

Abstract
Many biomaterials are designed to regulate the interactions between artificial and natural surfaces. However, when materials are inserted through the cell membrane itself the interface formed between the interior edge of the membrane and the material surface is not well understood and poorly controlled. Here we demonstrate that by replicating the nanometer-scale hydrophilic-hydrophobic-hydrophilic architecture of transmembrane proteins, artificial “stealth” probes spontaneously insert and anchor within the lipid bilayer core, forming a high-strength interface. These nanometer-scale hydrophobic bands are readily fabricated on metallic probes by functionalizing the exposed sidewall of an ultrathin evaporated Au metal layer rather than by lithography. Penetration and adhesion forces for butanethiol and dodecanethiol functionalized probes were directly measured using atomic force microscopy (AFM) on thick stacks of lipid bilayers to eliminate substrate effects. The penetration dynamics were starkly different for hydrophobic versus hydrophilic probes. Both 5- and 10 nm thick hydrophobically functionalized probes naturally resided within the lipid core, while hydrophilic probes remained in the aqueous region. Surprisingly, the barrier to probe penetration with short butanethiol chains (Eo,5 nm = 21.8kbT, Eo,10 nm = 15.3kbT) was dramatically higher than longer dodecanethiol chains (Eo,5 nm = 14.0kbT, Eo,10 nm = 10.9kbT), indicating that molecular mobility and orientation also play a role in addition to hydrophobicity in determining interface stability. These results highlight a new strategy for designing artificial cell interfaces that can nondestructively penetrate the lipid bilayer.
Keywords: atomic force microscopy, biophysics, membranes, proteins
The ability to specifically and nondestructively incorporate inorganic structures into or through biological membranes is a key step toward realizing full bioinorganic integration, such as arrayed on-chip patch-clamps, drug delivery, and biosensors. However, molecular delivery and interfaces to inorganic objects, such as patch-clamp pipettes, generally rely upon destructive formation of membrane holes and serendipitous adhesion (1), rather than selective penetration and attachment into the bilayer itself. Because a key aspiration of biomaterials is seamlessly interfacing artificial materials with natural components, a more benign means to penetrate through the cell membrane is required. While surface modification techniques have been highly successful at controlling cell mobility, proliferation, and differentiation on two-dimensional surfaces (2–4), bridging across the cell membrane itself has been much less studied. A prime example of such a system is membrane proteins, whose outer surface is designed to specifically interact with the interior of the cell membrane lipid bilayer. The tight junction between the lipid and protein eliminates constitutive ion or protein leakage, allowing membrane proteins to regulate the chemical flux through the bilayer.
While most gene and drug carrier particles appear to enter the cell by endocytotic mechanisms (5), materials such as cationic polymers (6, 7) and nanoparticles (8) have been shown to directly penetrate the membrane. However, these highly charged species can create holes leading to significant cytotoxicity, and the underlying lipid-cation interaction mechanism is still poorly understood (9, 10). New materials delivery systems such as DNA functionalized nanowires pierce the membrane and have had some success delivering cargo, but cells are unable to survive longer than several days following penetration (11). Recent experiments with sub-6nm diameter thiol-functionalized Au nanoparticles (8) have discovered that molecular-scale phase segregation of hydrophilic and hydrophobic components apparently enables direct translocation through the membrane without hole-formation. This provides some optimism that defined surfaces with nm-scale heterogeneity may dictate interactions with the lipid core, yet how to extend this architecture to larger structures is still unknown.
Ideally, interaction between a probe and the bilayer interior could be achieved by modifying the material’s surface characteristics. A necessary trait of these “artificial membrane proteins” is the capacity to specifically insert into the bilayer core and form a strong interface, mimicking endogenous transmembrane proteins. A broad variety of molecular functionalizing agents are available, including small molecules, peptides, and polymers. Peptide interactions with the hydrophobic membrane core has been studied at length and are generally well-described by an empirical hydrophobicity scale (12). However, these thermodynamic guidelines provide little insight into the protein-lipid dynamics important for kinetic processes such as bilayer penetration. Other characteristics of molecular agents such as entropy, crystallinity, orientation, and spatial patterning may also play important roles and enable property tuning beyond what is currently available with proteins.
Here we present a simple microfabricated architecture based on metallic multilayer probes that allows probe fusion into a lipid bilayer core and systematic control of lateral bilayer-material interactions (Fig. 1). The probes consist of a metallic post with a thin, 2–10 nm hydrophobic band designed to fuse into the core of the lipid bilayer. These “stealth” probes are designed to mimic two essential transmembrane protein characteristics: The transmembrane regions are mostly hydrophobic with hydrophilic groups on either side, and the thickness of the hydrophobic domain should be commensurate with the bilayer thickness (13). These characteristics are extremely encouraging for engineered biomimetic systems, because no special protein interactions or molecular-scale configurations are necessary. Instead, creating a ∼3–5 nanometer hydrophobic band on an otherwise hydrophilic structure is the critical design feature.
