

Abstract
Generation of induced pluripotent stem (iPS) cells holds a great promise for regenerative medicine and other aspects of clinical applications. Many types of cells have been successfully reprogrammed into iPS cells in the mouse system; however, reprogramming human cells have been more difficult. To date, human dermal fibroblasts are the most accessible and feasible cell source for iPS generation. Dental tissues derived from ectomesenchyme harbor mesenchymal-like stem/progenitor cells and some of the tissues have been treated as biomedical wastes, for example, exfoliated primary teeth and extracted third molars. We asked whether stem/progenitor cells from discarded dental tissues can be reprogrammed into iPS cells. The 4 factors Lin28/Nanog/Oct4/Sox2 or c-Myc/Klf4/Oct4/Sox2 carried by viral vectors were used to reprogram 3 different dental stem/progenitor cells: stem cells from exfoliated deciduous teeth (SHED), stem cells from apical papilla (SCAP), and dental pulp stem cells (DPSCs). We showed that all 3 can be reprogrammed into iPS cells and appeared to be at a higher rate than fibroblasts. They exhibited a morphology indistinguishable from human embryonic stem (hES) cells in cultures and expressed hES cell markers SSEA-4, TRA-1-60, TRA-1-80, TRA-2-49, Nanog, Oct4, and Sox2. They formed embryoid bodies in vitro and teratomas in vivo containing tissues of all 3 germ layers. We conclude that cells of ectomesenchymal origin serve as an excellent alternative source for generating iPS cells.
Introduction
The foundation of cell-based therapy lies in the technologies of procuring cells, especially stem cells. Pluripotent embryonic stem (ES) cells are the most promising cell source for cell-based therapy in regenerative medicine as they give rise to cells of all germ layers and their supply is potentially unlimited. Recent development of generating induced pluripotent stem (iPS) cells by introducing 4 factors: c-Myc, Klf4, Oct4, and Sox2 [1–2] or Lin28, Nanog, Oct 4, and Sox2 [3] into somatic cells has shed light on the possibility of obtaining autologous pluripotent embryonic-like stem cells circumventing the need of dealing with nuclear transfer and embryos [1–3]. The initial establishment of human iPS cells was based on the reprogramming of dermal fibroblasts (DFs) with the understanding that dermal tissue is easy to access. Other types of cells in the mouse system such as subpopulation of neural stem cells have been found to be easily reprogrammed with <4 factors [4–6]. However, from the perspective of clinical applications, neural stem cells are not easily accessible if autologous human iPS cells are to be generated. Because the introduction of these factors has been via viral vectors, significant efforts have been put into removing the vectors from cells after they are being reprogrammed into iPS cells [7–11]. Nonetheless, any approach that involves the use of vector systems, even after they are removed, poses some uncertainty on their safety.
To completely circumvent the use of vectors, delivery of recombinant protein-based 4 factors to generate iPS cells in the mouse and human system has been reported [12–13]. Another alternative is not to use these genes and their products at all but to use by chemical stimulation. Small molecule screening by a established mouse cell line carrying a reporter gene (eg, Oct4-GFP) was able to filter out a list of chemicals that are able to induce pluripotency by substituting some of the 4 genes [5–6,14]. Although the evidence showing a total replacement of the 4 factors by chemicals has not been reported, it is anticipated that this will emerge in the very near future.
Once the vector system is no longer an issue, the next questions will be the search of an alternative or better source of cells for iPS cell generation. Most studies used a mouse system in which iPS cells are much easier to derive. Several types of mouse cells have been used to generate iPS cells successfully including DFs, β-cells, liver cells, gut epithelial cells, neural stem cells, mouse adult bone marrow mononuclear cells, and B cells [1,4,15–20]. In human system, DFs, amniotic fluid-derived cells, skin keratinocytes, ES cell-derived fibroblasts (ES-Fs), CD34 blood cells, mesenchymal stem cells (MSCs), and so on, have been shown to be reprogrammed to iPS cells [2,21–25]. However, it appears that, in general, it is easier to reprogram more immature cells than adult cells. It would even require the addition of hTERT (telomerase reverse transferase) and SV40 large T to turn MSCs into iPS cells [22]. Human skin keratinocytes appear to be easy to reprogram with a high success rate [24]. One type of human stem cells that are feasible and accessible is dental stem cells originally derived from ectomesenchyme. There are several types of human dental stem cells that can be easily obtained from shed primary teeth or extracted permanent teeth including stem cells from human exfoliated deciduous teeth (SHED), stem cells from apical papilla (SCAP), and dental pulp stem cells (DPSCs). SHED are isolated from the pulp tissue of the shed primary teeth [26]. SCAP are derived from the developing tissue at the apex of a tooth root named apical papilla in our previous reports [27,28]. This tissue is considered the precursor of dental pulp. DPSCs were the first type of dental stem cells to be characterized and are obtained from dental pulp tissue of permanent teeth [29,30]. In this present study, we found that all these dental stem cells can be successfully reprogrammed into iPS cells.
Materials and Methods
Cell cultures
Human SHED, SCAP, and DPSCs primary cultures were established as previously described [26–29,31]. In brief, pulp tissue from shed primary teeth (for SHED) or extracted permanent teeth (for DPSCs) and apical papilla (for SCAP) from immature permanent teeth were removed from teeth, minced, and digested in a solution of 3 mg/mL collagenase type I and 4 mg/mL dispase (Sigma-Aldrich, St. Louis, MO) for 30–60 min at 37°C. The digested mixtures were passed through a 70-mm cell strainer (Falcon, BD Labware, Franklin Lakes, NJ) to obtain single cell suspensions. Cells were seeded into 6-well plates and cultured with α-minimum essential medium (α-MEM; GIBCO/Invitrogen, Carlsbad, CA) supplemented with 15% fetal bovine serum (FBS; Gemini Bio-Products, Inc., Woodland, CA), 2 mM l-glutamine, 100 μM l-ascorbic acid-2-phosphate, 100 U/mL penicillin-G, 100 mg/mL streptomycin, and 0.25 mg/mL fungizone (Gemini Bio-Products, Inc.) and maintained in 5% CO2 at 37°C. Colony formation units of fibroblastic cells (CFU-F) were normally observed within 1–2 weeks after cell seeding and were passaged at 1:3 ratio when they reached ∼70%–80% confluence. Heterogeneous populations of SHED, SCAP, and DPSCs were frozen and stored in liquid nitrogen at passages 0–2. Cells were thawed and expanded for experimentation. These heterogeneous populations of adherent, clonogenic dental stem/progenitor cells were routinely tested for their cell surface marker expression with flow cytometry and they were positive for STRO-1, CD146, CD73, CD90, and CD105, and negative for CD14, CD34, and CD45, typical of mesenchymal cell type [32].
