

Abstract
Protein-chromophore interactions in photoreceptors often shift the chromophore absorbance maximum to a biologically relevant spectral region. A fundamental question regarding such spectral tuning effects is how the electronic ground state S0 and excited state S1 are modified by the protein. It is widely assumed that changes in energy gap between S0 and S1 are the main factor in biological spectral tuning. We report a generally applicable approach to determine if a specific residue modulates the energy gap, or if it alters the equilibrium nuclear geometry or width of the energy surfaces. This approach uses the effects that changes in these three parameters have on the absorbance and fluorescence emission spectra of mutants. We apply this strategy to a set of mutants of photoactive yellow protein (PYP) containing all 20 side chains at active site residue 46. While the mutants exhibit significant variation in both the position and width of their absorbance spectra, the fluorescence emission spectra are largely unchanged. This provides strong evidence against a major role for changes in energy gap in the spectral tuning of these mutants and reveals a change in the width of the S1 energy surface. We determined the excited state lifetime of selected mutants and the observed correlation between the fluorescence quantum yield and lifetime shows that the fluorescence spectra are representative of the energy surfaces of the mutants. These results reveal that residue 46 tunes the absorbance spectrum of PYP largely by modulating the width of the S1 energy surface.
Keywords: photoreceptor, protein-chromophore interactions, wavelength regulation
Light-driven proteins, consisting of a protein-chromophore complex, employ two general strategies to optimize the biological effectiveness of the position of their absorbance spectra. First, covalent modifications of a chromophore can shift its absorbance maximum 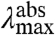 . An important example is the strong red-shift in the absorbance spectrum of bacteriochlorophyll compared to plant chlorophyll (1). Second, specific protein-chromophore interactions can shift the absorbance spectrum of the protein-bound chromophore. A classic example of this spectral tuning phenomenon is color vision in vertebrates (2, 3): the same retinal chromophore can absorb in the blue, green, and red, depending on the amino acid sequence of the rhodopsin to which it is bound. Spectral tuning provides an example of protein-ligand interactions that tune the properties of the cofactor to biologically relevant values.
. An important example is the strong red-shift in the absorbance spectrum of bacteriochlorophyll compared to plant chlorophyll (1). Second, specific protein-chromophore interactions can shift the absorbance spectrum of the protein-bound chromophore. A classic example of this spectral tuning phenomenon is color vision in vertebrates (2, 3): the same retinal chromophore can absorb in the blue, green, and red, depending on the amino acid sequence of the rhodopsin to which it is bound. Spectral tuning provides an example of protein-ligand interactions that tune the properties of the cofactor to biologically relevant values.
Two main approaches have been used to unravel the factors involved in spectral tuning: determining (i) which amino acids in the protein contribute to spectral tuning, and (ii) what type of protein-chromophore interactions cause spectral tuning. Interactions that alter the degree of charge delocalization over the chromophore are of particular importance. These two issues have been explored extensively in animal visual rhodopsins and the archaeal rhodopsins (4–7). Here we study spectral tuning in a more recently discovered photoreceptor, photoactive yellow protein (PYP) (8, 9), and use it to develop and explore a third approach to investigate spectral tuning. Central in this approach are the effects of an individual residue on the shape of the S0 ground state and S1 excited state energy surfaces. PYP offers a highly accessible system (10) to study spectral tuning. We use it here to test a widely used assumption that has dominated the field of biological spectral tuning: that changes in the energy gap ΔE between the S0 and S1 energy surfaces are the major factor in spectral tuning.
PYP is a blue light receptor from the halophilic photosynthetic eubacterium Halorhodospira halophila (8, 9). It functions as the photoreceptor for negative phototaxis in H. halophila (11). The purified protein exhibits a photocycle (9, 12) initiated by the photoisomerization (13) of its p-coumaric acid (pCA) chromophore (14, 15) by a flip in the position of its carbonyl group (16). PYP consists of an antiparallel 6-stranded β-sheet flanked by five α-helices (17). Its pCA chromophore is fully buried within the protein and its deprotonated phenolic oxygen forms short hydrogen bonds with the side chains of Tyr42 and Glu46 (Fig. 1A). Site-directed mutagenesis studies (summarized in ref. 18) have identified Glu46 as a key factor in the spectral tuning of PYP (19–21).
Fig. 1.

Spectral tuning in PYP. (A) Structure of the pCA chromophore in the native state of PYP with active site residues Tyr42 and Glu46. (B) Amplitude-normalized absorbance spectra for free pCA, pCA attached to fully unfolded PYP in its neutral (pU) and ionized (pU-) state, and PYP in its native pG state. All four samples are in aqueous solution. Note that besides the absorbance maximum also the shape and width of the absorbance band is affected.