Fig. 1.

Diagram of stealth probe integration with a lipid bilayer. (A). Surface-surface interactions regulated by tethering molecules to the substrate. (B) The “stealth probe” structure with a hydrophobic domain designed to interact specifically with the hydrophobic membrane core through selective surface functionalization. (C) A functionalized band several nanometers thick is defined by selective self-assembly of molecules. (D) A hydrophobic functionalized band interacts specifically with the hydrophobic core of the lipid bilayer, similar to the behavior of membrane proteins.
Lithographically defining 3–5 nm features on nonplanar structures (such as a cylindrical probe) is beyond even state-of-the-art lithography equipment. However, hydrophobic bands of these dimensions can be readily formed by molecularly functionalizing the exposed sidewall of an evaporated metal layer. Fig. 1 shows a schematic of a stealth probe that uses self-assembly of hydrophobic molecules on the edge of an Au layer sandwiched between hydrophilic metals. The height of the functional band is determined by the thickness of the Au, which can be controlled to 1–2 nm using current electron-beam evaporation or sputtering techniques (14). This technique provides a flexible platform to examine how both architecture and various molecular agents influence adhesion within the bilayer itself and could be integrated into a number of biointerface systems, such as deep brain implants, neural prosthetics, and patch-clamp devices.
Here we use atomic force microscopy (AFM) to directly measure the location, penetration force, and adhesion strength of stealth probes functionalized with different molecular fusion agents within the bilayer. Previous studies have found that AFM is an excellent tool to measure bilayer thickness, force penetration barriers, and the effects of homogeneous tip functionalization (15, 16). In addition, we find that the dynamic characteristics of AFM penetration through a series of bilayers reveal the preferential probe localization within a bilayer. The adhesion force of different probe functionalities was measured from the probe retraction force from the bilayer, and it was discovered that different length alkanes had significantly different adhesion strengths despite very similar hydrophobicities. These measurements show that the stealth probe architecture is a straightforward means to control integration and penetration of inorganic microstructures into lipid bilayers and may provide a flexible platform for nondestructive integration into cells.
Results and Discussion
Stealth probes were fabricated directly upon silicon AFM tips in order to measure the probe force and displacement during probe penetration through a lipid bilayer as shown in Fig. 2. Initially, conical Si cantilever tips are milled into a roughly 500 nm diameter cylinder in a focused ion beam microscope (FIB) (Fig. 2 B and G). The cantilevers are then transferred to an electron-beam metal evaporator and layered Cr-Au-Cr stacks of either 10-5-10 nm or 10-10-10 ± 0.5 nm are deposited. After metal deposition, the post sidewalls have a thin covering of Cr (Fig. 2 C and H) that inhibits self-assembly onto the gold layer. A second FIB milling is performed to remove the sidewall deposition and reduce the post diameter to ∼200 nm, exposing the edges of the individual metal layers. Transmission electron microscopy (TEM) confirmed the Au layer has a clean sidewall free of Cr (Fig. 2 I and J). The Au edge was then functionalized by self-assembly in 5 mM butanethiol or dodecanethiol solutions in ethanol for 12 h, rinsed with ethanol, and blown dry. To ensure this treatment did not functionalize the Cr layer we measured the contact angles of planar Cr films after immersion in the alkane thiol solutions, which were uniformly hydrophilic with a water contact angle < 20°. Planar Au surfaces had contact angles of 110° and 110.5° for butanethiol and dodecanethiol, respectively. Similar stealth probes could also be fabricated on Si wafers by e-beam lithography to define the post, followed by metal stack evaporation and lift-off.
Fig. 2.

AFM post probe fabrication. (A) A Si AFM tip is used to fabricate stealth probes. (B) Using the FIB, the tip was shaped into a post ∼500 nm in diameter. (C) Cr-Au-Cr metal films were evaporated onto the entire cantilever and post, covering the top and sidewalls of the milled post. (D) Tips were remilled in the FIB to trim the excess metal from the sidewalls of the post and expose the edge of the Au layer. Final tip diameter is ∼200 nm. (E) Fabricated tips were subsequently functionalized using alkanethiol self-assembly. (F–H) SEM images of corresponding fabrication step (A–C). (I) TEM image of final stealth probe. (J) TEM image of layered metal stack at tip of stealth probe. The 10 nm Au band (Dark, Central Band) is visible between the two Cr layers, with a clean edge profile.