Tooth sample collection to obtain dental stem/progenitor cells conformed to approved protocols by the Medical Institutional Review Boards at the University of Maryland (H-29892) and University of Southern California (HS-06-00458). Primary human gingival fibroblasts (hGFs) were isolated from freshly collected gingival tissues and maintained in the same medium for dental stem/progenitor cells. Primary human foreskin fibroblasts (hFFs) from American Type Culture Collection (ATCC, Manassas, VA) were maintained in DMEM supplemented with 10% FBS. Cells at passage 3 or below were used for experimentation. Human embryonic stem cells H1 (WA01) were obtained from WiCell Research Institute (Madison, WI) and maintained in conditions following an NIH standard protocol (http://stemcells.nih.gov/research/NIHresearch/scunit/culture.asp). Feeder cells were mouse embryonic fibroblasts (MEFs) obtained from ATCC or GlobalStem Inc. (Rockville, MD).
Transduction and reprogramming of SHED/SCAP/DPSCs
Heterogeneous primary human dental stem/progenitor cells at passages 2 and 3 were used for reprogramming. Our first attempt to reprogram cells was by using the 4 factor genes (c-Myc, Klf4, Oct4, and Sox2) each subcloned into the pLenti6.2/C-Lumio/V5-DEST vector system containing a CMV promoter (Invitrogen, www.invitrogen.com/site/us/en/home.html). Approximately 30%–50% of transduced cells underwent cell death in the first few days. The survived cells proliferated faster than before the transduction and began morphological changes (fibroblastic to epithelial cell-like). The cells were seeded onto feeder cells within 7 days following transduction to allow reprogramming. Within 2 weeks, a few cell colonies similar to ES cell colonies emerged. These colonies were passaged to new feeder cells but later all underwent cell death. Several attempts were made and the same results occurred. Subsequently, lentiviral vectors pSin-EF2-gene-Pur carrying 1 of the 4 factors Lin28, Nanog, Oct4, and Sox2 were obtained from Addgene (www.addgene.org) and virus was produced [3]. Cells were seeded into wells (1 × 105/well) of 12-well plates and grown to ∼70% confluent. Virus carrying each factor was added at equal amounts (0.5–1 × 107 transduction unit/well) to the cell cultures with the presence of polybrene (4 μg/mL). Two to three days after the transduction, 1 × 104 dental stem cells were passaged onto the feeder MEFs plated in a 10-cm dish in the presence of human embryonic stem (hES) cell medium containing 4–10 ng/mL of bFGF. The medium was refreshed every 2 days until ES cell-like colonies emerged (on average before 3 weeks). We also subcloned human c-Myc, Klf4, Oct4, and Sox2 into the vector pMXs and produced retrovirus to transducer cells with the same approaches as the above.
Subcloning of emerged ES cell-like colonies
Emerged ES cell-like colonies were considered at passage 0. Each colony was manually subcloned into wells of 12-well plates seeded with MEFs and maintained separately from other subcloned colonies. The expanded passage 1 colonies were then passaged into wells of 6-well plates for further expansion until sufficient number of colonies was reached for experimental analyses. From passage 1, cell colonies were maintained in hES cell medium containing 100 ng/mL of bFGF.
Provirus integration
Genomic DNA of ES cell-like colonies was isolated using FlexiGene DNA kit (Qiagen, Valencia, CA). Primers specific for each transgene were used to amplify the 4 proviral transgenes using a standard PCR method [3]. A specific forward primer for each gene and a common reverse primer SP3 located in the backbone of the pSin-EF2-gene-Pur were used: forward primers, LIN28: 5′-AAGCGCAGATCAAAAGGAGA-3′ (target size 518 bp); Nanog: 5′-CAGAAGGCCTCAGCACCTAC-3′ (732 bp); Oct4: 5′-CAGTGCCCGAAACCCACAC-3′ (656 bp); Sox2: 5′-TACCTCTTCCTCCCACTCCA-3′ (467 bp); reverse primer (SP3), 5′-AGAGGAACTGCTTCCTTCACGACA-3′. PCR products were size fractionated on 2% agarose gels stained with ethidium bromide.
Karyotyping
Standard G-banding chromosome analysis was carried out in the Cytogenetics Lab at Columbia University Med Center (CUMC). Selected iPS cell clones were analyzed.
Immunocytofluorescence
iPS cell colonies grown on MEFs in 12-well plates were fixed in 100% ice cold methanol, incubated in blocking buffer [32.5 mM NaCl, 3.3 mM Na2HPO4, 0.76 mM KH2PO4, 1.9 mM NaN3, 0.1% (w/v) bovine serum albumin (BSA), 0.2% (v/v) Triton X-100, 0.05% (v/v) Tween 20, and 5% goat serum] for 30 min followed by addition of the following antibodies for 1 h at room temperature: mouse anti-human SSEA-4, TRA-1-60, TRA-1-80, TRA-2-49 (these 4 antibodies were from Chemicon Int Inc., www.chemicon.com), and Oct4; goat anti-human Nanog; and rabbit anti-human Sox2 (these 3 antibodies were from Santa Cruz Biotech., Inc., http://www.scbt.com). After washing, cultures were incubated with anti-mouse, -goat, or -rabbit antibodies Alexa Fluor 488 or 594 (Invitrogen for 1 h at room temperature and the cell nuclei stained with DAPI (Invitrogen). Images were analyzed under a fluorescence microscope.
Reverse transcription polymerase chain reaction (RT-PCR)
Specific primers to amplify endogenous 4 factor genes and exogenous 4 factor genes were used to conduct RT-PCR and their levels compared [3]. Cells were harvested for RNA isolation using RNeasy Mini Kit (Qiagen, Valenicia, CA). cDNA was synthesized with SuperScript RT-PCR kit (Invitrogen, Carlsbad, CA) followed by PCR using Taq DNA polymerase (Invitrogen) to detect the gene expression.
For detection of transgene and endogenous 4 factor gene expression, the following genes were examined with specific primers [3]: Nanog (NM_024865), first set that detects both transgene (exogenous) and endogenous gene expression, forward, 5′-CAGAAGGCCTCAGCACCTAC-3′; reverse, 5′-ATTGTTCCAGGTCTGGTTGC-3′ (111 bp); second set that detects only endogenous gene expression, forward, 5′-TTGGAAGCTGCTGGGGAAG-3′, reverse, 5′-GATGGGAGGAGGGGAGAGGA-3′ (193 bp); Oct4 (NM_002701), first set (exogenous + endogenous), forward, 5′-CAGTGC CCGAAACCCACAC-3′, reverse, 5′-GGAGACCCAGCAGC CTCAAA-3′ (161 bp); second set (endogenous), forward, 5′-AGTTTGTGCCAGGGTTTTTG-3′, reverse, 5′-ACTTCACCTTCCCTCCAACC-3′ (113 bp); and GAPDH, forward, 5′-CAA GGC TGA GAA CGG GAA GC-3′; reverse, 5′-AGG GGG CAG AGA TGA TGA CC-3′ (194 bp). The primers that only detect exogenous are the same as in provirus integration experiment.
