

Abstract
Anthropogenic activities, dominated by emissions of sulfur dioxide (SO2), have perturbed the global sulfur (S) cycle. Uncertainties in timescales of S transport and chemistry in the atmosphere lead to uncertainties in the predicted impact of S emissions. Measurements of cosmogenic 35S may potentially be used to resolve existing uncertainties in the photochemical and chemical transformation of S in the environment. The lack of a simple, effective, and highly sensitive technique to measure 35S activity in samples with low activities may explain the scarcity of published measurements. We present a set of new sample handling and measurement procedures optimized for the measurement of 35S in natural samples with activities as low as 0.20 dpm above background (2σ, integration time = 2 hr). We also report simultaneous measurements of aerosol (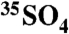 ) and gas phase (
) and gas phase (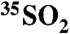 ) collected at inland and coastal locations; the range of observed activities corresponds to SO2 residence lifetimes of 0.2 ± 0.04 (coastal) - 22.3 d ± 0.04 (inland). These optimized techniques offer the potential for resolving atmospheric processes that occur on 6–12-hour timescales as well as resolving transport phenomena such as stratospheric mixing into the troposphere.
) collected at inland and coastal locations; the range of observed activities corresponds to SO2 residence lifetimes of 0.2 ± 0.04 (coastal) - 22.3 d ± 0.04 (inland). These optimized techniques offer the potential for resolving atmospheric processes that occur on 6–12-hour timescales as well as resolving transport phenomena such as stratospheric mixing into the troposphere.
Keywords: aerosol sulfate, sulfur cycle, dry deposition, sulfur dioxide residence times
According to the Intergovernmental Panel on Climate Change (1) anthropogenic emissions of sulfur dioxide have perturbed the sulfur cycle on local, regional, and global scales. These emissions and their oxidized end products lead to increases in acid rain (H2SO4) and aerosol sulfate (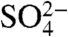 ) concentrations that play a significant role in climate and the global sulfur cycle. Sulfur-containing aerosol particles serve as cloud condensation nuclei affecting cloud formation and the hydrological cycle. Our knowledge of the chemical and photochemical processes that govern the chemical transformations and transport of sulfur compounds in the atmosphere is incomplete due to the complex, multivalent nature of sulfur and uncertainties in the understanding of aerosol chemistry. Sulfur in the atmosphere exists simultaneously as a solid and gas, further complicating matters. The development of new and/or improved analytical techniques to study the sulfur cycle on short timescales (hours to days) is, therefore, of considerable importance. Here we describe significant advances in the detection sensitivity of the cosmogenic isotope 35S that can be used as a tracer of gas and aerosol phase lifetimes and turnover kinetics with high time and aerosol size resolutions.
) concentrations that play a significant role in climate and the global sulfur cycle. Sulfur-containing aerosol particles serve as cloud condensation nuclei affecting cloud formation and the hydrological cycle. Our knowledge of the chemical and photochemical processes that govern the chemical transformations and transport of sulfur compounds in the atmosphere is incomplete due to the complex, multivalent nature of sulfur and uncertainties in the understanding of aerosol chemistry. Sulfur in the atmosphere exists simultaneously as a solid and gas, further complicating matters. The development of new and/or improved analytical techniques to study the sulfur cycle on short timescales (hours to days) is, therefore, of considerable importance. Here we describe significant advances in the detection sensitivity of the cosmogenic isotope 35S that can be used as a tracer of gas and aerosol phase lifetimes and turnover kinetics with high time and aerosol size resolutions.
Stable isotopic measurements of atmospheric species such as nitrate (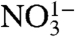 ) and sulfate (
) and sulfate (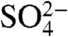 ) have recently been used to provide strong constraints on the oxidative processing of their precursors, NOx and SOx, in the atmosphere (2–6). The radionuclide 35S (β-decay to 35Cl, t1/2 = 87.4 d) is continuously produced in the atmosphere by the interaction of cosmic rays with 40Ar and provides an additional opportunity for tracing atmospheric processes. Upon production, 35S rapidly oxidizes to 35SO (lifetime < 1 ms) and to
) have recently been used to provide strong constraints on the oxidative processing of their precursors, NOx and SOx, in the atmosphere (2–6). The radionuclide 35S (β-decay to 35Cl, t1/2 = 87.4 d) is continuously produced in the atmosphere by the interaction of cosmic rays with 40Ar and provides an additional opportunity for tracing atmospheric processes. Upon production, 35S rapidly oxidizes to 35SO (lifetime < 1 ms) and to 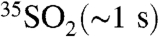 (7, 8). In the atmosphere,
(7, 8). In the atmosphere, 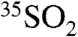 undergoes wet deposition (removal below and within clouds), dry deposition (gravimetric settling or interactions with surfaces), or may be oxidized to sulfate (
undergoes wet deposition (removal below and within clouds), dry deposition (gravimetric settling or interactions with surfaces), or may be oxidized to sulfate (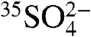 ) and incorporated onto aerosol particles (which eventually undergo dry and wet deposition). The sink reactions occur on timescales of a few hours to a few days, depending on the local atmospheric environment; thus the concentration of
) and incorporated onto aerosol particles (which eventually undergo dry and wet deposition). The sink reactions occur on timescales of a few hours to a few days, depending on the local atmospheric environment; thus the concentration of 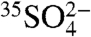 and
and 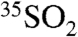 is expected to vary significantly as a function of time, meteorology, humidity, and location. These variables can vary on timescales of hours to days, which highlights the need of the present work of developing high time resolution measurement capabilities.
is expected to vary significantly as a function of time, meteorology, humidity, and location. These variables can vary on timescales of hours to days, which highlights the need of the present work of developing high time resolution measurement capabilities.
The chemical properties of 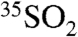 and
and 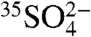 are expected to be nearly identical to SO2 and
are expected to be nearly identical to SO2 and 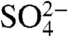 , respectively. Measurements of 35S in aerosol sulfate may be used to better resolve aerosol aging and chemistry, lifetimes of aerosol
, respectively. Measurements of 35S in aerosol sulfate may be used to better resolve aerosol aging and chemistry, lifetimes of aerosol 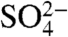 and gas phase SO2, and provide a better measure of boundary layer dynamics (9, 10). Previous 35S measurements have been used to calculate SO2 fluxes (9, 11–14) and depositional rates (wet and dry) (9, 12, 15, 16) and trace atmospheric sulfate deposition into lakes, rivers (17), and catchments (18–22). Some of these papers have included measurements of
and gas phase SO2, and provide a better measure of boundary layer dynamics (9, 10). Previous 35S measurements have been used to calculate SO2 fluxes (9, 11–14) and depositional rates (wet and dry) (9, 12, 15, 16) and trace atmospheric sulfate deposition into lakes, rivers (17), and catchments (18–22). Some of these papers have included measurements of 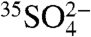 in bulk aerosols (9, 23).
in bulk aerosols (9, 23).