Butanethiol and dodecanethiol were chosen as hydrophobic functionalizing agents to examine how molecular hydrophobicity and mobility influenced the probe-lipid interface. Butanethiol has a short, 4-carbon chain with high surface mobility on Au films, while dodecanethiol has a 12-carbon chain that forms crystalline monolayers (17, 18). The hydrophobicity of these two species are similar (110° and 110.5°, respectively), but planar dodecanethiol monolayers are crystalline, while the liquid-like butanethiol monolayers adopt more random conformations. These functionalized tips were compared to control samples with unfunctionalized Cr probes, which were uniformly hydrophilic.
The forces and dynamics of probe penetration through a lipid bilayer were measured by advancing the probe into a stack of lipid bilayers using an AFM in force-testing mode. Previous AFM lipid penetration tests have used one or a few bilayers supported on a solid surface (19, 20). However, single or double supported bilayers are not ideal for examining penetration behavior because the stealth probe tip will come into contact with the underlying substrate. This issue was avoided by creating thick, pancake-like stacks of hundreds to thousands of bilayers, allowing penetration through a large number of membranes without probe-substrate interactions. The thick bilayer stacks used here also differ from previous lipid experiments that hydrated a subregion of a larger stacked membrane structure (21).
A force-displacement curve for penetration of an unfunctionalized hydrophilic tip through the top 39 bilayers of a stack is shown in Fig. 3A (see Fig. S1A and B for force-displacement curves for 5 nm Au bands functionalized with butanethiol or dodecanethiol). Upon contact the stack is compressed an average of 16.9 ± 6.7% (smooth approach curve from z = 300 to 450 nm), until the applied force is sufficient to penetrate the bilayer, an average force of 58.3 ± 3.0 nN at a loading rate of 3.6 nN/s. Each sawtooth deflection corresponds to the tip breaking through a single bilayer. The vertical step-height of each sawtooth corresponds to the distance the probe tip moves when breaking through the bilayer (“breakthrough distance”), while the failure force is the highest force value before rupture. The lipid stacks produce high-quality breakthrough data, as seen from the uniformity of the force and distance.
Fig. 3.

(A) Representative AFM curve of penetration through a bilayer stack with an unfunctionalized probe (Red: approach, Black: withdrawal). Each of the 39 vertical drops corresponds to breaking through the hydrophobic core of a single lipid bilayer. (B) Histograms of stacked bilayer penetration distances for unfunctionalized, 5 nm Au/butanethiol, and 5 nm Au/dodecanethiol functionalized probes. (C) Schematic of breakthrough regions for each membrane probe. Hydrophilic, unfunctionalized probes breakthrough the hydrophobic bilayer core only, while hydrophobic functionalized probes jump from bilayer center to center. (D) Tip penetration behavior as a function of z-piezo displacement. Unfunctionalized probes have sharp jumps corresponding to breaking through the hydrophobic core, followed by relaxation through the hydrophilic region. Hydrophobic functionalized probes jump from hydrophobic core to hydrophobic core, with breakthrough distances corresponding to the lamellar repeat distance.
Surprisingly, we discovered that the breakthrough distance the tip moves after rupturing the bilayer is indicative of the probe’s relative resting position within the bilayer. This is highly advantageous because traditional techniques such as optical microscopy, x-ray diffraction, or TEM cannot measure the position of an individual probe within the bilayer. Previous measurements with hydrophilic pyramidal AFM tips found that the breakthrough distance was equivalent to the thickness of the hydrophobic bilayer core, which acts as a barrier to penetration due to the interfacial energy of contact (20). In this case, the hydrophilic tip pushes against the top of the hydrophobic layer until enough force is applied to rupture the membrane, at which point the tip jumps through the hydrophobic core and stops at the aqueous headgroup region. Measurements with the hydrophilic control tips confirmed these results, with a breakthrough distance of 2.9 ± 0.3 nm (Fig. 3B). This agrees quantitatively with the 3.1 nm hydrophobic core thickness of 2∶1 SOPC:cholesterol bilayers as used here, including the bilayer compression before breakthrough (19, 22, 23). The hydrophilic probe thus normally resides within the hydrophilic gaps or membrane headgroups before penetration.