For detection of gene expression in embryoid bodies (EBs), the following genes were examined with specific primers [21]: ectoderm-specific genes, Nestin (NM_006617), forward, 5′-GCGTTGGAACAGAGGTTGGA-3′, reverse, 5′-TGGGAGCAAAGATCCAAGAC-3′ (target size 327 bp); NCAM (NM_000615), forward, 5′-ATGGAAACTCTATTAA AGTGAACCTG-3′, reverse, 5′-TAGACCTCATACTCA GCATTCCAGT-3′ (178 bp); mesoderm-specific, Brachyury (NM_003181), forward, 5′-ACCCAGTTCATAGCG GTGAC-3′, reverse, 5′-CAATTGTCATGGGATTGCAG-3′ (392 bp); RUNX1 (NM_001001890), forward, 5′-CCCTAGGG GATGTTCCAGAT-3′ reverse, 5′-TGAAGCTTTTCCCTCTT CCA-3′ (162 bp); endoderm-specific genes, α-fetoprotein (AFP, NM_001134), forward, 5′-AGCTTGGTGGTGGATGA AAC-3′, reverse, 5′-CCCTCTTCAGCAAAGCAGAC-3′ (248 bp); GATA4 (NM_002052), forward, 5′-CTAGACCGTG GGTTTTGCAT-3′, reverse, 5′-TGGGTTAAGTGCCCCT GTAG-3′ (275 bp).
Promoter DNA methylation profiles
Bisulfite genomic sequencing analyses were performed. Conversion of unmethylated cytosines to uracil of purified genomic DNA was carried out as described in EZ DNA Methylation-Gold Kit (ZYMO, Orange, CA). Five hundred micrograms of genomic DNA was treated in each reaction, and the elution was used for PCRs. The promoter regions of Oct4 and Nanog were amplified by PCR (Taq Polymerase, Invitrogen) using Oct4 primer set 7 and Nanog primer set 2 [33] with the following parameters: denaturing, 94°C for 2 min; 38 cycles of 94°C for 20 s, 54°C for 20 s, and 68°C for 1 min, and followed by 68°C for 10 min. Bisulfite sequencing primers are in the following: Oct4–7: forward: 5′-TAGTTGGGATGTGTAGAGTTTGAGA-3′; reverse: 5′-TAAACCAAAACAATCCTTCTACTCC-3′. Nanog-2: forward: 5′-GAGTTAAAGAGTTTTGTTTTTAAAAATTAT-3′; reverse: 5′-TCCCAAATCTAATAATTTATCATATCTTTC-3′. The PCR products were confirmed with electrophoresis and cloned into pCR-Blunt II-TOPO vector (Invitrogen) and sequenced with Sp6 primer.
Telomerase activity assay
Telomerase activity was examined using Telo TAGGG Telomerase PCR ELISA kit (Roche Applied Science, Indianapolis, IN) according to the manufacture’s protocol. In brief, 0.2 × 106 cells were lysed and added to a premix solution containing biotin-labeled synthetic P1-TS primer and P2 primer. The resultant extended products are subsequently amplified by PCR. The generated PCR products were hybridized with a digoxigenin-(DIG)-labeled, telomeric repeat-specific detection probe and immobilized to a streptavidin-coated microplate. The immobilized PCR products were then detected with an antibody against digoxigenin (anti-DIG-POD) that were conjugated to peroxidase. Subsequently, the probe was visualized by virtue of peroxidase metabolizing TMB to form a colored reaction product. As positive control, 293T cells were used and the cell extracts heated at 85°C for 10 min were served as negative control.
Embryoid body (EB) formation
iPS or hES cell colonies on feeders were grown to a size 1 day passed the splitting time. Colonies were treated with 0.2–0.5 mg/mL dispase for 20–30 min until the colonies completely detached from the plate and transferred to a 15-mL conical tube to allow precipitation. Cells were washed once with hES cell media. The precipitated colonies were then transferred to wells of 6-well plates containing ES cell media without bFGF. The media were changed every day until EBs processed for analysis.
Neurogenic differentiation in vitro
EB-mediated neurogenic differentiation of iPS cells was performed. iPS cell colonies grown on Matrigel were treated with dispase (1 mg/mL) for 7 min to detach the cell colonies, resuspended in DMEM/F12 (supplemented with 20% FBS and 2 mM glutamine), and plated into Petri dishes. After 4 days in cultures, EBs were collected and plated into Matrigel-coated wells of 4-well plates. After cell attachment, the medium was changed to DMEM supplemented with B27 and 20 ng/mL bFGF. Cells were incubated in the differentiation medium for 8 days and subsequently stained for neuronal marker Tuj1 (Sigma-Aldrich, St. Louis, MO). Cells were fixed in 4% paraformaldehyde for 15 min and washed with PBS 3 times. Triton X-100 (0.1%) was applied to permeabilize the cells for 15 min followed by PBS washing for 3 times. Fixed cells were blocked with 5% goat serum for 1 h. Primary mouse monoclonal antibody to human Tuj1 at 1:1,000 was applied and incubated at 4°C overnight. After 3 times of PBS washing, secondary goat anti-mouse Alexa Fluor 488 (1:1,000) antibodies and Hoechst 33342 (1:1,000) (Sigma-Aldrich) were added and incubated at room temperature for 1 h. Staining was examined under a fluorescent microscope.
Teratoma formation
ES cell-like colonies ∼70% confluent in 2–6 wells of 6-well plates were harvested by collagenase IV or 0.05% Trypsin–EDTA treatment, collected into tubes, centrifuged, and the pellets suspended in DMEM/F12 and Matrigel (3:1). The resuspended cells were injected intramuscularly into the right and/or left hind leg of a SCID mouse (SCID, NOD.CB17-Prkdc-scid/J; Jackson Laboratory, Bar Harbor, ME). Nine to eleven weeks after injection, tumors were dissected and fixed with PBS containing 4% paraformaldehyde. Paraffin-embedded tissue was sectioned and stained with hematoxylin and eosin (H&E). All animal procedures followed a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Columbia University (protocol# AC-AAAB1141).
Results
Formation of ES cell-like colonies and expansion
Heterogeneous population of dental tissue-derived stem/progenitor cells were transduced with the 4 factors bearing viral vectors. ES cell-like colonies began emerging on the feeder layer ∼2–3 weeks after gene transduction. In 6 independent experiments of various approaches, 15 SHED-, 4 SCAP-, and 65 DPSC-ES cell-like clones were obtained (Table 1). Double infection increased the success rate. The rate of formation of ES cell-like colonies from double-transduced DPSCs or SHED during reprogramming is ∼1/1,000, which increased by up to 5-fold compared to single transduction. This rate is 4–10 times greater than the reported using DFs (0.01%–0.02%) [1,3]. In experiment 1, only one colony was formed after 5 × 104 cells were seeded into a 10-cm dish for reprogramming. Overcrowded non-ES cell-like colonies may have obscured the ES cell-like colony formation. Reducing the number of seeded cells onto MEFs for reprogramming improved the success of ES cell-like colony formation. Representative clones at passage 0 are shown in Figure 1 top panels. Several clones were selected for expansion and further analysis (Fig. 1 bottom panels). Both hGFs and hFFs yielded no ES cell-like colonies from these experiments.
Table 1.