Three major factors contribute to spectral tuning in PYP (22) (Fig. 1B). First, the formation of the thioester bond between the pCA and Cys69 results in a red-shift from 284 to 335 nm. Second, the deprotonation of the phenolic oxygen of the pCA causes a further red-shift to 400 nm. The strong downshift in the pKa of the pCA from 8.8 in solution (22) to 2.8 in PYP (8, 23) ensures that the chromophore in PYP is deprotonated under physiological conditions. Finally, specific protein-chromophore interactions result in the final red-shift to 446 nm. These interactions have been studied by both site-directed mutagenesis (18–21) and computational approaches (24–27) (SI Text). These studies have revealed Tyr42 and Glu46 as the two dominant side chains in the spectral tuning of PYP (18–21, 24–28). Weakening of the Glu46-pCA hydrogen bond (29, 30) in the E46Q mutant results in a red-shift to 460 nm, while complete disruption of the hydrogen bond in the E46V mutant results in a further red-shift to 478 nm (21). Based on computational studies it has been proposed that hydrogen bonding, charge-charge interactions, and burial of the pCA in the hydrophobic protein interior are factors in the spectral tuning of PYP (24, 25).
Here we propose a straightforward approach to probe if spectral tuning alters not only the ΔE but also the equilibrium nuclear geometry Re or the width W of the S0 and S1 energy surfaces. We find that mutations of active site residue Glu46 in PYP affect spectral tuning not by a change in ΔE but by a change in W of the S1 energy surface.
Results and Discussion
A Strategy for Probing the Changes in S0 and S1 Energy Surface that Cause Spectral Tuning.
We propose a strategy to examine the effects of a selected residue on the spectral tuning of a protein-chromophore complex by changes in the S0 and S1 energy surfaces. The approach is to measure not only absorbance spectra but also fluorescence emission spectra for a set of substitution mutants at this position. Substitution mutations at the residue under study in principle can affect three independent properties of the S0 and S1 energy surfaces: ΔE, Re, and W. The expected consequences of changes in these three properties can be formulated as described below. The electronic structure of PYP has been examined by a number of computational studies (24–27), but the details of the shape of the S0 and S1 energy surfaces and Franck-Condon factors for the pCA in its active site are difficult to obtain with high accuracy. However, general trends in changes in the position and shape of the absorbance and emission fluorescence maxima as a result of changes in ΔE, Re, and W can be derived. These qualitative considerations allow for the approach reported here. We make the approximation that electronic transitions are initiated from the vibrational ground state of S0 and that fluorescence emission occurs from the vibrational ground state of S1. This assumption is supported by the relatively small contribution of vibrational cooling during the lifetime of the S1 state in PYP as experimentally detected by ultrafast changes in the chromophore fluorescence emission spectrum of PYP (31). For the sake of simplicity we first describe the spectra based on harmonic energy potentials for the S0 and S1 states and consider the changes in Franck-Condon factors for absorption and fluorescence emission expected for changes in ΔE, Re, and W of these two energy surfaces.
(Case 1) A change in 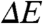 . In the case that the substitutions alter only the ΔE (Fig. 2A), the position of the
. In the case that the substitutions alter only the ΔE (Fig. 2A), the position of the 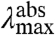 and fluorescence emission maxima
and fluorescence emission maxima 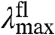 will be shifted in the same direction and by the same amount (on a cm-1 scale), while the width and shape of the absorbance and fluorescence emission spectra (when plotted on an energy scale) remain unchanged. The correlation between
will be shifted in the same direction and by the same amount (on a cm-1 scale), while the width and shape of the absorbance and fluorescence emission spectra (when plotted on an energy scale) remain unchanged. The correlation between 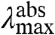 and
and 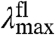 in the substitution mutants will therefore be linear with a slope of 1 (see Fig. 5). This is referred to below as ΔE-tuning.
in the substitution mutants will therefore be linear with a slope of 1 (see Fig. 5). This is referred to below as ΔE-tuning.
Fig. 2.

Strategy for probing excited state surfaces in spectral tuning. Three archetypal changes in energy surfaces by mutations and the resulting spectral tuning effects for absorbance and fluorescence emission spectra are shown. The energy surfaces, electronic transitions, and spectra for both the wild-type protein (black) and mutant protein (red) are indicated schematically for each of the three cases. Changes in both peak position and shape of the absorbance spectra (solid lines) and fluorescence emission spectra (dotted lines) are depicted. Shifts in the positions of the absorbance and fluorescence emission maxima are indicated by horizontal blue arrows. In case 1, which represents the classical explanation of spectral tuning, the energy gap ΔE between the electronic ground state S0 and the first electronically excited state S1 is altered. In case 2 the difference in nuclear equilibrium geometry ΔRe is altered. In case 3 the width W of the S1 energy surface is altered.