Because the atmospheric aerosol chemistry of sulfur depends on the type, size, and number density of aerosol particles, one of the principal goals of this work is to improve existing measurement techniques so as to obtain particle-size-resolved measurements of 35S in aerosol sulfate without the need to collect samples for long periods of time that may mask short-term variability.
Recently, a technique using low-level liquid scintillation spectroscopy (LSS) was developed to measure 35S in lake water (17). Our review of this technique revealed that the methods used introduced high backgrounds, prohibiting their use for determining the activities of natural samples with low 35S/S ratios. Here we present improved sample handling and analysis techniques employing LSS and our first field results. The improvements include unique sample preparation procedures, identification and correction of previously unreported backgrounds, as well as a method for optimizing the integration of scintillation spectra. Our improved methods allow for the measurement of the abundance of 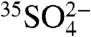 as a function of aerosol size as well as
as a function of aerosol size as well as 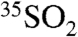 with sample collection times as short as 12 hr at a coastal and an inland location at similar altitudes and latitudes. Using these measurements, we estimated the overall lifetime of SO2 at these two locations.
with sample collection times as short as 12 hr at a coastal and an inland location at similar altitudes and latitudes. Using these measurements, we estimated the overall lifetime of SO2 at these two locations.
The increased sensitivity to 35S concentrations presented in this paper should allow for the study of a broader range of environmental processes such as boundary layer dynamics, evolution of sulfate in aircraft plumes in the lower stratosphere, deposition of sulfate into the hydrological cycle, kinetics of SO2 oxidation, and aerosol dynamics (including the transport and evolution of sulfur in remote locations such as Antarctica). In summary, the enhanced sensitivity of our method expands the range of biogeochemical processes that can now be explored.
Results and Discussion
Method Testing.
A Wallac Quantalus 1220 Ultra Low-Level Liquid Scintillation Counter was used for all the measurements and optimization tests that we report here. This instrument minimizes cosmic ray backgrounds using passive and active shielding, multiple multichannel analyzers, and anticoincidence counting techniques. A 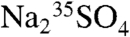 standard (MP Biomedical) was used to optimize the sample preparation techniques. The instrument’s detection efficiency (εD) was determined using a calibrated (absolute) 14C activity standard from Perkin Elmer (dpm = 96,200)* (24). By comparing the instrument reported count rate (counts per minute, cpm) and the expected activity (disintegrations per minute, dpm) for the absolute standard, the instrument’s overall detection efficiency (
standard (MP Biomedical) was used to optimize the sample preparation techniques. The instrument’s detection efficiency (εD) was determined using a calibrated (absolute) 14C activity standard from Perkin Elmer (dpm = 96,200)* (24). By comparing the instrument reported count rate (counts per minute, cpm) and the expected activity (disintegrations per minute, dpm) for the absolute standard, the instrument’s overall detection efficiency (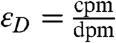 ) was determined to be 0.946. This efficiency is an upper limit to the overall efficiency of the detection technique presented here, since natural samples will also incur losses in their measured activity by being lost during preparation. Given the high activity of the 14C standard, this efficiency determination is unaffected by electronic noise or sample vial backgrounds.
) was determined to be 0.946. This efficiency is an upper limit to the overall efficiency of the detection technique presented here, since natural samples will also incur losses in their measured activity by being lost during preparation. Given the high activity of the 14C standard, this efficiency determination is unaffected by electronic noise or sample vial backgrounds.
Energy channel optimization.
The detection system bins photon pulse events into one of 1,024 energy channels, with the higher channels corresponding to highest energies. The β-decay spectrum of 35S is well defined with a maximum decay energy of 0.1675 MeV (15). If the energy spectra of different cosmogenic species are sufficiently distinct, partial-to-complete discrimination between decay events of 35S and those of other radio-nuclides present in the sample (e.g. 14C) can be achieved by selective integration of the energy spectra. Fig. S1 displays the spectra measured for 35S, 14C, and radioactive isotopes of barium. While each channel contributes noise to the integrated count rate, integrating counts only in the energy range where 35S decay events occur results in improved signal to noise ratios. In collected aerosol and gas samples, organic molecules containing 14C may contribute to the background activity, and our optimization considers these contributions. Using the spectra shown in Fig. S1, the ratio of the time- and channel-integrated signal [Sm(ch1,ch2)] and background [Bm(ch1,ch2)] ratio can be used to optimize the counting of samples and is defined as
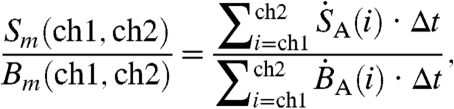 |
[1] |
where ch1 and ch2 are the start and end channels of integration, respectively, and 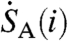 and
and 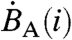 are the observed count rates in channel i of 35S and interfering backgrounds (including 14C, radioactive barium isotopes, and/or scintillation gel). This ratio is plotted separately for the backgrounds expected from barium, the scintillation gel, and 14C as a function of ch2 in Fig. S2. The ratio of
are the observed count rates in channel i of 35S and interfering backgrounds (including 14C, radioactive barium isotopes, and/or scintillation gel). This ratio is plotted separately for the backgrounds expected from barium, the scintillation gel, and 14C as a function of ch2 in Fig. S2. The ratio of 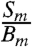 reaches a maximum value close to the channel where the 35S decay spectrum reaches its maximum (ch2 ∼ 450). Integration between 1 (ch1) to 445 (ch2) and 455 (ch2) captures 95% and 97% of all decay events under the 35S spectrum shown in Fig. S2 while minimizing background contributions.
reaches a maximum value close to the channel where the 35S decay spectrum reaches its maximum (ch2 ∼ 450). Integration between 1 (ch1) to 445 (ch2) and 455 (ch2) captures 95% and 97% of all decay events under the 35S spectrum shown in Fig. S2 while minimizing background contributions.
Taking measurements of β- activities using LSS requires using a scintillation gel that fluoresces when excited by emitted β- particles as well as a vial to hold the sample-gel mixture. The choice of scintillation vial material, preparation techniques, and the amount of scintillation gel volume used can affect the sensitivity of measurements in LSS.
Major gains in sensitivity were attained by minimizing the background activities of scintillation vials and reagents used in the preparation of samples. The most significant background reductions were obtained by using plastic vials (Fisherbrand, 20 mL, ∼0.145 dpm) as opposed to glass vials (Wheaton Glass-20 mL, 0.45 dpm). Similarly, glass fiber filters (Whatman GF/B), like those recommended by Hong and Kim (17) to isolate and count lake water sulfate as BaSO4, also possessed relatively high activities (∼2.6 dpm per 2 × 2 inch section). These activities likely originate from the decay 40K present in borosilicate glass and, as a result, we avoided the use of glass vials and fiber paper in our subsequent tests and natural sample measurements. Microquartz filters were not tested in this study.