Summary of the Reprogramming Experiments
| Experiment | Cells | Infection condition | No. of transduced cells seeded onto MEFs | No. of ES cell-like colonies | % (No. of ES cell-like/no. of seeded) |
|---|---|---|---|---|---|
| 1 | Single infection | 5 × 104 (10-cm dish) | |||
| SHED | 0 | 0 | |||
| DPSCs | 1 | 0.002 | |||
| GFs | 0 | 0 | |||
| 2 | Single infectiona | 1 × 104 (10-cm dish) | |||
| SHED | 2 | 0.02 | |||
| DPSCs | 2 | 0.02 | |||
| GFs | 0 | 0 | |||
| 3 | Double infectionb | 4 × 103 (1 well of 6-well plate) | |||
| SHED | 13 (4 wells) | 0.08 | |||
| DPSCs | 19 (4 wells) | 0.1 | |||
| GFs | 0 (4 wells) | 0 | |||
| 4 | Single infection | 3 × 103 (1 well of 6-well plate) | |||
| SCAP | 4 (2 wells) | 0.07 | |||
| DPSCs | 2 (2 wells) | 0.03 | |||
| hGFs | 0 (2 wells) | 0 | |||
| hFFs | 0 (2 wells) | 0 | |||
| 5 | Double infectionc | 2 × 104 (10-cm dish) | |||
| DPSCs | 23 | 0.1 | |||
| 6 | Double infectionc | 2 × 104 (10-cm dish) | |||
| DPSCs | 18 | 0.1 |
Experiments 1–4, pSin-EF2-gene-Pur vector set (carrying Lin28, Nanog, Oct4, or Sox2) was used; experiments 5 and 6, pMXs vector set (carrying c-Myc, Klf4, Oct4, or Sox2) was used.
aRemaining transduced cells from experiment 1 were frozen, thawed, and seeded onto mouse embryonic fibroblasts (MEFs) for reprogramming.
bRemaining transduced cells from experiment 1 were frozen, thawed, infected again with the same viral vector set, and seeded onto MEFs for reprogramming.
cCells were infected with virus once followed by a second infection 24 h later.
Abbreviations: SHED, stem cells from exfoliated deciduous teeth; SCAP, stem cells from apical papilla; DPSC, dental pulp stem cell; hGF, human gingival fibroblast; hFF, human foreskin fibroblast.
FIG. 1.
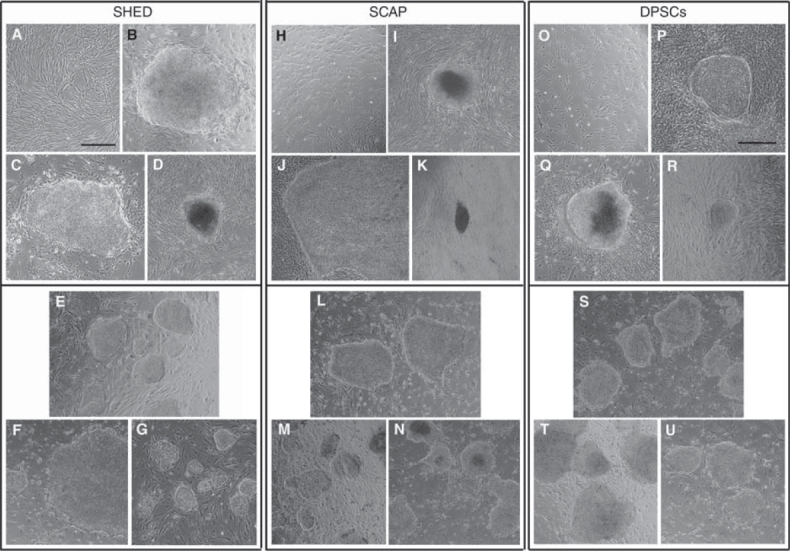
Emerging of embryonic stem (ES) cell-like colonies and expansion. Stem cells from exfoliated deciduous teeth (SHED) (A–G), stem cells from apical papilla (SCAP) (H–N), and dental pulp stem cells (DPSCs) (O–U) were transduced with the 4 factors (Lin28, Nanog, Oct4, and Sox2) and seeded onto mouse embryonic fibroblasts (MEFs) until ES cell-like colonies emerged (representative clones). Top panels, from pre-transduction (A, H, and O) to emerging of ES cell-like colonies on MEFs at passage 0 (B–D, I–K, and P–R). Lower panels, ES cell-like colonies at passage 0 were hand-picked and passaged to new MEFs for expansion. (B) Clone 1 to (E) at passage 3; (C) clone bix2 to (F) at passage 4; (D) clone 2 to (G) at passage 2; (I) clone 1 to (L) at passage 4; (J) clone d1–1 to (M) at passage 13; (K) clone d2–2 to (N) at passage 9; (P) clone 1 to (S) at passage 35; (Q) clone 2 to (T) at passage 4; (R) clone bix1 to (U) at passage 10. Scale bars = 500 μm [the bar in (A) represents all the images except (P)].
Protein expression of ES cell gene markers
Immunocytofluorescent analysis indicate that DPSC-, SHED-, SCAP-iPS cells expressed hES cell markers SSEA-4, TRA-1-60, TRA-1-80, TRA-2-49, Nanog, Oct4, and Sox2 as indicated in the representative data in Figure 2. These markers in untransduced cells were not detected by the same staining procedures (data not shown).
FIG. 2.

Embryonic stem (ES) cell-associated gene expression by the putative induced pluripotent stem (iPS) cell clones. (A) Dental pulp stem cells (DPSCs)-iPS cell clone 1 at passage 7 were fixed and stained with antibodies against human embryonic stem (hES) cell-associated genes and examined under fluorescence microscopy. (B) Stem cells from exfoliated deciduous teeth (SHED)-iPS cell clone 2 bix at passage 6. (C) Stem cells from apical papilla (SCAP)-iPS cell d2-clone 1 at passage 6. Expressed genes stained either in red or green fluorescence; DAPI, nuclear stain. Scale bars = 100 μm.
Provirus integration
To examine the presence of transgenes in the genome of those iPS cell clones, genomic DNA was isolated and primers specific for each transgene were used to amplify 4 proviral transgenes. The 4 factors were all integrated into the genome of the transduced SHED/SCAP/DPSC-iPS cells (Fig. 3).
FIG. 3.
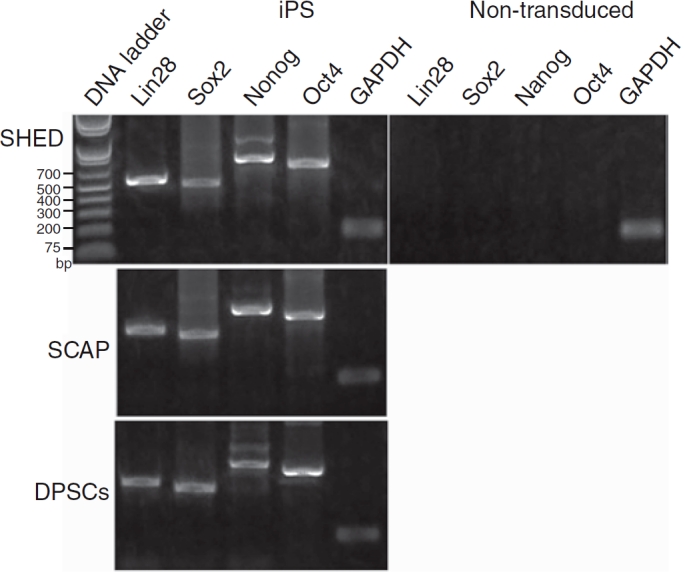
Proviral integration and gene expression analysis by PCR. Stem cells from exfoliated deciduous teeth (SHED), stem cells from apical papilla (SCAP)-induced pluripotent stem (iPS) cells, and dental pulp stem cells (DPSCs)-iPS cells were harvestedfor DNA isolation and subjected to PCR for the detection of Lin28, Nanog, Oct4, and Sox2 transgenes in the genome. Passage numbers at cell harvesting: SHED-iPS cells, p10; SCAP-iPS cells, p8; DPSC-iPS cells, p12.
