyzes’ 5′-pdGTT oligomerization in solution. FEBS Lett. 1992;298:57–60. doi: 10.1016/0014-5793(92)80021-8. [DOI] [PubMed] [Google Scholar]
- 22.Saladino R, Crestini C, Cicirielloc F, Costanzo G, Di Mauro E. Formamide chemistry and the origin of informational polymers. Chem Biodivers. 2007;4:694–720. doi: 10.1002/cbdv.200790059. [DOI] [PubMed] [Google Scholar]
- 23.Joyce G, et al. Chiral selection in poly(C)-directed synthesis of oligo(G) Nature. 1984;310:602–604. doi: 10.1038/310602a0. [DOI] [PubMed] [Google Scholar]
- 24.Joyce GF, Inoue T, Orgel LE. Non-enzymatic template-directed synthesis on RNA random copolymers—poly(C,U) templates. J Mol Biol. 1984;176:279–306. doi: 10.1016/0022-2836(84)90425-x. [DOI] [PubMed] [Google Scholar]
- 25.Weimann BJ, Lohrmann R, Orgel LE, Schneider-Bernloehr H, Sulston JE. Template-directed synthesis with adenosine-5′-phosphorimidazolide. Science. 1968;161:387. doi: 10.1126/science.161.3839.387. [DOI] [PubMed] [Google Scholar]
- 26.Kawamura K, Okamoto F. Cyclization and dimerization of hexanucleotides containing guanine and cytosine with water-soluble carbodiimide. Viva Origino. 2001;29:162–167. doi: 10.1093/nass/44.1.217. [DOI] [PubMed] [Google Scholar]
- 27.Eichinger BE. Cyclization in reversible and irreversible step-growth polymerizations. Comput Theor Polym S. 2000;10:83–88. [Google Scholar]
- 28.Mills JB, Vacano E, Hagerman PJ. Flexibility of single-stranded DNA: Use of gapped duplex helices to determine the persistence lengths of poly(dT) and poly(dA) J Mol Biol. 1999;285:245–257. doi: 10.1006/jmbi.1998.2287. [DOI] [PubMed] [Google Scholar]
- 29.Kebbekus P, Draper DE, Hagerman P. Persistence length of RNA. Biochemistry. 1995;34:4354–4357. doi: 10.1021/bi00013a026. [DOI] [PubMed] [Google Scholar]
- 30.Porschke D. Elementary steps of base recognition and helix-coil transitions in nucleic acids. Mol Biol Biochem Biophys. 1977;24:191–218. doi: 10.1007/978-3-642-81117-3_5. [DOI] [PubMed] [Google Scholar]
- 31.Hud NV, Jain SS, Li X, Lynn DG. Addressing the problems of base pairing and strand cyclization in template-directed synthesis—A case for the utility and necessity of ‘molecular midwives’ and reversible backbone linkages for the origin of proto-RNA. Chem Biodivers. 2007;4:768–783. doi: 10.1002/cbdv.200790063. [DOI] [PubMed] [Google Scholar]
- 32.Jain SS, Anet FAL, Stahle CJ, Hud NV. Enzymatic behavior by intercalating molecules in a template-directed ligation reaction. Angew Chem Int Ed Engl. 2004;43:2004–2008. doi: 10.1002/anie.200353155. [DOI] [PubMed] [Google Scholar]
- 33.Joung IS, Çetinkol ÖP, Hud NV, Cheatham TE. Molecular dynamics simulations and coupled nucleotide substitution experiments indicate the nature of A•A base pairing and a putative structure of the coralyne-induced homo-adenine duplex. Nucleic Acids Res. 2009;37:7715–7727. doi: 10.1093/nar/gkp730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Çetinkol ÖP, Hud NV. Molecular recognition of poly(A) by small ligands: An alternative method of analysis reveals nanomolar, cooperative and shape-selective binding. Nucleic Acids Res. 2009;37:611–621. doi: 10.1093/nar/gkn977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Persil Ö, Santai CT, Jain SS, Hud NV. Assembly of an antiparallel homo-adenine DNA duplex by small-molecule binding. J Am Chem Soc. 2004;126:8644–8645. doi: 10.1021/ja0492891. [DOI] [PubMed] [Google Scholar]
- 36.Xing F, Song G, Ren J, Chaires JB, Qu X. Molecular recognition of nucleic acids: Coralyne binds strongly to poly(A) FEBS Lett. 2005;579:5035–5039. doi: 10.1016/j.febslet.2005.07.091. [DOI] [PubMed] [Google Scholar]
- 37.Jain SS, Polak M, Hud NV. Controlling nucleic acid secondary structure by intercalation: Effects of DNA strand length on coralyne-driven duplex disproportionation. Nucleic Acids Res. 2003;31:4608–4615. doi: 10.1093/nar/gkg648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polak M, Hud NV. Complete disproportionation of duplex poly(dT)•poly(dA) into triplex poly(dT)•poly(dA)•poly(dT) and poly(dA) by coralyne. Nucleic Acids Res. 2002;30:983–992. doi: 10.1093/nar/30.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Çetinkol ÖP, Engelhart AE, Nanjunda RK, Wilson WD, Hud NV. Submicromolar, selective G-quadruplex ligands from one pot: Thermodynamic and structural studies of human telomeric DNA binding by azacyanines. ChemBioChem. 2008;9:1889–1892. doi: 10.1002/cbic.200800234. [DOI] [PubMed] [Google Scholar]