The scintillation gel contributes to the overall activity of a sample but is a strictly necessary reagent for measuring samples because of its role in converting the energy loss from a decay event into photons that are detected by the photomultiplier tubes of the Quantulus. The use of optimized integration channels (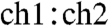 ) resulted in a 30% reduction in the background contributed by the scintillation cocktail for a given gel volume (VG). The activity measurements reported below use the optimized integration channels. Because measurements of empty vials yielded negligible (< 0.01 dpm) activities, the observed background activity in scintillation vials filled with variable amounts of gel must come from the gel itself. This gel-specific background (
) resulted in a 30% reduction in the background contributed by the scintillation cocktail for a given gel volume (VG). The activity measurements reported below use the optimized integration channels. Because measurements of empty vials yielded negligible (< 0.01 dpm) activities, the observed background activity in scintillation vials filled with variable amounts of gel must come from the gel itself. This gel-specific background (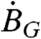 ) is a nonlinear function of VG and can be fit by a second-order polynomial with respect to VG as
) is a nonlinear function of VG and can be fit by a second-order polynomial with respect to VG as
| [2] |
Using a 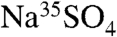 (aq.) standard (MP Biomedical, ∼4,000 dpm/mL of solution of H2O) to ensure that
(aq.) standard (MP Biomedical, ∼4,000 dpm/mL of solution of H2O) to ensure that 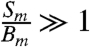 , we determined the counting efficiency of the liquid scintillation technique for aqueous samples of volume 0.1 mL and 1 mL using various values of VG. These counting efficiencies are shown in Fig. 1. Polynomial fits to these data yield the following counting efficiencies as a function of gel volume:
, we determined the counting efficiency of the liquid scintillation technique for aqueous samples of volume 0.1 mL and 1 mL using various values of VG. These counting efficiencies are shown in Fig. 1. Polynomial fits to these data yield the following counting efficiencies as a function of gel volume:
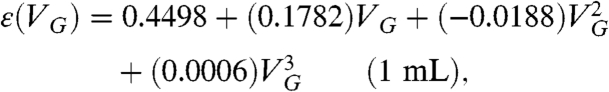 |
[3] |
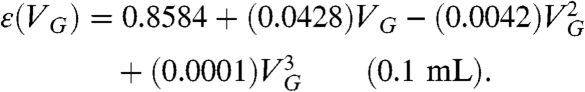 |
[4] |
Fig. 1.

35S decay event detection efficiency [ε(VG)] as a function of gel volume (VG) used for 0.1 mL and 1 mL aqueous samples. Note that the counting efficiency exceeds 90% for smaller amounts of gel compared to aqueous samples with larger volumes.
Optimizing VG to maximize sensitivity in weak samples.
We determined the optimal scintillation gel volume as follows: We assumed that an aqueous sample of volume VSA [0.1 or 1 mL of NaSO4 (aq.)] is to be counted. Because we want to determine the sample’s unknown activity 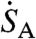 (dpm) in a gel volume VG, we need to know what amount of gel volume maximizes our signal to noise. The expected signal, Sm, that is reported by the scintillation system during an integration time Δt is given by
(dpm) in a gel volume VG, we need to know what amount of gel volume maximizes our signal to noise. The expected signal, Sm, that is reported by the scintillation system during an integration time Δt is given by
| [5] |
where εD is the overall detection efficiency of the Quantulus detection system (∼0.94) and εV(VG) is the gel volume dependence of the detection efficiency (e.g. Eq. 3 or 4) for an aqueous sample, and εprep is the sample preparation efficiency. We note that in our tests with laboratory standards, the sample preparation efficiency was 100% (εprep = 1). The noise in the Quantalus scintillation spectroscopic system is dominated by fluctuations in the total number of events detected in the same time interval and is given by
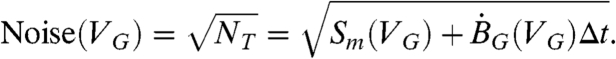 |
[6] |
The ratio 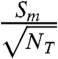 is plotted as a function of VG in Fig. 2 for a weak sample with
is plotted as a function of VG in Fig. 2 for a weak sample with 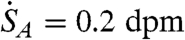 and Δt = 1 and 2 hr. Examination of Fig. 2 reveals that the signal to noise (and hence the minimum detection limit, MDL) can be maximized for 1 mL aqueous samples when VG = 2.5 mL. Even more important, however, is the observation that smaller aqueous volumes, in this case 0.1 mL, yield substantially improved MDLs. The main reason for this behavior is that the efficiency of detection [εV(VG)] as a function of VG for 0.1 mL aqueous samples is already close to 90% when VG = 1. Additional gains in efficiency obtained by using larger gel volumes are marginal and are offset by the higher backgrounds associated with the scintillation gel. Thus, contrary to previous method descriptions, we find that minimizing the gel volume can significantly improve the MDL for the LSS of 35S.
and Δt = 1 and 2 hr. Examination of Fig. 2 reveals that the signal to noise (and hence the minimum detection limit, MDL) can be maximized for 1 mL aqueous samples when VG = 2.5 mL. Even more important, however, is the observation that smaller aqueous volumes, in this case 0.1 mL, yield substantially improved MDLs. The main reason for this behavior is that the efficiency of detection [εV(VG)] as a function of VG for 0.1 mL aqueous samples is already close to 90% when VG = 1. Additional gains in efficiency obtained by using larger gel volumes are marginal and are offset by the higher backgrounds associated with the scintillation gel. Thus, contrary to previous method descriptions, we find that minimizing the gel volume can significantly improve the MDL for the LSS of 35S.
Fig. 2.

Signal/noise (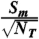 ) for 0.1 and 1 mL aqueous samples as a function of gel volume. Note that smaller amounts of water lead to drastic improvements in the minimum detection limit.
) for 0.1 and 1 mL aqueous samples as a function of gel volume. Note that smaller amounts of water lead to drastic improvements in the minimum detection limit.
Background interferences from barium isotopes.
See SI Methods.
Preparation efficiency.
See SI Methods.
Natural Sample Results.
We collected and measured size-segregated (SI Methods) atmospheric aerosol and SO2 samples at the Scripps Institute of Oceanography (SIO, latitude = 32.867°N, longitude = 117.257°W) and San Fernando Valley (SFV, latitude = 34.212°N, longitude = 118.061°W) during September 2007. These samples were processed before the optimized techniques described here were developed, and their activities were measured as BaSO4. Size-segregated aerosol sulfate and gaseous SO2 samples (SI Methods) were also collected during July 2008 and were measured as Na2SO4(aq). The uncertainties, determined by counting statistics, for the 2007 samples are larger than those associated with the 2008 samples due to the difficulties in preparing and measuring these samples as BaSO4, as we have discussed in our measurement techniques. Overall, the background corrected activities we observed ranged from 0 (within the measurement uncertainty) to 22.20 ± 1.22 dpm (see Tables 1 and 2). All samples were recounted once the naturally present 35S had fully decayed and were spiked with 0.1 mL of the 35S standard. This procedure allowed us to correct for sample self-absorption of scintillation events. The corrected count rates are reported in Tables 1 and 2.