mRNA expression and promoter methylation status
Endogenous gene and transgene expression of Nanog and Oct4 in iPS cells and their non-transduced control counter parts was analyzed by RT-PCR. Exogenous Nanog expression in iPS cells was higher than the endogenous, whereas endogenous Oct4 expression is as high as the exogenous (Fig. 4A). Endogenous gene expression in non-transduced control was detectable but in low levels. The exogenous genes were not silenced after reprogramming in our system, which may be due to the use of vector type—lentiviral vector [3] versus retroviral vector [1–2].
FIG. 4.

RT-PCR analyses and bisulfite sequencing. (A) RT-PCR detection of gene expression of Nanog and Oct4 (pLentiviral vector) in non-transduced control cells (C) and iPS cells. Endo, endogenous gene expression; exo, exogenous transgene expression. RNA was isolated from stem cells from exfoliated deciduous teeth (SHED)-iPS cells at passage 15; stem cells from apical papilla (SCAP)-iPS cells at passage 8, and dental pulp stem cells (DPSCs)-induced pluripotent stem (iPS) cells at passage 12. (B) The DNA methylation status of Nanog and Oct4 promoter regions were examined by bisulfite sequencing. The closed circles indicate methylated sites of the analyzed regions.
The methylation status of cytosine guanine dinucleotides (CpG) in the promoter regions of Nanog and Oct4 was examined using bisulfite DNA sequencing method. Selected DPSCs- and SHED-iPS cell clones were examined and the results showed that Nanog promoter had similar or slightly higher number of methylated sites in SHED- and DPSC-iPS cells than their non-transduced counterparts (Fig. 4B). The Oct4 promoter had less methylated sites in SHED-/DPSC-iPS cells than the non-transduced cells, which corresponds to the findings of Oct4 mRNA expression in cells. The methylation status of Nanog promoter did not correspond to the expression levels of endogenous mRNA of Nanog in non-transduce control cells and their iPS cells, suggesting other mechanism is controlling the endogenous expression, or other sites of the promoter regions are more responsible for this regulation.
Normal karyotypes of dental stem cell-derived iPS cells
Cytogenetic analysis of selected iPS cells showed a normal karyotype of a male (SHED-iPS cells) or two female (SCAP- and DPSC-iPS cells) karyotype (Fig. 5). The findings are in agreement with those reported to date that reprogramming using a vector system to introduce the factors does not alter the cell karyotype.
FIG. 5.
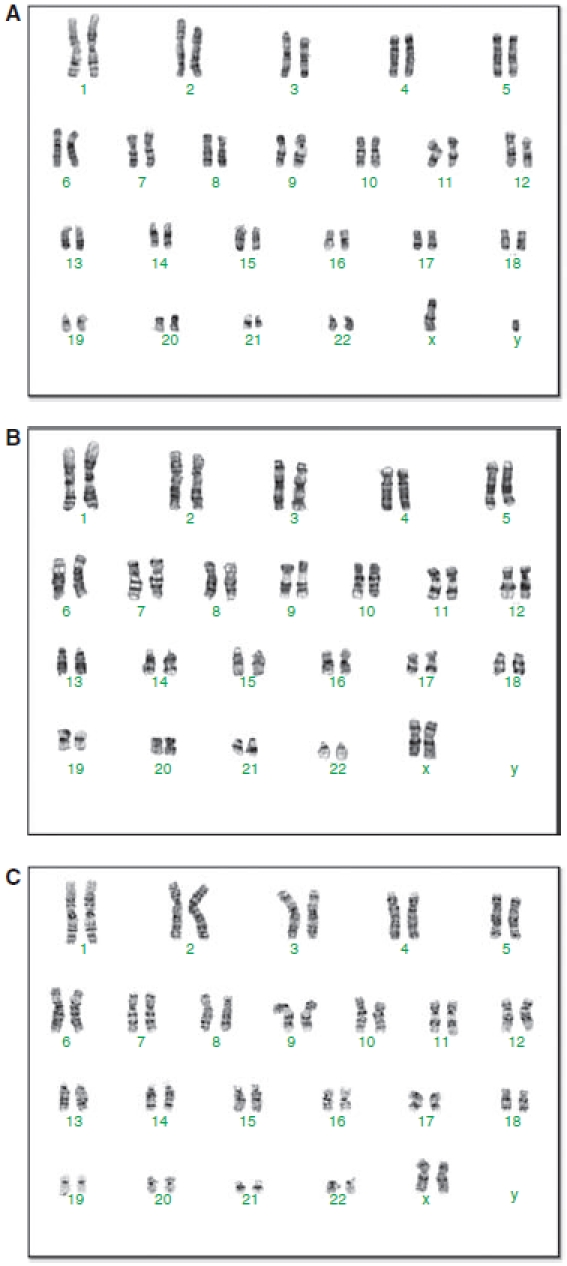
Karyotyping of induced pluripotent stem (iPS) cells cells. (A) stem cells from exfoliated deciduous teeth (SHED)-iPS clone B2 at passage 11. Original SHED from a ∼10-year-old male. (B) stem cells from apical papilla (SCAP)-iPS d2-clone 1 at passage 18. Original SCAP from a 16-year-old female. (C) dental pulp stem cells (DPSCs)-iPS clone 1 at passage 12. Original DPSCs from a 19-year-old female.
Increased telomerase activities in iPS cells
Pluripotent stem cells usually have high telomerase activity to maintain the integrity of chromosome structure. After reprogramming, the telomerase activities of SHED-, SCAP-, and DPSC-iPS cells increased dramatically in comparison to their non-transduced counterparts, and were as high as those of hES cells (Fig. 6).
FIG. 6.