Fig. 5.

Analysis of spectral tuning by residue 46 in PYP based on the correlation between absorbance maxima and fluorescence emission maxima. The predicted correlations for spectral tuning by a change in ΔE (red), ΔRe (green), and S1 W (blue) are shown together with the experimentally observed correlation (black).
(Case 2) A change in 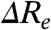 . Substitutions that increase the difference in Re between the S0 and S1 states (
. Substitutions that increase the difference in Re between the S0 and S1 states (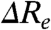 ) will blue-shift the position of the
) will blue-shift the position of the 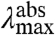 but in contrast will red-shift the
but in contrast will red-shift the 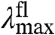 (Fig. 2B). The width and shape of both absorbance and fluorescence emission spectra will most likely be altered, and to a similar extent (in the case of similar W of the S0 and S1 energy surfaces). Therefore,
(Fig. 2B). The width and shape of both absorbance and fluorescence emission spectra will most likely be altered, and to a similar extent (in the case of similar W of the S0 and S1 energy surfaces). Therefore, 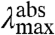 and
and 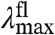 will be anticorrelated, with a slope near -1. We refer to this as Re-tuning. In general, when
will be anticorrelated, with a slope near -1. We refer to this as Re-tuning. In general, when 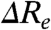 is close to zero only the Franck-Condon factors for the 0-0 transition will be nonzero, resulting in narrow absorbance and emission spectra and a small Stokes shift. Since the absorbance spectrum of PYP is relatively broad (∼2,000 cm-1 at 3/4 height), a significant
is close to zero only the Franck-Condon factors for the 0-0 transition will be nonzero, resulting in narrow absorbance and emission spectra and a small Stokes shift. Since the absorbance spectrum of PYP is relatively broad (∼2,000 cm-1 at 3/4 height), a significant 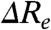 is involved in electronic excitation of the pCA chromophore in PYP.
is involved in electronic excitation of the pCA chromophore in PYP.
(Case 3) A change in W. The third possibility is that the substitutions change the W of either the S0 or S1 energy surface. Predictions for the spectral changes caused by a change in both the W of the S0 or S1 state can be derived. Since our experimental data indicate (see below) a change in W of S1 in the PYP mutants studied here, below we examine the consequences of a change in W of the S1 state, while the S0 energy surface remains unchanged (Fig. 2C). A change in W of the S1 state will cause all Franck-Condon factors contributing to the absorbance spectrum to be altered. This will lead to a change in both 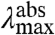 and the shape of the absorbance spectrum. Fluorescence emission from the vibrational ground state of S1 to the unchanged S0 energy surface will be largely unaffected, both with respect to
and the shape of the absorbance spectrum. Fluorescence emission from the vibrational ground state of S1 to the unchanged S0 energy surface will be largely unaffected, both with respect to 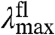 and the shape of the emission spectra. The wave function of the lowest vibrational level of the S1 state will be slightly altered by the change in W, and this will have a small effect on the fluorescence emission spectrum. However, this effect is much smaller than the other spectral changes discussed here, and is therefore neglected. This S1 W-tuning case will therefore result in mutants that exhibit changes in
and the shape of the emission spectra. The wave function of the lowest vibrational level of the S1 state will be slightly altered by the change in W, and this will have a small effect on the fluorescence emission spectrum. However, this effect is much smaller than the other spectral changes discussed here, and is therefore neglected. This S1 W-tuning case will therefore result in mutants that exhibit changes in 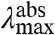 but an unaltered
but an unaltered 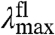 . Since photoexcitation of pCA causes the transition of an electron in a bonding π orbital to an antibonding π∗ orbital, the energy surface (considered with respect to the chemically and functionally relevant C = C torsional angle coordinate) for the S1 state with a C - C single bond will likely be significantly broader than that of the S0 state with a C = C double bond (Fig. 2C). Thus, below we will consider the case that the W of the S1 state is broader than that of the S0 state.
. Since photoexcitation of pCA causes the transition of an electron in a bonding π orbital to an antibonding π∗ orbital, the energy surface (considered with respect to the chemically and functionally relevant C = C torsional angle coordinate) for the S1 state with a C - C single bond will likely be significantly broader than that of the S0 state with a C = C double bond (Fig. 2C). Thus, below we will consider the case that the W of the S1 state is broader than that of the S0 state.