Table 1.
35S activity of aerosol 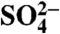 found in particles > 1.5 μm in size at Scripps Institution of Oceanography and San Fernando Valley
found in particles > 1.5 μm in size at Scripps Institution of Oceanography and San Fernando Valley
| Sample | dpm per sample | 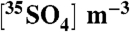 |
dpm/μmol S | dpm/μmol nss-S | %NSS |
| SFV 9/3/2007* | 4.90 ± 3.83 | 453 ± 347 | 0.21 ± 0.16 | 0.34 ± 0.26 | 60.2% |
| SFV 9/7/2007* | -2.06 ± 3.61 | - 187 ± 328 | -0.05 ± 0.09 | -0.12 ± 0.21 | 42.9% |
| SFV 9/16/2007* | -0.88 ± 3.11 | -80 ± 282 | -0.02 ± 0.09 | -0.05 ± 0.19 | 45.9% |
| SFV 7/21/2008 | 16.47 ± 1.06 | 1494 ± 96 | 0.43 ± 0.03 | 1.23 ± 0.08 | 34.9% |
| SFV 7/25/2008 | -0.30 ± 1.33 | -27 ± 121 | -0.01 ± 0.03 | -0.02 ± 0.11 | 29.4% |
| SFV 7/28/2008 | 5.62 ± 1.22 | 510 ± 111 | 0.16 ± 0.03 | 1.01 ± 0.22 | 15.5% |
| SVF 8/1/2008 | 3.84 ± 1.09 | 349 ± 99 | 0.09 ± 0.02 | 0.22 ± 0.06 | 39.8% |
| SIO 9/4/2007* | -0.021 ± 0.643 | -2 ± 58 | 0 ± 0.01 | - | 0 |
| SIO 9/7/2007* | -0.122 ± 0.653 | -11 ± 59 | 0 ± 0.01 | - | 0 |
| SIO 9/11/2007* | -0.138 ± 0.691 | -16 ± 63 | 0 ± 0.007 | - | 0 |
| SIO 7/21/2008 | 2.16 ± 0.84 | 196 ± 76 | 0.36 ± 0.14 | - | 0 |
| SIO 7/25/2008 | 3.06 ± 0.86 | 278 ± 78 | 0.10 ± 0.03 | - | 0 |
| SIO 7/28/2008 | -1.62 ± 0.74 | -147 ± 67 | -0.06 ± 0.03 | - | 0 |
| SIO 8/1/2008 | 2.26 ± 0.77 | 205 ± 70 | 0.07 ± 0.02 | - | 0 |
Samples measured as BaSO4(s) are indicated using *. All other samples (2008) were measured as Na2SO4(aq). Air volume per sample (Vair) was equal to 2,000 m3 and the total number of 35S(35N) in each sample was determined using the relationship 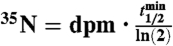 , where
, where 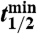 is the radioactive-decay half-life of 35S expressed in minutes and dpm is the measured activity of the sample in units of disintegrations per minute at the time of collection. The volumetric concentrations of 35S were calculated as
is the radioactive-decay half-life of 35S expressed in minutes and dpm is the measured activity of the sample in units of disintegrations per minute at the time of collection. The volumetric concentrations of 35S were calculated as 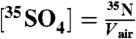 . Non-sea-salt sulfate was determined using anion and cation measurements of aerosol samples and the molar ratio of sulfate to sodium (0.0604) in sea-salt spray.
. Non-sea-salt sulfate was determined using anion and cation measurements of aerosol samples and the molar ratio of sulfate to sodium (0.0604) in sea-salt spray.
Table 2.
35S activity of aerosol 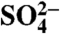 found in particles < 1.5 μm in size at Scripps Institution of Oceanography and San Fernando Valley
found in particles < 1.5 μm in size at Scripps Institution of Oceanography and San Fernando Valley
| Sample | dpm per sample | 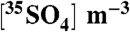 |
dpm/μmol S | dpm/μmol nss-S | %NSS |
| SFV 9/3/2007* | 10.84 ± 2.87 | 984 ± 260 | 0.18 ± 0.05 | 0.28 ± 0.07 | 64.1% |
| SFV 9/7/2007* | 13.63 ± 2.84 | 1236 ± 258 | 0.27 ± 0.06 | 0.40 ± 0.08 | 68.9% |
| SFV 9/16/2007* | 13.18 ± 2.59 | 1196 ± 235 | 0.18 ± 0.03 | 0.27 ± 0.05 | 66.4% |
| SFV 7/21/2008 | 3.76 ± 1.06 | 341 ± 96 | 0.03 ± 0.01 | 0.03 ± 0.01 | 88.3% |
| SFV 7/25/2008 | 20.43 ± 1.33 | 1854 ± 121 | 0.10 ± 0.01 | 0.12 ± 0.01 | 88.7% |
| SFV 7/28/2008 | 22.20 ± 1.22 | 2013 ± 111 | 0.16 ± 0.01 | 0.19 ± 0.01 | 84.7% |
| SFV 8/1/2008 | 9.80 ± 1.09 | 889 ± 99 | 0.04 ± 0.00 | 0.04 ± 0.00 | 92.7% |
| SIO 9/4/2007* | -1.09 ± 2.53 | -99 ± 230 | - 0.01 ± 0.03 | -0.01 ± 0.03 | 75.9% |
| SIO 9/7/2007* | -1.94 ± 2.29 | -176 ± 207 | -0.04 ± 0.05 | -0.06 ± 0.07 | 67.3% |
| SIO 9/11/2007* | 3.91 ± 2.45 | 355 ± 223 | 0.08 ± 0.05 | 0.13 ± 0.08 | 61.8% |
| SIO 7/21/2008 | 11.46 ± 0.97 | 1040 ± 88 | 0.55 ± 0.05 | 0.85 ± 0.07 | 64.0% |
| SIO 7/25/2008 | 3.26 ± 0.91 | 295 ± 83 | 0.03 ± 0.01 | 0.04 ± 0.01 | 81.4% |
| SIO 7/28/2008 | 0 ± 0.90 | 0 ± 82 | 0.00 ± 0.01 | 0.00 ± 0.02 | 72.2% |
| SIO 8/1/2008 | 6.39 ± 1.10 | 580 ± 99 | 0.06 ± 0.01 | 0.15 ± 0.03 | 37.0% |
Samples measured as BaSO4(s) are indicated using *. All other samples (2008) were measured as Na2SO4(aq). Air volume per sample (Vair) was equal to 2,000 m3 and the total number of 35S(35N) in each sample was determined using the relationship 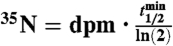 , where
, where 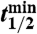 is the radioactive-decay half-life of 35S expressed in minutes and dpm is the measured activity of the sample in units of disintegrations per minute. The air volume concentrations of 35S were calculated as
is the radioactive-decay half-life of 35S expressed in minutes and dpm is the measured activity of the sample in units of disintegrations per minute. The air volume concentrations of 35S were calculated as 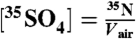 . Non-sea-salt sulfate was determined using anion and cation measurements of aerosol samples and the ratio of sulfate to sodium (0.0604) in sea-salt spray.