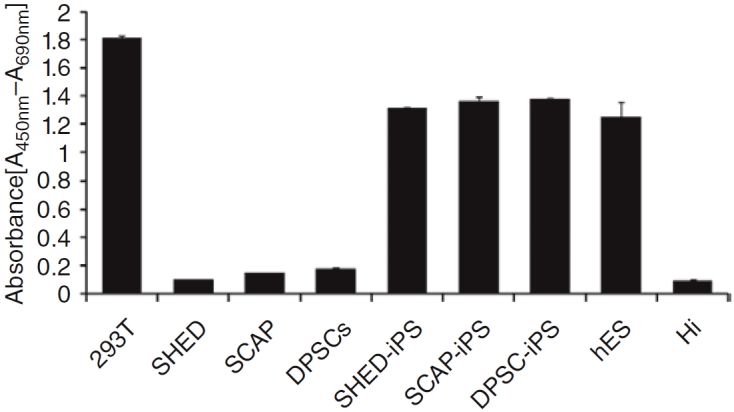
Telomerase activity analysis. Telomerase activity is presented in the form of relative absorbance. human embryonic stem (hES) cell or induced pluripotent stem (iPS) cells were grown in Matrigel-coated feeder-free wells and harvested for analysis. The iPS cell clones (stem cells from exfoliated deciduous teeth (SHED) at passage 33; stem cells from apical papilla (SCAP) at 27; dental pulp stem cells (DPSCs) at 41) used for this assay were the same as those for karyotyping. Their non-transduced counterparts were all at passage 3. 293T cells were used as positive control; heated 293T cell extract was negative control (HI).
In vitro neural differentiation
DPSC-iPS cells were tested for their neurogenic potentials with an EB-mediated approach. Under the stimulation of a neurogenic medium, cells from the EBs developed into neural rosette-like morphology. At a higher magnification, individual cells with elongated cellular processes were observed (Fig. 7A and 7B). To determine whether these cells that morphologically resembling neuronal cells express neuronal genes, cells were stained with a neuronal precursor marker Tuj1. As demonstrated in Figure 7D, the elongated cytoplasmic processes are positive for Tuj1 staining, indicating the neurogenic potential of these iPS cells.
FIG. 7.

Neural differentiation of induced pluripotent stem (iPS) cells derived from dental pulp stem cells (DPSCs). After 4 days, Embryoid body (EB) were plated into Matrigel-coated wells of 4-well plates. Neurogenic medium described in Materials and Methods was added to cultures after cell attachment. Differentiated iPS cells developed into a morphology resembling neural rosettes on day 6 after neurogenic stimulus (A) and subsequently extended elongated cell cytoplasmic processes resembling neurons on day 12 after stimulation (B). (C–E) These cells and their processes showed positive staining for the neuronal precursor marker Tuj1 (C, phase; D, Tuj1 staining in green; E, Hoechst 33342 nuclear stain in blue).
In vitro embryoid body (EB) analysis
EB formation allows iPS cells to differentiate into various cell types. We first tested the expression of genes in cells representing all 3 germ layers. On day 8 during EB formation, EBs from SHED-, SCAP-, and DPSC-iPS cells were harvested and total RNA isolated for RT-PCR analysis. The results in Figure 8 show that these genes were expressed in the EBs. At 3–4 weeks, EBs formed cystic cavities that could be observed under the inverted microscope (Fig. 8Ba and 8Be). At 6th week, some iPS cell-derived EBs were processed for histological analysis. Presence of cavities or cystic space in the EBs was verified. Cells in the EBs differentiated into different cell types, some of which formed tissue-like structures resembling those of ectodermal, mesodermal, or endodermal origin. Neuroepithelial-like structures and neural rosettes were observed (Fig. 8Bc and 8Bd). Inner cystic bodies may be encircled by a layer of epithelial-like cells resembling primitive endodermal epithelium (Fig. 8Bg and 8Bh). The cavity may also be walled by an outer layer epithelial-like cells and the space was structured by loose connective tissue (Fig. 8Bb). Areas of cartilagenius matrix may appear in the EBs (Fig. 8Bf). Amorphous eosinophilic granular materials were seen in areas of the EB mass suggesting apoptotic activities as a process of cell differentiation and tissue modeling. EB formation from hES cell was also performed in parallel as a comparison (Fig. 8Bi to 8Bi).
FIG. 8.
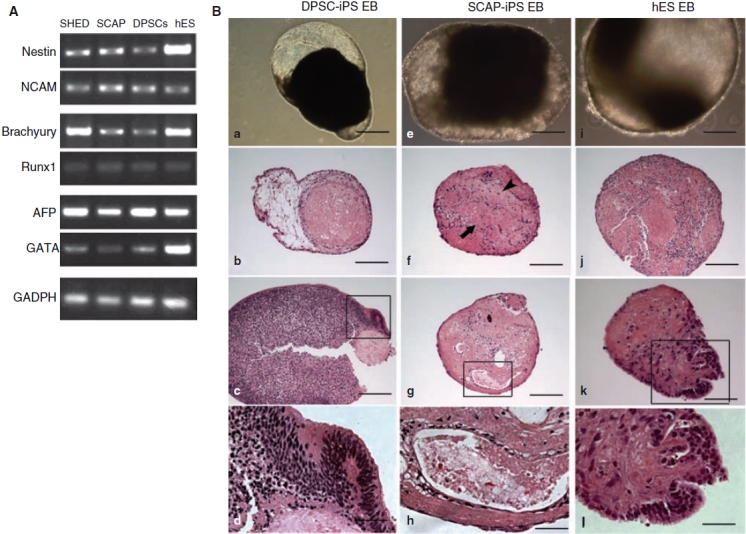
Embryoid body (EB) formation. (A) RT-PCR analysis of gene expression in EBs from stem cells from exfoliated deciduous teeth (SHED)-, stem cells from apical papilla (SCAP), and dental pulp stem cells (DPSCs)-induced pluripotent stem (iPS) cells clones on day 8. Genes representing different germ layers were examined. Ectoderm: Nestin and NCAM; mesoderm: Brachyury and Runx1; endoderm: AFP and GATA. (B) Morphological and histological analysis of EBs. (a–d) DPSC-iPS cell-derived EBs. (e–h) SCAP-iPS cell-derived EBs. (i–l) human embryonic stem (hES) cell-derived EBs. (a, e, i) EBs grown in cultures 3–4 weeks. (b–d, f–h, i–l) H&E stain of fixed EBs after 6 wks (DPSC-iPS and SCAP-iPS cells) or 4 wks (hES cells) of EB formation in cultures. (d, h, l) Magnified views of the respective boxed areas in (c, g, k). (f) Arrowhead indicates cartilaginous matrix; arrow indicates area of granular materials. Passage numbers of iPS cells when EB formation initiated: SHED-iPS, p18; SCAP-iPS, p15; DPSC-iPS, p25. Scale bars = 500 μm (a); (b, e–g, i, j) 200 μm; (k) 100 μm; (d, h, l) 50 μm.
Teratoma formation
Ultimately, fully reprogrammed cells must be able to form teratomas in vivo containing tissue cell types of all 3 germ layers in order to be considered truly ES cell-like iPS cells. As demonstrated in Figure 9, all 3 types of dental iPS cells (SHED, SCAP, and DPSCs) formed teratomas in SCID mice. The teratomas are highly cystic containing multilobular structures filled with tissue fluid or blood. Histological analysis showed that SHED/SCAP/DPSC-iPS cells developed into primitive tissues representing all germ layers including neural tissues (ectoderm), cartilage (mesoderm), and glandular or respiratory epithelial layers (endoderm). There were numerous ectodermal neuroepithelial-like tissues including pigmented retinal epithelium-like tissues as well as glandular structures. Some glandular tissues exhibited branching from the wall of the cyst into the lumen (Fig. 9L). The mesodermal-derived tissues were filling in the space between ectoderm- and endoderm-derived tissues including cartilage and fibrous connective tissues. A few small bone spicules were observed in some samples.