However, for hydrophobic stealth probes the band is expected to reside within the center of the bilayer to minimize interfacial energy. The breakthrough distance in this case should then equal the center-to-center distance between bilayers due to the band translating from the middle of one membrane to the next to avoid contact with the aqueous regions. Indeed, 5 nm Au bands functionalized with butanethiol or dodecanethiol give breakthrough distances of 5.6 ± 0.6 nm and 5.7 ± 0.6 nm, respectively (Fig. 3B), nearly double the hydrophilic result. The breakthrough histograms for these two molecules are extremely similar, indicating both probe functionalizations reside in the same well-defined location. The breakthrough distances agree with the measured equilibrium lamellar spacing of 6.46 nm (24) after accounting for the 16.7% compression of the bilayer stack, which reduces the expected spacing to 5.4 ± 0.4 nm. The behavior of probes with 10 nm Au bands is similar to that of 5 nm Au bands, with breakthrough distances of 5.7 ± 0.3 nm and 5.9 ± 0.2 nm for butanethiol and dodecanethiol functionalizations, respectively (Fig. S2). Thus, both 5- and 10- nm thick bands functionalized with butane and dodecane thiol reside in the middle of the hydrophobic bilayer core.
The most striking difference between hydrophobic and hydrophilic functionalized surfaces is revealed in the bilayer penetration dynamics (Fig. 3D and Figs. S3 and S4). For the hydrophilic Cr probe the tip motion consists of short, abrupt 3 nm drops corresponding to membrane core penetration, followed by linear motion (Fig. 3D, Green Curve) as the AFM cantilever moves vertically through the bilayer stack. During the linear motion the force also increases linearly (Fig. 3A), and the total distance traversed before breakthrough is equal to the thickness of the hydration layer plus the headgroup thickness. We believe this results from the hydrophilic probe contacting the top of an unbroken bilayer and expelling the water layer beneath it as the probe is advanced. This causes the first two bilayers underneath the probe to be in close proximity right before rupture. Following breakthrough of the top bilayer, the probe is then immediately in contact with the next bilayer and the process repeats.
In contrast, both butanethiol and dodecanethiol stealth probes exhibit abrupt, 5–6 nm drops corresponding to membrane core to membrane core transitions, followed by extremely flat plateaus indicating very little motion within the bilayer with increasing force (Red, Blue Curves). For either functionalization the average motion between breakthroughs was 0.2 nm, indicating the probes had a very strong affinity for the center of the bilayer. No intermediate states were observed during the breakthrough events, which might indicate metastable locations for the hydrophobic band. The dramatic difference between the two mechanisms enables facile identification of where the hydrophobic band naturally resides.
The adhesion strength of the probes within the bilayer was measured by a combination of the AFM force-displacement curves during probe withdrawal (Fig. 4, Black Curve) and dynamic force-tests of penetration. Unfunctionalized tips (Fig. 4A) show lipid-tether formation as typically observed with hydrophilic probes, characterized by the multiple step-like failure events with zero-withdrawal force plateaus on withdrawal (25, 26). These thin membrane tubules nonspecifically bind to the probe and are extruded from the main bilayer with low resistance, often extending tens of microns in length (27). As each tether either snaps or breaks free of the probe, rapid jumps in tip deflection occur.
Fig. 4.

Representative AFM force curves for 3 different stealth probes (Red: approach, Black: withdrawal). (A) Unfunctionalized probe. (B) 10 nm Au/butanethiol functionalized probe. (C) 10 nm Au/dodecanethiol functionalized probe. (Insets) Timescale of interface rupture for the tear-off event.
The functionalized stealth probes (10 nm Au band) exhibit no penetration force for the first bilayer on approach and only a single pull-off event, signifying spontaneous insertion and formation of a single bilayer-probe interface without membrane tethers (Fig. 4B and C). These failure events occurred over a smaller range of distances than the tethers and with much longer timescales (Fig. 4 insets). The duration of the rupture event for the unfunctionalized, butanethiol, and dodecanethiol probes was 0.5 ± 0.26 ms, 7.95 ± 5.5 ms, and 1.87 ± 0.81 ms, respectively. The longer rupture times are consistent with a failure mechanism that involves rearrangement of a number of lipids, as would be expected for the approximately 628 nm lipid-probe contact perimeter (∼1800 lipids). The adhesion force as a function of withdrawal distance (Fig. 5) also shows that the butanethiol probe adheres strongly within the bilayer. While unfunctionalized probes have the same rupture force at all distances and large force plateaus consistent with lipid tethers, butanethiol tips have a distinctly nonlinear response that increases with distance. As the probe is withdrawn, the bilayers create a restoring force that increases with distance. An equivalent response has been observed during extraction of membrane proteins that are attached to the underlying cytoskeleton (28). Similarly, the butanethiol probe must embed firmly enough to cause the subsurface bilayers to mechanically deform before failure. In contrast, the dodecanethiol tips failed almost immediately and no trend in adhesion force could be ascertained. In order to verify that these results were not probe-related artifacts, a dodecanethiol probe was cleaned with UV-ozone, rinsed in ethanol to remove any gold oxide, then refunctionalized with butanethiol. This yielded nonlinear results equivalent to the other butanethiol probes.