. Non-sea-salt sulfate was determined using anion and cation measurements of aerosol samples and the ratio of sulfate to sodium (0.0604) in sea-salt spray.
While detailed modeling of the observed absolute activities (dpm) is beyond the scope of this work, examination of the 35S activity in the gas and aerosol phases points to the potential of the methods developed here to address present uncertainties in our understanding of the global S budget. We found, with the exception of a few days, that the activities contained within aerosol particles < 1.5 μm were generally higher than those within particles of diameter > 1.5 μm. Despite their proximate latitudes and longitudes, the activities in both size fractions were generally higher at SFV than at SIO. The fraction of non-sea-salt sulfate (NSS) was measured and calculated (SI Methods) for each sample using the total concentration of sulfate and sodium and the molar ratio of sulfate to sodium (0.0604) found in seawater. The SIO samples had a much higher fraction of the total sulfate derived from sea salt, with the coarse aerosols dominated by sea-salt sulfate. The lower 35S activity in these samples is consistent with these aerosol particles being fresh sea-salt aerosols without any measurable amounts of secondary sulfate produced by the oxidation of 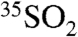 . A few coarse samples at SIO have an activity > 0, yet have Na+ abundances that indicate that 100 ± 8% of the sulfate is from sea-salt spray. An estimate of the amount of non-sea-salt sulfate that would be needed to explain this apparent discrepancy was obtained using the molar ratio of 35S/S ratio from SO2 collected at SIO, and this estimate indicates that the activity falls within the total measurement uncertainty of the ion-chromatographic method used to determine the cation and anion concentrations.
. A few coarse samples at SIO have an activity > 0, yet have Na+ abundances that indicate that 100 ± 8% of the sulfate is from sea-salt spray. An estimate of the amount of non-sea-salt sulfate that would be needed to explain this apparent discrepancy was obtained using the molar ratio of 35S/S ratio from SO2 collected at SIO, and this estimate indicates that the activity falls within the total measurement uncertainty of the ion-chromatographic method used to determine the cation and anion concentrations.
To provide the proper context to relate our 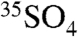 and
and 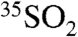 concentration measurements (reported as activities) to atmospheric processes, we follow a treatment similar to that of Ref. 12 in relating the concentration of
concentration measurements (reported as activities) to atmospheric processes, we follow a treatment similar to that of Ref. 12 in relating the concentration of 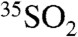 to the overall lifetime of SO2 (τoverall) in an atmospheric box model. For a stable boundary layer and ignoring mixing from outside of this box, the time-dependent concentration of
to the overall lifetime of SO2 (τoverall) in an atmospheric box model. For a stable boundary layer and ignoring mixing from outside of this box, the time-dependent concentration of 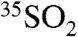 can be expressed as
can be expressed as
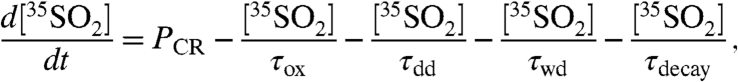 |
[7] |
where PCR is the cosmic ray production rate of 35S. The timescales for sinks of 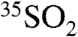 are represented by an oxidative lifetime (τox), a dry deposition lifetime (τdd), the lifetime with respect to wet deposition (τwd), and the radioactive lifetime (τdecay) of 35S (radioactive mean-life = 126 days). Combining all sink terms proportional to
are represented by an oxidative lifetime (τox), a dry deposition lifetime (τdd), the lifetime with respect to wet deposition (τwd), and the radioactive lifetime (τdecay) of 35S (radioactive mean-life = 126 days). Combining all sink terms proportional to 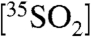 , defining
, defining 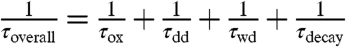 , assuming steady-state conditions within the box (
, assuming steady-state conditions within the box (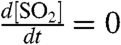 ), and solving the resulting expression for τoverall yields
), and solving the resulting expression for τoverall yields
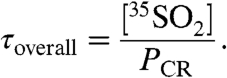 |
[8] |
The production rate of 35S in the troposphere reported by Ref. 13 of 4.75 × 10-4 atoms m-3 s-1 was used to determine the integrated production in a 10 km high volume. The results are presented in Table 3.
Table 3.
Summary of 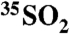 concentrations and τoverall estimated using Eq. [9] and
concentrations and τoverall estimated using Eq. [9] and 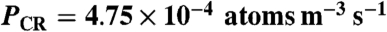
| Sample date | dpm per sample | 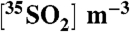 |
τoverall (days) |
| SFV | |||
| 7/21/2008 | 4.80 ± 0.19 | 436 ± 17 | 10.6 ± 0.4 |
| 7/25/2008 | 9.03 ± 0.18 | 820 ± 16 | 20.0 ± 0.4 |
| 7/28/2008 | 6.21 ± 0.17 | 565 ± 16 | 13.8 ± 0.4 |
| 8/1/2008 | 10.07 ± 0.19 | 915 ± 18 | 22.3 ± 0.4 |
| SIO | |||
| 7/21/2008 | 0.10 ± 0.19 | 9 ± 17 | 0.2 ± 0.4 |
| 7/25/2008 | 1.19 ± 0.17 | 108 ± 15 | 2.6 ± 0.4 |
| 7/28/2008 | 2.02 ± 0.19 | 184 ± 17 | 4.5 ± 0.4 |
| 8/1/2008 | 1.81 ± 0.14 | 165 ± 12 | 4.0 ± 0.3 |
Air volume per sample (Vair) was equal to 2,000 m3 and the total number of 35S(35N) in each sample was determined using the relationship: 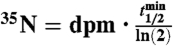 , where
, where 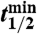 is the radioactive-decay half-life of 35S expressed in minutes and dpm is the measured activity of the sample in units of disintegrations per minute. The air volume concentrations of 35S were calculated as
is the radioactive-decay half-life of 35S expressed in minutes and dpm is the measured activity of the sample in units of disintegrations per minute. The air volume concentrations of 35S were calculated as 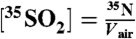 .
.