- 40.James KD, Ellington AD. Surprising fidelity of template-directed chemical ligation of oligonucleotides. Chem Biol. 1997;4:595–605. doi: 10.1016/s1074-5521(97)90245-3. [DOI] [PubMed] [Google Scholar]
- 41.Sievers D, von Kiedrowski G. Self replication of complementary nucleotide-based oligomers. Nature. 1994;369:221–224. doi: 10.1038/369221a0. [DOI] [PubMed] [Google Scholar]
- 42.von Kiedrowski G. A self-replicating hexadeoxynucleotide. Angew Chem Int Ed Engl. 1986;25:932–935. [Google Scholar]
- 43.Orgel LE, Lohrmann R. Prebiotic chemistry and nucleic acid replication. Acc Chem Res. 1974;7:368–377. [Google Scholar]
- 44.Zielinski WS, Orgel LE. Oligoaminonucleoside phosphoramidates. Oligomerization of dimers of 3′-amino-3′-deoxy-nucleotides (GC and CG) in aqueous solution. Nucleic Acids Res. 1987;15:1699–1715. doi: 10.1093/nar/15.4.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolli M, Micura R, Eschenmoser A. Pyranosyl-RNA: chiroselective self-assembly of base sequences by ligative oligomerization of tetra nucleotide-2′,3′-cyclophosphates (with a commentary concerning the origin of biomolecular homochirality) Chem Biol. 1997;4:309–320. doi: 10.1016/s1074-5521(97)90074-0. [DOI] [PubMed] [Google Scholar]
- 46.Arnott S, Bond PJ, Chandrasekaran R. Visualization of an unwound DNA duplex. Nature. 1980;287:561–563. doi: 10.1038/287561a0. [DOI] [PubMed] [Google Scholar]
- 47.Bond PJ, Langridge R, Jennette KW, Lippard SJ. X-Ray fiber diffraction evidence for the neighbor exclusion binding of a platinum metallointercalation reagent to DNA. Proc Natl Acad Sci USA. 1975;72:4825–4829. doi: 10.1073/pnas.72.12.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crothers DM. Calculation of binding isotherms for heterogeneous polymers. Biopolymers. 1968;6:575–584. doi: 10.1002/bip.1968.360060411. [DOI] [PubMed] [Google Scholar]
- 49.Shieh H-S, Berman HM, Dabrow M, Neidle S. The structure of a drug-deoxydinucleoside phosphate complex; generalized conformational behavior of intercalation complexes with RNA and DNA fragments. Nucleic Acids Res. 1980;8:85–97. doi: 10.1093/nar/8.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horowitz ED, Hud NV. Ethidium and proflavine binding to a 2′,5′-linked RNA duplex. J Am Chem Soc. 2006;128:15380–15381. doi: 10.1021/ja065339l. [DOI] [PubMed] [Google Scholar]
- 51.Horowitz ED, Lilavivat S, Holladay BW, Germann MW, Hud NV. Solution structure and thermodynamics of 2′,5′ RNA intercalation. J Am Chem Soc. 2009;131:5831–5838. doi: 10.1021/ja810068e. [DOI] [PubMed] [Google Scholar]
- 52.Schöning KU, et al. Chemical etiology of nucleic acid structure: The alpha-threofuranosyl-(3′- > 2′) oligonucleotide system. Science. 2000;290:1347–1351. doi: 10.1126/science.290.5495.1347. [DOI] [PubMed] [Google Scholar]
- 53.Eschenmoser A. Searching for nucleic acid alternatives. Chimia. 2005;59:836–850. [Google Scholar]
- 54.Groebke K, et al. Why pentose- and not hexose-nucleic acids? Purine-purine pairing in homo-DNA: Guanine, isoguanine, 2,6-diaminopurine, and xanthine. Helv Chim Acta. 1998;81:375–474. [Google Scholar]
- 55.Battersby TR, Albalos M, Friesenhahn MJ. An unusual mode of DNA duplex association: Watson–Crick interaction of all-purine deoxyribonucleic acids. Chem Biol. 2007;14:525–531. doi: 10.1016/j.chembiol.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 56.Heuberger BD, Switzer C. An alternative nucleobase code: characterization of purine-purine DNA double helices bearing guanine-isoguanine and diaminopurine-7-deaza-xanthine base pairs. ChemBioChem. 2008;9:2779–2783. doi: 10.1002/cbic.200800450. [DOI] [PubMed] [Google Scholar]
- 57.Crick FHC. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 58.Mansy SS, et al. Template-directed synthesis of a genetic polymer in a model protocell. Nature. 2008;454:122–125. doi: 10.1038/nature07018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakata M, et al. End-to-end stacking and liquid crystal condensation of 6-to 20-base pair DNA duplexes. Science. 2007;318:1276–1279. doi: 10.1126/science.1143826. [DOI] [PubMed] [Google Scholar]
- 60.Huang KS, Haddadin MJ, Olmstead MM, Kurth MJ. Synthesis and reactions of some heterocyclic azacyanines. J Org Chem. 2001;66:1310–1315. doi: 10.1021/jo001484k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.