Fig. 5.

Adhesion force as a function of pull-off distance. Unfunctionalized probes show behavior consistent with tether formation, namely a constant adhesion force with increasing pull-off distance. Butanethiol functionalized probes exhibit and increase in force with increasing pull-off distance. Dodecanethiol functionalized probes display reduced adhesion compared to both butanthiol and unfunctionalized probes. Dotted lines to guide eye.
While the locations of the butanethiol and dodecanethiol probes within the bilayer are virtually identical, their adhesion strengths differed considerably, revealing hydrophobicity alone does not determine the interfacial strength. The average maximum adhesion force for the three different probes shows unfunctionalized probes have an average of 1.05 ± 0.11 nN, butanethiol probes an average of 1.90 ± 0.20 nN, and dodecanethiol probes an average of 0.45 ± 0.08 nN (see Fig. S5 for histograms). However, it should be noted that rupture forces and rates are intrinsically linked to the rate at which the bond is loaded. Numerous AFM experiments have shown this dependence, including single-molecule force-clamp experiments (29, 30) and bilayer penetration experiments (16, 31). Therefore, it may be possible that the observed differences are simply due to differing dependencies on loading rate.
In order to address this question, we measured the energy barrier for bilayer penetration using dynamic force spectroscopy (DFS) experiments. The data was acquired in a force-clamp mode (29) that was performed by rapidly applying a large force load, halting, and allowing the cantilever to relax by breaking through the bilayer stack. Fig. 6A shows a typical force-displacement curve, clearly showing the distinct stair-steps corresponding to bilayer failure. The failure rate (breakthroughs per second) at each force value was then compiled and fit to a standard Langevin model (32) for barrier crossing with applied force:
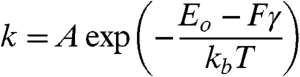 |
[1] |
where k is the failure rate at a given force, A is the attempt frequency (the cantilever resonance frequency of ∼6 kHz was used) (16), Eo is the unstressed energy barrier height, F is the applied force, T is the absolute temperature, and γ is the location of the energy barrier. Fig. 6B and C show ln(k) vs F for 5 and 10 nm band thicknesses for both functionalizations and the corresponding linear fits. Both 5- and 10 nm Au bands functionalized with butanethiol exhibit significantly larger energy barriers (Eo,5 nm = 21.8kbT, Eo,10 nm = 15.3kbT, γ5 nm = 1.01 pm, γ10 nm = 0.43 pm), than probes functionalized with dodecanethiol, (Eo,5 nm = 14.0kbT, Eo,10 nm = 10.9kbT, γ5 nm = 0.43 pm; γ10 nm = 0.19 pm), confirming the trend observed during adhesion testing. The adhesion strength for the 5 nm thick bands was higher for either functionalization, likely reflecting the strain required for a single bilayer to conform to a 10 nm band (also see Fig. S3).
Fig. 6.

(A) Force-clamp testing. The force is ramped to a high force load (60 nN) on top of the bilayer stack, then the position of the z-piezo is fixed, and the probe relaxes by breaking though the bilayers. Each stair step corresponds to a single bilayer breakthrough. The breakthrough rate is then measured as a function of the applied force. (B and C) Linear fits to ln(k) for the 5 nm (B) and 10 nm (C) band thicknesses for dynamic force spectroscopy reveal butanethiol has a higher adhesion energy than dodecanethiol.
Interestingly, the difference between the two functionalized probes agrees with pioneering studies on general anesthetics (33, 34). Haydon et al. examined the anesthetic properties of n-alkanes infiltrating and swelling lipid bilayers, concluding that homologues shorter than heptane readily adsorb horizontally into the space between leaflets, thereby thickening the bilayer. Subsequent research using neutron scattering (35), x-ray diffraction, and differential scanning calorimetry (36) confirmed hexane partitions into the midplane with a horizontal orientation, while longer chain alkanes reside perpendicular to the midplane, parallel to the lipid acyl chains. These results qualitatively agree with the different adhesion forces for the two functionalizations, which are ostensibly oriented in-plane relative to the lipid bilayer and perpendicular to the lipid tails. Butanethiol should prefer the horizontal orientation and indeed has a high adhesion force. Dodecanethiol should favor the vertical orientation, yet it is likely unable to do so from its crystalline structure (17). The large free energy of the terminal methyl group makes the dodecane-lipid tailgroup intersection quite weak and a likely location to nucleate a defect. Chain mobility may similarly play a role, as butanethiol exists in a disordered, fluid-like phase on planar surfaces (17, 18) compared to the crystalline dodecane, allowing the chain to reorient as necessary.