Inspection of Table 3 reveals significant and systematic differences in τoverall between the inland (SFV) and oceanic (SIO) sites that likely reflect differences in the predominant mechanisms affecting the lifetime of SO2 at these two locations. Given the absence of precipitation events during the sample collection times, we ignore the contribution of wet deposition (τwd → ∞). Surface resistance models indicate that large differences in deposition velocities (vdd) exist for SO2 over land (0.2–0.4 cm s-1) and seawater (∼0.8 cm s-1) (25). For a mixing height of 1000 m, these vdd values translate to deposition lifetimes of about 5.78–2.89 and 1.5 d for SFV and SIO, respectively. An additional factor that determines τoverall at each of these locations is the removal of SO2 by OH. This lifetime is given by
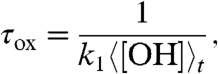 |
[9] |
where k1 is the effective rate constant for the reaction 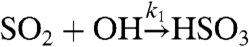 (k1 ∼ 3 × 10-12 cm3 s-1) (26), and 〈[OH]〉t is the time-averaged (over 24 hr) concentration of OH per cm3. Inserting values typical of polluted environments (〈[OH]〉t ∼ 1 × 106 cm-3) leads to estimates for τox ∼ 3–4 d, although the oxidation lifetime of SO2 in the free troposphere may be longer. At this time, we do not know why the SFV and SIO lifetimes are longer than expected based on oxidative and dry deposition considerations, but these differences may be due to boundary layer shifts and/or mixing of air from the free troposphere. Detailed modeling of these factors, in the future, should help in clarifying their importance.
(k1 ∼ 3 × 10-12 cm3 s-1) (26), and 〈[OH]〉t is the time-averaged (over 24 hr) concentration of OH per cm3. Inserting values typical of polluted environments (〈[OH]〉t ∼ 1 × 106 cm-3) leads to estimates for τox ∼ 3–4 d, although the oxidation lifetime of SO2 in the free troposphere may be longer. At this time, we do not know why the SFV and SIO lifetimes are longer than expected based on oxidative and dry deposition considerations, but these differences may be due to boundary layer shifts and/or mixing of air from the free troposphere. Detailed modeling of these factors, in the future, should help in clarifying their importance.
A similar analysis can be made to interpret the 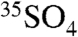 concentration data presented in Tables 1 and 2. To begin, we express the overall rate of change of
concentration data presented in Tables 1 and 2. To begin, we express the overall rate of change of 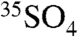 as
as
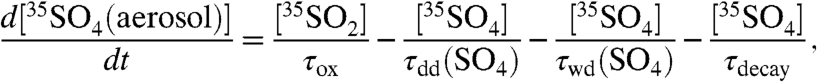 |
[10] |
where τox is again the overall lifetime of SO2, τdd(SO4) and τwd(SO4) are the dry and wet deposition timescales of aerosol sulfate, and τdecay is defined as before. Given the ability to discriminate size-dependent activities in aerosols, the activity in aerosol sulfate can be subdivided into two size bins and expressed as
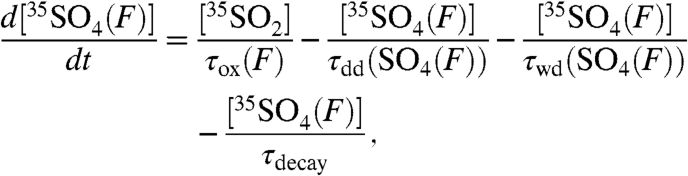 |
[11] |
and
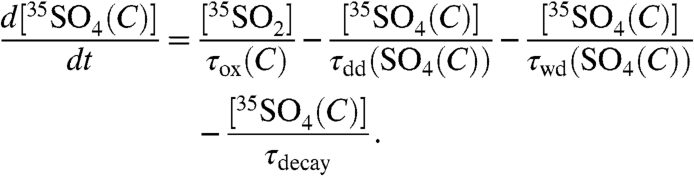 |
[12] |
Here, the oxidative [τox(F,C)], dry deposition [τdd(F,C)], and wet deposition [τwd(F,C)] lifetimes are broken up into two components to correspond to fine (F) and coarse (C) aerosols. Again, if we assume steady-state conditions and combine the sink terms into one overall timescale for fine and coarse aerosols (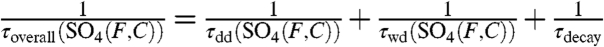 ), we find that
), we find that
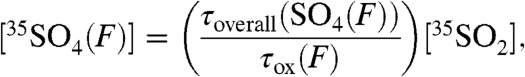 |
[13] |
and
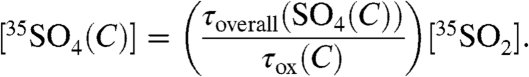 |
[14] |
The higher (lower) activities per unit volume observed in fine (coarse) aerosol particles, therefore, are expected to depend on (1) the concentration of 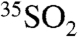 , (2) the ratio of the overall residence time for fine (coarse) aerosol sulfate, and (3) the rate of SO2 oxidation and incorporation into fine (coarse) aerosol particles. In this discussion, we neglect the possibility that 35S found in fine aerosol particles may end up in coarse aerosol particles due to aerosol size growth dynamics, although these timescales (hours to days) are expected to depend on total particle concentrations and characteristic particle sizes (27), and this process may be important for very polluted environments such as SFV. If coagulation were the dominant mechanism affecting the relative ratio of activity in fine and coarse aerosols, however, we would expect to see the same ratio of 35S to total S in both of these sizes. Given the above discussion, it is interesting to note that the days with nonzero activities in SIO coarse particles in 2008 also corresponded to days with the lowest (shortest τoverall) concentrations of
, (2) the ratio of the overall residence time for fine (coarse) aerosol sulfate, and (3) the rate of SO2 oxidation and incorporation into fine (coarse) aerosol particles. In this discussion, we neglect the possibility that 35S found in fine aerosol particles may end up in coarse aerosol particles due to aerosol size growth dynamics, although these timescales (hours to days) are expected to depend on total particle concentrations and characteristic particle sizes (27), and this process may be important for very polluted environments such as SFV. If coagulation were the dominant mechanism affecting the relative ratio of activity in fine and coarse aerosols, however, we would expect to see the same ratio of 35S to total S in both of these sizes. Given the above discussion, it is interesting to note that the days with nonzero activities in SIO coarse particles in 2008 also corresponded to days with the lowest (shortest τoverall) concentrations of 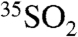 . These observations are consistent with an increase of the heterogeneous processing of SO2 onto coarse aerosol particles and not to changes in the deposition velocities of SO2, which would only lead to a decrease in the
. These observations are consistent with an increase of the heterogeneous processing of SO2 onto coarse aerosol particles and not to changes in the deposition velocities of SO2, which would only lead to a decrease in the 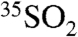 concentration. Given the prevalence of sea-salt particles as a medium for the aqueous phase oxidation of SO2, we suggest that these observations may be consistent with enhanced uptake and aqueous phase oxidation of
concentration. Given the prevalence of sea-salt particles as a medium for the aqueous phase oxidation of SO2, we suggest that these observations may be consistent with enhanced uptake and aqueous phase oxidation of 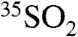 during these time periods. Future work with larger datasets and detailed modeling of the coupled gas and aerosol phase chemistry and ambient meteorological conditions will be needed to fully interpret these types of results and their variation. We emphasize that without the optimized methods that we have developed, an in-depth examination of short timescale (∼6–12 hours) processes would not be possible.
during these time periods. Future work with larger datasets and detailed modeling of the coupled gas and aerosol phase chemistry and ambient meteorological conditions will be needed to fully interpret these types of results and their variation. We emphasize that without the optimized methods that we have developed, an in-depth examination of short timescale (∼6–12 hours) processes would not be possible.