FIG. 9.

Histological analysis of teratomas containing multi-differentiated tissues derived from induced pluripotent stem (iPS) cells. (A–D) stem cells from exfoliated deciduous teeth (SHED)-iPS cell (passage 4)-derived teratoma; (E–H) stem cells from apical papilla (SCAP)-iPS cell (passages 7–9)-derived; (I–L) dental pulp stem cells (DPSCs)-iPS cell (passages 20–29)-derived. (A, D, E, I, H) Mainly primitive neural tissues, neural rosettes, and retinal epithelium (ectoderm); (B, F, J) mainly cartilage (mesoderm); (C, G, K, L) mainly glandular tissue or respiratory epithelium (endoderm). Scale bars: (A, D–H, K, L) 200 μm; (B, C, I, J) 50 μm. Asterisk in (H, L) indicates external space outside of the teratomas; asterisk in (K) indicates the space of a cavity inside the teratoma. Non-transduced cells did not form teratomas (data not shown).
Discussion
Human dermal fibroblasts were the first type of cells being reprogrammed into iPS cells. Other human cell types that may be potentially easier to reprogram have been tested [24–25]. Although human MSCs were considered to be easier to reprogram, the evidence indicates the opposite [22]. Our results revealed that unlike other developmentally mature somatic cells such as neonatal foreskin fibroblasts, adult MSCs, and adult DFs that needed the introduction of hTERT and SV40 large T to succeed the reprogramming [22], dental tissue-derived mesenchymal-like stem cells SHED, SCAP, and DPSCS can easily be reprogrammed into iPS cells at relatively higher rates. We also simultaneously transduced hFFs from ATCC and hGFs in parallel with dental stem cells under the same experimental setting and infection protocols, no ES cell-like colonies have emerged from these fibroblasts, suggesting that dental stem cells are more readily to be reprogrammed into iPS cells.
Dental stem cells are mesenchymal-like stem cells and are different from MSCs in many aspects [32]. The frequency of colony-forming cells derived from dental pulp tissue (22–70 colonies/104 cells plated) is higher than that of MSCs from bone marrow (2.4–3.1 colonies/104 cells plated) [29]. They proliferate rapidly in culture with an average population doubling (PD) time of ∼20 h [34] and can reach a PD of up to 100 or more before senescence [26]. In addition, from our unpublished work along with the reported, DPSCs and SHED already express a number of ES cell-associated genes such as c-Myc, Oct4, Nanog, SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 at low levels [35,36], which may facilitate the reprogramming.
Although many types of adult stem cells are multipotent including dental stem cells used in this study, their sources and life spans are limited. They are insufficient in number to regenerate sizable tissues or organs. iPS cells may one day resolve this issue when proven to be safe and controllable for clinical applications. Since the generation of iPS cells, much attention has been focused on the avoidance of the use of viral vectors and the characterization of iPS cells in comparison to ES cells. Rapid progress in these areas of research has led to the realization that vector system is no longer a needed approach [12,13], and iPS cells behave identically to ES cells in terms of the ability to support full-term development of tetraploid blastocyst-complemented embryos [37]. Although these progresses have been made in the mouse system, they help project the great potential of human iPS cells for various clinical applications. To further advance the iPS cell research, efforts should also be put into human iPS cell studies as human cells have been known to be more difficult to reprogram than mouse cells.
While various types of discarded or easily obtainable normal human tissues are potentially a cell source to generate iPS cells, it is unknown at present whether iPS cells derived from different types of cells behave in the same manner. More importantly, iPS cells from different cell types may also be different in their abilities to undergo guided differentiation into specialized cells compared to embryonic stem cells [38]. Therefore, iPS cells should be generated from various easily accessible human tissues and characterized thoroughly. Because the dental stem cells derive from ectomesenchyme [39,40], it is possible that iPS cells derived from cells of this tissue have the propensity to differentiate into oral, craniofacial, and especially odontogenic tissues under the appropriate stimulus. It has been noted that differences occur between the iPS cells generated from different cell types, for example, mesenchymal versus endodermal origin, in terms of the kinetics of reprogramming and the outcomes of the generated chimeric mice [41].
SHED are from shed primary teeth of children at ages 6–12. SCAP and DPSCs from third molars are from young individuals ages 16–22. These age groups contain more immature cells suitable for reprogramming purposes. Furthermore, tooth banks and dental stem cell banks are emerging in the industrial domain and the infrastructure to establish the banking system is relatively easy. Tooth is a small organ and the cell isolation process from the dental tissues is also relatively simple. Utilizing these dental somatic cells for regenerative purposes and establishing dental stem cell-derived iPS cell bank can be a near-future possibility. Our studies represent the first step toward establishing protocols to generate iPS cells using an easily accessible alternative cell source. Further investigation that is critical to reaching the goals of utilizing iPS cells for clinical applications includes: (i) generating dental stem cell-derived iPS cells using non-vector approaches, (ii) characterizing of these dental stem cell-derived iPS cells in comparison with iPS cells from other cell sources and hES cells, (iii) establishing reliable protocols to guide iPS cells toward differentiated cells for specific tissue regeneration, and (iv) determining the genomic stability of iPS cells in long-term cultures.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health R01 DE019156–01 (G.T.-J.H.), RO1 DE17449 (S.S.), and by the NIAMS/NIH Intramural Research Program Z01 AR41131 (R.S.T). We wish to thank Dr. Barbara Mallon (NIH, Stem Cell Unit, Bethesda, MD), Dr. Qi-Long Ying (Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell Research at USC, Los Angeles, CA), Dr. Shinya Yamanaka (Center for iPS cell research and application, Kyoto University, Japan), and Dr. Junying Yu (University of Wisconsin, Madison, WI) for providing assistance and advice in human ES cell or iPS cell cultures.
Contributor Information
Xing Yan, Section of Oral and Diagnostic Sciences, Division of Endodontics, College of Dental Medicine, Columbia University, New York, New York..
Haiyan Qin, Center for Craniofacial Molecular Biology, University of Southern California School of Dentistry, Los Angeles, California..
Cunye Qu, Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell Research, Keck School of Medicine, University of Southern California, Los Angeles, California..
Rocky S. Tuan, Cartilage Biology and Orthopaedics Branch, National Institute of Arthritis, and Musculoskeletal and Skin Diseases, Department of Health and Human Services, National Institutes of Health, Bethesda, Maryland..
Songtao Shi, Center for Craniofacial Molecular Biology, University of Southern California School of Dentistry, Los Angeles, California..
George T.-J. Huang, Section of Oral and Diagnostic Sciences, Division of Endodontics, College of Dental Medicine, Columbia University, New York, New York. Cartilage Biology and Orthopaedics Branch, National Institute of Arthritis, and Musculoskeletal and Skin Diseases, Department of Health and Human Services, National Institutes of Health, Bethesda, Maryland. Department of Endodontics, Prosthodontics, and Operative Dentistry, College of Dental Surgery, Dental School, University of Maryland, Baltimore, Maryland.