The implications from the interplay between molecular mobility and hydrophobicity in terms of interface formation extend the possible applications to particle and drug delivery. While surfaces with a mobile, hydrophobic surface functionalization create a strong adhesive bond, restricting mobility decreases the adhesion strength, allowing for short-term association followed by permeation. This agrees with the results of Verma et al. (8), who found that nanoparticles with ordered rings of hydrophilic and hydrophobic domains penetrate cellular membranes.
Conclusion
The stealth probe design demonstrates that by recreating the nanometer-scale architecture of transmembrane proteins through a simple microfabrication technique, bioengineered structures can specifically insert into the bilayer core and form a strong interface. The adhesion strength with the bilayer interior depended upon the hydrophobicity and molecular mobility of nanoscale hydrophobic bands, allowing tuning with different molecular species. As suggested by earlier calorimetry studies (37), these results indicate the hydrophobic effect is only one factor in material stability within the lipid bilayer, and that mobility and/or chain orientation are other primary attributes. In general, tailoring the bilayer-probe interface characteristics should be possible by altering the probe geometry, including band thickness, number of bands, and molecular functionalization. Probe penetration dynamics through stacks of lipid bilayers were found to provide detailed insight into the preferential location of the probe, lipid organization, and adhesion strength. Because the stealth probe architecture can be fabricated onto atomic force microscope tips or in large arrays on solid substrates, both fundamental studies of the mechanics of molecular membrane penetration as well as technological applications such as arrays of on-chip patch clamps are exciting opportunities for further research.
Materials and Methods
Stealth Probe Fabrication.
Standard commercial AFM cantilevers with a nominal spring constant of 0.08 N/m (CSC-38/Au BS, MikroMasch USA) were mounted vertically in a focused ion beam microscope (FIB) (FEI Strata 235DB). The tips were milled to a post shape approximately 500 nm in diameter and 600 nm long using a 30 kV, 10 pA Ga-beam, followed by a 90° rotation onto their sides and subsequent milling to complete the post geometry (Fig. 2B and G). A layered Cr-Au-Cr structure (each Cr metal layer = 10 nm thick, Au layer = 5 or 10 nm) was deposited by e-beam metal evaporation (Temescal) on the modified AFM cantilevers at a rate of 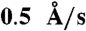 (Fig. 2C and H). Thicknesses are ± 5% and were calibrated using x-ray reflectivity. Following metal deposition, the cantilevers were remilled in the FIB to a final diameter of approximately 200 nm using the same milling procedure, with the exception of the beam current being reduced to 1 pA (Fig. 2D and I).
(Fig. 2C and H). Thicknesses are ± 5% and were calibrated using x-ray reflectivity. Following metal deposition, the cantilevers were remilled in the FIB to a final diameter of approximately 200 nm using the same milling procedure, with the exception of the beam current being reduced to 1 pA (Fig. 2D and I).
Formation of Lipid Bilayer Stacks.
Stacks of 30–2000 lipid bilayers were formed by gentle hydration of a dried lipid cake. Glass coverslips (VWR, 25 mm dia.) were cleaned for 30 min in Piranha etch. Coverslips were then rinsed thoroughly with deionized water and dried under nitrogen. Ten microliter drops of 10 mg/mL of a 2∶1 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) and cholesterol (Avanti Polar Lipids) solution in chloroform were deposited on the clean coverslips, dried under a stream of nitrogen, and desiccated under vacuum for at least 4 hrs. Desiccated coverslips were mounted in an Asylum Research closed fluid cell. A strip of PTFE was used to manually spread the dried lipid over the coverslip into a thin layer. Following spreading, 1 mL of 1.6% NaCl solution was added to the fluid cell and allowed to sit for approximately 2 h.
Force Testing Procedure.
Membrane probes were functionalized for at least 12 hrs in 5 mM ethanolic solutions of either 1-butanethiol (Alfa Aesar) or 1-dodecanethiol (Sigma-Aldrich). Previously used tips could be refunctionalized with different molecules following a 30-min UV-ozone cleaning (UVO Cleaner, Jetlight Company Inc.). After UV-ozone treatment, tips were soaked in pure ethanol (Sigma-Aldrich) for 30 min to remove any gold oxide.