In contrast to SIO coarse particles (∼0–3 dpm per 2000 m3), the SFV coarse aerosol size fraction had significantly higher (∼0–16 dpm per 2000 m3) 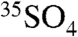 concentrations. In light of Eqs. 13 and 14, these differences may be explained in part by the higher concentrations of
concentrations. In light of Eqs. 13 and 14, these differences may be explained in part by the higher concentrations of 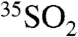 seen at SFV (∼4–10 dpm per 2000 m3) compared to SIO (∼0–2 dpm per 2000 m3) and/or substantial differences in the aerosol age (lifetime).
seen at SFV (∼4–10 dpm per 2000 m3) compared to SIO (∼0–2 dpm per 2000 m3) and/or substantial differences in the aerosol age (lifetime).
We compare the 35S measurements presented here to the pioneering work of Tanaka and Turekian (9), which reported 35S activities in aerosols collected weekly in 1992 at New Haven, Connecticut. Air samples collected during July and August of 1992 were reported to have activities ranging from 1.1 × 10-3–14.5 × 10-3 dpm/μmol S for SO4 and 6 × 10-4–2.2 × 10-3 dpm/μmol S for SO2. We note that these values are significantly lower than our measurements, which are summarized in Table 1. These differences in the total-sulfur normalized values reported by Tanaka and Turekian (9) and the present values may result from the dilution generated by larger regional SO2 fluxes emitted by coal burning in the eastern United States. These high sulfur coal emissions in essence dilute the 35S activity when normalized to the total mass of S in aerosols. In contrast, California does not operate coal burning electric plants or have any other large sources of SO2 that would dilute 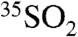 by a comparable factor. These results highlight the utility of the 35S, as first shown by Tanaka and Turekian (12), to understand differences in regional atmospheric chemistry and transport. Reporting 35S activity with respect to sampled air volume minimizes the dilution effect described above, but normalization has other uses, and thus reporting in both units is advisable.
by a comparable factor. These results highlight the utility of the 35S, as first shown by Tanaka and Turekian (12), to understand differences in regional atmospheric chemistry and transport. Reporting 35S activity with respect to sampled air volume minimizes the dilution effect described above, but normalization has other uses, and thus reporting in both units is advisable.
Conclusion
This work presents improved and optimized methods for measuring 35S in natural samples. These methods have reduced the limit of detection down to 0.200 dpm or 36,000 35S atoms for a one-hour integration and have been shown to be suitable for measurements of 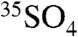 in size-segregated aerosols and
in size-segregated aerosols and 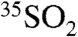 . We have identified sensitivity-limiting backgrounds in chemical reagents used for sample preparation and these findings should be considered in future 35S measurements. In light of this work, we suggest that reports of excess 35S in aerosols, in comparison to 7Be or 32P, may be erroneously high due to the high backgrounds that may be introduced by barium and suggests that the conclusions of Osaki et al. (23) may need to be revised. We recommend that 35S activity measurements should be made as NaSO4 rather than BaSO4 in weakly radiogenic samples.
. We have identified sensitivity-limiting backgrounds in chemical reagents used for sample preparation and these findings should be considered in future 35S measurements. In light of this work, we suggest that reports of excess 35S in aerosols, in comparison to 7Be or 32P, may be erroneously high due to the high backgrounds that may be introduced by barium and suggests that the conclusions of Osaki et al. (23) may need to be revised. We recommend that 35S activity measurements should be made as NaSO4 rather than BaSO4 in weakly radiogenic samples.
In the present work we have also simultaneously measured 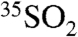 and aerosol
and aerosol 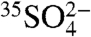 in two distinct locations. The 35S activity differences observed between the two locations, as well as the differences observed in the two aerosol size fractions, illustrates the ability of 35S to further elucidate the local and regional cycling of SO2 and aerosol SO4. Given the observed 35S concentrations and the minimum detection limits presented here, use of these techniques should allow for high time resolution studies of SO2 and sulfate. While this work focused specifically on measurements of aerosol
in two distinct locations. The 35S activity differences observed between the two locations, as well as the differences observed in the two aerosol size fractions, illustrates the ability of 35S to further elucidate the local and regional cycling of SO2 and aerosol SO4. Given the observed 35S concentrations and the minimum detection limits presented here, use of these techniques should allow for high time resolution studies of SO2 and sulfate. While this work focused specifically on measurements of aerosol 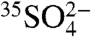 and
and 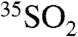 , the techniques can be readily applied to measure 35S in fog, rain, and snow. For example, given the production rates of 35S (1.5 times that in troposphere) and the residence time of SO4 in the stratosphere (∼2.5 years) (28), we estimate that the concentration of
, the techniques can be readily applied to measure 35S in fog, rain, and snow. For example, given the production rates of 35S (1.5 times that in troposphere) and the residence time of SO4 in the stratosphere (∼2.5 years) (28), we estimate that the concentration of 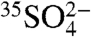 in the stratosphere,
in the stratosphere, 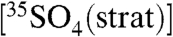 , is
, is 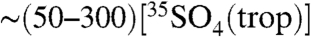 , or
, or 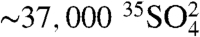 molecules per m-3, based on the range of concentrations we report here. The enhanced sensitivities and techniques we present here could be used to measure
molecules per m-3, based on the range of concentrations we report here. The enhanced sensitivities and techniques we present here could be used to measure 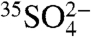 in snow, and enhanced concentrations of
in snow, and enhanced concentrations of 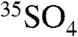 may be used as a proxy for stratospheric intrusions in Antarctica and in samples from other remote field locations.
may be used as a proxy for stratospheric intrusions in Antarctica and in samples from other remote field locations.
Because of the length of biological cycles, this technique offers a unique way to measure 35S in fast growing plants, phytoplankton, and bacteria and therefore could provide a unique radioactive tracer to understand these processes.
Methods
Low-level liquid scintillation spectroscopy, combined with a high activity 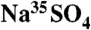 (aq.) standard, was used to develop the optimized 35S sample preparation protocols. A multistage high-volume sampler was used for the aerosol and gas-phase S collections. Complete details can be found in Results and Discussion and in SI Text.
(aq.) standard, was used to develop the optimized 35S sample preparation protocols. A multistage high-volume sampler was used for the aerosol and gas-phase S collections. Complete details can be found in Results and Discussion and in SI Text.
Supplementary Material
ACKNOWLEDGMENTS.