Author Disclosure Statement
There are no commercial associations that might create a conflict of interest in connection with this article.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Kim JB, Sebastiano V, Wu G, Araúzo-Bravo MJ, Sasse P, Gentile L, Ko K, Ruau D, Ehrich M, van den Boom D, Meyer J, Hübner K, Bernemann C, Ortmeier C, Zenke M, Fleischmann BK, Zaehres H, Schöler HR. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009;136:411–419. doi: 10.1016/j.cell.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y, Do JT, Desponts C, Hahm HS, Scholer HR, Ding S. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell. 2008;2:525–528. doi: 10.1016/j.stem.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, Desponts C, Do JT, Hahm HS, Scholer HR, Ding S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, Isacson O, Jaenisch R. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hamalainen R, Cowling R, Wang W, Liu P, Gertsenstein M, Kaji K, H-K Sung, Nagy A. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez F, Barragan Monasterio M, Tiscornia G, Montserrat Pulido N, Vassena R, Batlle Morera L, Rodriguez Piza I, Izpisua Belmonte JC. Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proc Natl Acad Sci USA. 2009;106:8918–8922. doi: 10.1073/pnas.0901471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, Siuzdak G, Scholer HR, Duan L, Ding S. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim D, C-H Kim, J-I Moon, Y-G Chung, M-Y Chang, B-S Han, Ko S, Yang E, Cha KY, Lanza R, K-S Kim. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyssiotis CA, Foreman RK, Staerk J, Garcia M, Mathur D, Markoulaki S, Hanna J, Lairson LL, Charette BD, Bouchez LC, Bollong M, Kunick C, Brinker A, Cho CY, Schultz PG, Jaenisch R. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc Natl Acad Sci USA. 2009;106:8912–8917. doi: 10.1073/pnas.0903860106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stadtfeld M, Brennand K, Hochedlinger K. Reprogramming of pancreatic beta cells into induced pluripotent stem cells. Curr Biol. 2008;18:890–894. doi: 10.1016/j.cub.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanna J, Markoulaki S, Schorderet P, Carey BW, Beard C, Wernig M, Creyghton MP, Steine EJ, Cassady JP, Foreman R, Lengner CJ, Dausman JA, Jaenisch R. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264. doi: 10.1016/j.cell.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JB, Zaehres H, Wu G, Gentile L, Ko K, Sebastiano V, Arauzo-Bravo MJ, Ruau D, Han DW, Zenke M, Scholer HR. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454:646–650. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- 18.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoi T, Yae K, Nakagawa M, Ichisaka T, Okita K, Takahashi K, Chiba T, Yamanaka S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 20.Kunisato A, Wakatsuki M, Kodama Y, Shinba H, Ishida I, Nagao K. Generation of induced pluripotent stem (iPS) cells by efficient reprogramming of adult bone marrow cells. Stem Cells Dev. 2009 doi: 10.1089/scd.2009.0149. [DOI] [PubMed] [Google Scholar]
- 21.Loh YH, Agarwal S, Park IH, Urbach A, Huo H, Heffner GC, Kim K, Miller JD, Ng K, Daley GQ. Generation of induced pluripotent stem cells from human blood. Blood. 2009;113:5476–5479. doi: 10.1182/blood-2009-02-204800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotech. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 24.Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, Edel M, Boue S, Belmonte JCI. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotech. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Zhou J, Shi G, Ma Y, Yang Y, Gu J, Yu H, Jin S, Wei Z, Chen F, Jin Y. Pluripotency can be rapidly and efficiently induced in human amniotic fluid-derived cells. Hum Mol Genet. 2009;18:4340–4349. doi: 10.1093/hmg/ddp386. [DOI] [PubMed] [Google Scholar]
- 26.Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, Shi S. SHED: Stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci USA. 2003;100:5807–5812. doi: 10.1073/pnas.0937635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonoyama W, Liu Y, Fang D, Yamaza T, Seo BM, Zhang C, Liu H, Gronthos S, Wang CY, Shi S, Wang S. Mesenchymal stem cell-mediated functional tooth regeneration in Swine. PLoS ONE. 2006;1:e79. doi: 10.1371/journal.pone.0000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonoyama W, Liu Y, Yamaza T, Tuan RS, Wang S, Shi S, Huang GTJ. Characterization of the apical papilla and its residing stem cells from human immature permanent teeth: a pilot study. J Endod. 2008;34:166–171. doi: 10.1016/j.joen.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA. 2000;97:13625–13630. doi: 10.1073/pnas.240309797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, DenBesten P, Robey PG, Shi S. Stem cell properties of human dental pulp stem cells. J Dental Res. 2002;81:531–535. doi: 10.1177/154405910208100806. [DOI] [PubMed] [Google Scholar]
- 31.Huang GT, Sonoyama W, Chen J, Park SH. In vitro characterization of human dental pulp cells: various isolation methods and culturing environments. Cell Tissue Res. 2006;324:225–236. doi: 10.1007/s00441-005-0117-9. [DOI] [PubMed] [Google Scholar]
- 32.Huang GT-J, Gronthos S, Shi S. Mesenchymal stem cells derived from dental tissues vs from other sources: the biology and role in regenerative medicine. J Dental Res. 2009;88:792–806. doi: 10.1177/0022034509340867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freberg CT, Dahl JA, Timoskainen S, Collas P. Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol Biol Cell. 2007;18:1543–1553. doi: 10.1091/mbc.E07-01-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang GTJ, Shagramanova K, Chan SW. Formation of odontoblast-like cells from cultured human dental pulp cells on dentin in vitro. J Endod. 2006;32:1066–1073. doi: 10.1016/j.joen.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Huang AH, Chen YK, Lin LM, Shieh TY, Chan AW. Isolation and characterization of dental pulp stem cells from a supernumerary tooth. J Oral Pathol Med. 2008;37:571–574. doi: 10.1111/j.1600-0714.2008.00654.x. [DOI] [PubMed] [Google Scholar]
- 36.Kerkis I, Kerkis A, Dozortsev D, Stukart-Parsons GC, Gomes Massironi SM, Pereira LV, Caplan AI, Cerruti HF. Isolation and characterization of a population of immature dental pulp stem cells expressing OCT-4 and other embryonic stem cell markers. Cells Tissues Organs. 2006;184:105–116. doi: 10.1159/000099617. [DOI] [PubMed] [Google Scholar]
- 37.Kang L, Wang J, Zhang Y, Kou Z, Gao S. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell. 2009;5:135–138. doi: 10.1016/j.stem.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Yamanaka S. A fresh look at iPS cells. Cell. 2009;137:13–17. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 39.Cobourne MT, Sharpe PT. Tooth and jaw: molecular mechanisms of patterning in the first branchial arch. Arch Oral Biol. 2003;48:1–14. doi: 10.1016/s0003-9969(02)00208-x. [DOI] [PubMed] [Google Scholar]
- 40.Chai Y, Jiang X, Ito Y, Bringas P, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- 41.Sridharan R, Plath K. Illuminating the black box of reprogramming. Cell Stem Cell. 2008;2:295–297. doi: 10.1016/j.stem.2008.03.015. [DOI] [PubMed] [Google Scholar]