Functionalized membrane probes were removed from solution, rinsed in ethanol, and mounted in an Asylum Research MFP-3D AFM. Spring constant calibration was done using the Sader and thermal methods. Stack penetration testing was performed at 500 nm/s, while adhesion force testing was performed at a rate of 2 μm/s. A dwell of 1 sec was used between extension and retraction, when the probe is in contact with the bilayers to allow for fusion to occur.
Force-clamp curves were obtained by initially bringing the probes into contact with a lipid stack at a rate of 4 μm/s. Loading was stopped when a force set point of 40–100 nN was reached. Once the set point was obtained, a 30–60 s dwell was triggered where the cantilever position was held constant. During this dwell session, the change in cantilever deflection was measured. Drift in the system was accounted for by leveling the low force/long time drift of the baseline.
Supplementary Material
Acknowledgments.
We acknowledge Ricardo Dolmetsch and John Huguenard for helpful conversations, and Evan Pickett for TEM assistance. We thank the National Science Foundation Center for Probing the Nanoscale (NSF CPN) Grant No. PHY-0425897 and Canon Inc. for support, and B.D.A acknowledges the NSF CPN for fellowship support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0909250107/DCSupplemental.
References
- 1.Sakmann B, Neher E. Single-Channel Recording. New York, NY: Plenum Press; 1995. [Google Scholar]
- 2.Benoit DSW, Schwartz MP, Durney AR, Anseth KS. Small functional groups for controlled differentiation of hydrogel-encapsulated human mesenchymal stem cells. Nat Mater. 2008;7(10):816–823. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saha K, Pollock JF, Schaffer DV, Healy KE. Designing synthetic materials to control stem cell phenotype. Curr Opin Chem Biol. 2007;11:381–387. doi: 10.1016/j.cbpa.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maheshwari G, Brown G, Lauffenburger DA, Wells A, Griffith LG. Cell adhesion and motility depend on nanoscale RGD clustering. J Cell Sci. 2000;113:1677–1686. doi: 10.1242/jcs.113.10.1677. [DOI] [PubMed] [Google Scholar]
- 5.Torchilin VP. Multifunctional nanocarriers. Adv Drug Delivery Rev. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Chung T-H, et al. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles in 3T3-L1 cells and human mesenchymal stem cells. Biomaterials. 2007;28:2959–2966. doi: 10.1016/j.biomaterials.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Harush-Frenkel O, Rozentur E, Benita S, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9:435–443. doi: 10.1021/bm700535p. [DOI] [PubMed] [Google Scholar]
- 8.Verma A, et al. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nat Mater. 2008;7:588–595. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leroueil PR, et al. Wide varieties of cationic nanoparticles induce defects in supported lipid bilayers. Nano Lett. 2008;8:420–424. doi: 10.1021/nl0722929. [DOI] [PubMed] [Google Scholar]
- 10.Hong S, et al. Interaction of polycationic polymers with supported lipid bilayers and cells: Nanoscale hole formation and enhanced membrane permeability. Bioconjugate Chem. 2006;17:728–734. doi: 10.1021/bc060077y. [DOI] [PubMed] [Google Scholar]
- 11.Kim W, Ng JK, Kunitake ME, Conklin BR, Yang P. Interfacing Silicon Nanowires with Mammalian Cells. J Am Chem Soc. 2007;129:7228–7229. doi: 10.1021/ja071456k. [DOI] [PubMed] [Google Scholar]
- 12.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 13.Lee AG. Lipid-protein interactions in biological membranes: a structural perspective. BBA-Biomembranes. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 14.Madou MJ. Fundamentals of Microfabrication: The Science of Miniaturization. 2nd Ed. CRC Press; 2002. [Google Scholar]
- 15.Künneke S, Krüger D, Janshoff A. Scrutiny of the failure of lipid membranes as a function of headgroups, chain length, and lamellarity measured by scanning force microscopy. Biophys J. 2004;86:1545–1553. doi: 10.1016/S0006-3495(04)74222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loi S, Sun G, Franz V, Butt H-J. Rupture of molecular thin films observed in atomic force microscopy. II. Experiment. Phys Rev E. 2002;66:031602. doi: 10.1103/PhysRevE.66.031602. [DOI] [PubMed] [Google Scholar]