We thank the anonymous reviewer and K. Turekian for their helpful and insightful comments that greatly improved this manuscript. We acknowledge Ivan Gaylor who donated the Quantulus and also D. Lal for insightful conversations. G.D. acknowledges the Camille and Henry Dreyfus Postdoctoral Program in Environmental Chemistry and the U.C. President’s Postdoctoral Fellowship Program for their support. L.B. and A.A. acknowledge Eloise and Russ Duff and the Kents for their financial support.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0901168107/DCSupplemental.
*The lack of an absolute activity standard for 35S and the significant overlap between the β- spectra of 14C and 35S allows us to make this a reasonable substitution.
References
- 1.Solomon S, et al. IPCC, 2007: Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge, UK: Cambridge Univ Press; 2007. p. 996. [Google Scholar]
- 2.Thiemens MH. History and Applications of Mass-Independent Isotope Effects. Annu Rev Earth Pl Sc. 2006;34(1):217–262. [Google Scholar]
- 3.Alexander B, et al. Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes. J Geophys Res. 2005;110:D10307. [Google Scholar]
- 4.Alexander B, Savarino J, Barkov NI, Delmas RJ, Thiemens MH. Climate driven changes in the oxidation pathways of atmospheric sulfur. Geophys Res Lett. 2002;29(14):1685. [Google Scholar]
- 5.Alexander B, Savarino J, Kreutz KJ, Thiemens MH. Impact of preindustrial biomass-burning emissions on the oxidation pathways of tropospheric sulfur and nitrogen. J Geophys Res. 2004;109:D08303. doi: 10.1029/2003JD004218. [Google Scholar]
- 6.Lee CCW, Thiemens MH. The δ17O and δ18O measurements of atmospheric sulfate from a coastal and high alpine region: A mass-independent isotopic anomaly. J Geophys Res-Atmos. 2001;106(D15):17359–17373. [Google Scholar]
- 7.Black G, Sharpless RL, Slanger TG. Rate coefficients at 298 K for SO reactions with O2, O3, and NO2. Chem Phys Lett. 1982;90(1):55–58. [Google Scholar]
- 8.Robertshaw JS, Smith IWM. Rate data for O + OCS → SO + CO and SO + O3 → SO2 + O2 by a new time-resolved technique. Int J Chem Kinet. 1980;12(10):729–739. [Google Scholar]
- 9.Tanaka N, Turekian KK. Determination of the dry deposition flux of SO2 using cosmogenic 35S and 7Be measurements. J Geophys Res-Atmos. 1995;100(D2):2841–2848. [Google Scholar]
- 10.Turekian KK, Graustein WC. Natural radionuclides in the atmosphere. In: Holland HD, Turekian KK, editors. Treatise on Geochemistry. Vol 4. Oxford: Elsevier-Pergamon; 2003. pp. 261–279. (The Atomosphere). (ed RF Keeling) [Google Scholar]
- 11.Junkermann W, Roedel W. Evidence for short SO2 lifetimes in the atmosphere: An in-situ measurement of atmospheric SO2 lifetime using cosmic ray produced Sulphur-38. Atmos Environ. 1983;17(12):2549–2554. [Google Scholar]
- 12.Tanaka N, Turekian KK. Use of cosmogenic 35S to determine the rates of removal of atmospheric SO2. Nature. 1991;352(6332):226–228. [Google Scholar]
- 13.Turekian KK, Tanaka N. The use of atmospheric cosmogenic 35S and 7Be in determining depositional fluxes of SO2. Geophys Res Lett. 1992;19(17):1767–1770. [Google Scholar]
- 14.Calvert JG, Chatfield RB, Delany AC, Martel EA. Evidence for short SO2 lifetimes in the atmosphere: An in-situ measurement of atmospheric SO2 lifetime using cosmic ray produced 38S. Atmos Environ. 1985;19(7):1205–1206. [Google Scholar]
- 15.Goel PS, Jha S, Lal D, Radhakrishna P, Rama P. Cosmic ray produced beryllium isotopes in rain water. Nucl Phys. 1956;1(3):196–201. [Google Scholar]
- 16.Lal D, Arnold JR, Honda M. Cosmic-ray production rates of 7Be in oxygen, and P32, P33, S35 in argon at mountain altitudes. Phys Rev. 1960;118(6):1626–1632. [Google Scholar]
- 17.Hong YL, Kim G. Measurement of cosmogenic 35S activity in rainwater and lake water. Anal Chem. 2005;77(10):3390–3393. doi: 10.1021/ac048128c. [DOI] [PubMed] [Google Scholar]
- 18.Kester C, Baron J, Turk J. Isotopic study of sulfate sources and residence times in a subalpine watershed. Environ Geol. 2003;43(5):606–613. [Google Scholar]
- 19.Novák M, Michel RL, Přechová E, Štěpánová M. The missing flux in a 35S budget for the soils of a small polluted catchment. Water Air Soil Poll. 2004;4(2–3):517–529. [Google Scholar]
- 20.Sueker JK, Turk JT, Michel RL. Use of cosmogenic 35S for comparing ages of water from three alpine-subalpine basins in the Colorado Front Range. Geomorphology. 1999;27(1-2):61–74. [Google Scholar]
- 21.Cape J. The use of 35S to study sulphur cycling in forests. Environ Geochem Hlth. 1993;15(2–3):113–118. doi: 10.1007/BF02627829. [DOI] [PubMed] [Google Scholar]
- 22.Michel RL, Turk JT, Campbell DH, Mast MA. Use of natural 35S to trace sulphate cycling in small lakes, Flattops Wilderness Area, Colorado, USA. Water Air Soil Poll. 2002;2(2):5–18. [Google Scholar]
- 23.Osaki S, Tagawa Y, Chijiiwa T, Sugihara S, Maeda Y. Atmospheric deposition of 35S. J Radioanal Nucl Ch. 1999;239(3):543–547. [Google Scholar]
- 24.Buckley JP. The use of carbon-14 as a secondary counting standard for sulphur-35. Int J Appl Radiat Is. 1970;22:41–42. doi: 10.1016/0020-708x(71)90157-8. [DOI] [PubMed] [Google Scholar]
- 25.Yiwen Y, Carmichael G. Modeling the dry deposition velocity of sulfur dioxide and sulfate in Asia. J Appl Meteorol. 1998;37:1084–1099. [Google Scholar]
- 26.Sander SP, et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies: Evaluation Number 15. Pasadena: Jet Propulsion Laboratory; 2006. JPL Publication 06-2. [Google Scholar]
- 27.Seinfeld JH, Pandis SN. Atmospheric Chemistry and Physics. New York: Wiley-Interscience; 2006. [Google Scholar]
- 28.van Velthoven PFJ, Kelder H. Estimates of stratosphere-troposphere exchange: Sensitivity to model formulation and horizontal resolution. J Geophys Res. 1996;101:1429–1434. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.