- 17.Bain CD, et al. Formation of monolayer films by the spontaneous assembly of organic thiols from solution onto gold. J Am Chem Soc. 1989;111:321–335. [Google Scholar]
- 18.Poirier GE, Tarlov MJ, Rushmeier HE. Two-dimensional liquid phase and the p × √3 phase of alkanethiol self-assembled monolayers on Au(111) Langmuir. 1994;10:3383–3386. [Google Scholar]
- 19.Pera I, Stark R, Kappl M, Butt H-J, Benfenati F. Using the atomic force microscope to study the interaction between two solid supported lipid bilayers and the influence of synapsin I. Biophys J. 2004;87:2446–2455. doi: 10.1529/biophysj.104.044214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider J, Barger W, Lee GU. Nanometer scale surface properties of supported lipid bilayers measured with hydrophobic and hydrophilic atomic force microscope probes. Langmuir. 2003;19:1899–1907. [Google Scholar]
- 21.Schäfer A, Salditt T, Rheinstädter MC. Atomic force microscopy study of thick lamellar stacks of phospholipid bilayers. Phys Rev E. 2008;77:021905. doi: 10.1103/PhysRevE.77.021905. [DOI] [PubMed] [Google Scholar]
- 22.Evans E, Ludwig F. Dynamic strengths of molecular anchoring and material cohesion in fluid biomembranes. J Phys-Condens Mat. 2000;12:A315–A320. [Google Scholar]
- 23.Rawicz W, Olbrich KC, McIntosh T, Needham D, Evans E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys J. 2000;79:328–339. doi: 10.1016/S0006-3495(00)76295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rand RP, Fuller N, Parsegian VA, Rau DC. Variation in hydration forces between neutral phospholipid bilayers: evidence for hydration attraction. Biochemistry. 1988;27:7711–7722. doi: 10.1021/bi00420a021. [DOI] [PubMed] [Google Scholar]
- 25.Maeda N, Senden TJ, di Meglio J-M. Micromanipulation of phospholipid bilayers by atomic force microscopy. BBA-Biomembranes. 2002;1564:165–172. doi: 10.1016/s0005-2736(02)00443-1. [DOI] [PubMed] [Google Scholar]
- 26.Sun M, et al. Multiple membrane tethers probed by atomic force microscopy. Biophys J. 2005;89:4320–4329. doi: 10.1529/biophysj.104.058180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koster G, Cacciuto A, Derényi I, Frenkel D, Dogterom M. Force barriers for membrane tube formation. Phys Rev Lett. 2005;94:068101. doi: 10.1103/PhysRevLett.94.068101. [DOI] [PubMed] [Google Scholar]
- 28.Afrin R, Ikai A. Force profiles of protein pulling with or without cytoskeletal links studied by AFM. Biochem Biophys Res Commun. 2006;348:238–244. doi: 10.1016/j.bbrc.2006.07.050. [DOI] [PubMed] [Google Scholar]
- 29.Oberhauser AF, Hansma PK, Carrion-Vazquez M, Fernandez JM. Stepwise unfolding of titin under force-clamp atomic force microscopy. Proc Natl Acad Sci USA. 2001;98(2):468–472. doi: 10.1073/pnas.021321798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schierf M, Li H, Fernandez JM. The unfolding kinetics of ubiquitin captured with single-molecule force-clamp techniques. Proc Natl Acad Sci USA. 2004;101(19):7299–7304. doi: 10.1073/pnas.0400033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butt H-J, Franz V. Rupture of molecular thin films observed in atomic force microscopy. I. Theory. Phys Rev E. 2002;66:031601. doi: 10.1103/PhysRevE.66.031601. [DOI] [PubMed] [Google Scholar]
- 32.Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997;72(4):1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haydon DA, Hendry BM, Levinson SR. The molecular mechanisms of anaesthesia. Nature. 1977;268:356–358. doi: 10.1038/268356a0. [DOI] [PubMed] [Google Scholar]
- 34.Haydon DA, Hendry BM, Levinson SR, Requena J. Anaesthesia by the n-alkanes. A comparative study of nerve impulse blockage and the properties of black lipid bilayer membranes. BBA-Biomembranes. 1977;470:17–34. doi: 10.1016/0005-2736(77)90058-x. [DOI] [PubMed] [Google Scholar]
- 35.White SH, King GI, Cain JE. Location of hexane in lipid bilayers determined by neutron diffraction. Nature. 1981;290:161–163. [Google Scholar]
- 36.McIntosh TJ, Simon SA, MacDonald RC. The organization of n-alkanes in lipid bilayers. BBA-Biomembranes. 1980;597:445–463. doi: 10.1016/0005-2736(80)90219-9. [DOI] [PubMed] [Google Scholar]
- 37.Rowe ES, Zhang F, Leung TW, Parr JS, Guy PT. Thermodynamics of membrane partitioning for a series of n-alcohols determined by titration calorimetry: Role of hydrophobic effects. Biochemistry. 1998;37(8):2430–2440. doi: 10.1021/bi9721602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.