We now consider the direction of the shift in 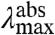 expected for an increase in the W of the S1 state. An increase in the W of S1 will result in an increase in the Franck-Condon factors for transitions to higher vibrational levels of S1 (SI Text), and thus in a blue-shift in the
expected for an increase in the W of the S1 state. An increase in the W of S1 will result in an increase in the Franck-Condon factors for transitions to higher vibrational levels of S1 (SI Text), and thus in a blue-shift in the 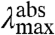 , together with a broadening of the absorbance spectrum. This argument therefore predicts a correlation between the
, together with a broadening of the absorbance spectrum. This argument therefore predicts a correlation between the 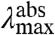 and width of the absorbance band, with more blue-shifted values of
and width of the absorbance band, with more blue-shifted values of 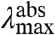 corresponding to a broader absorbance band (see below and Fig. S1).
corresponding to a broader absorbance band (see below and Fig. S1).
Three important factors may complicate the above analysis: anharmonicity of the energy surfaces, contributions of multiple vibrational modes to the optical transition, and inhomogeneous broadening of the spectra by heterogeneity in the configuration of the pCA. In the case of anharmonicity the exact values of the spectral shifts and the shape of the spectra would be slightly altered, but all of the above qualitative considerations would remain valid. The possible involvement of multiple vibrational modes is considered in SI Text and is found to leave the above qualitative predictions intact. For contributions of inhomogeneous broadening of the spectra to the spectral tuning effects we consider two cases. In the simplest case this would result in a similar broadening of all of the spectra; this would not alter the above conclusions and would not contribute to an explanation of spectral tuning (i.e., shifts in 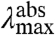 ). A more complex situation would arise if heterogeneity in pCA configuration would alter spectral peak positions and would do so in a manner that depends on the side chain placed at position 46. The regularities in the data presented below (Figs. 4 and 5 and Fig. S1) argue against this latter possibility (SI Text). These considerations validate the qualitative analysis of spectral tuning presented here based on harmonic energy potentials for the S0 and S1 states.
). A more complex situation would arise if heterogeneity in pCA configuration would alter spectral peak positions and would do so in a manner that depends on the side chain placed at position 46. The regularities in the data presented below (Figs. 4 and 5 and Fig. S1) argue against this latter possibility (SI Text). These considerations validate the qualitative analysis of spectral tuning presented here based on harmonic energy potentials for the S0 and S1 states.
Fig. 4.

Analysis of the variation in the absorbance and fluorescence emission spectra of the E46X mutants of PYP. Histograms are shown for the peak positions (A), widths at 3/4 height (B), and band asymmetries (C) of the absorbance spectra (broad gray bars) and fluorescence emission spectra (narrow black bars) of the mutants.
Based on these considerations, quite distinct spectral effects are predicted for changes in ΔE, ΔRe, and W. These three cases can therefore be experimentally distinguished by measuring the effect of mutations on the absorbance and fluorescence emission spectra of a protein. Below we describe such measurements for PYP mutants containing substitutions at active site position 46. All of these mutants remain folded, although the stability of most mutants is somewhat reduced (21).
UV/Vis Absorbance and Fluorescence Emission Spectra of the E46X Mutants Reveal W-Tuning.
The amplitude-normalized UV/Vis electronic absorbance spectra and fluorescence emission spectra of the 20 E46X mutants are shown in Fig. 3A. These data were used to extract the position and width of the absorbance and fluorescence emission spectra in the mutants (Fig. 3B and Table S1). The 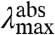 values in the mutants range from 441 to 478 nm, and the
values in the mutants range from 441 to 478 nm, and the 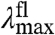 values from 500 to 511 nm. This analysis reveals that while the
values from 500 to 511 nm. This analysis reveals that while the 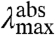 varies significantly for the 20 proteins (by 1,755 cm-1), their
varies significantly for the 20 proteins (by 1,755 cm-1), their 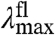 values are much less affected (421 cm-1) (Fig. 4A). The Stokes shift (difference between
values are much less affected (421 cm-1) (Fig. 4A). The Stokes shift (difference between 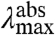 and
and 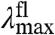 ) in the E46X mutants changes from 1,235 to 2,726 cm-1.
) in the E46X mutants changes from 1,235 to 2,726 cm-1.
Fig. 3.

Absorbance and fluorescence emission spectra of the 20 E46X mutants of PYP. (A) The amplitude-normalized absorbance and fluorescence emission spectra of the E46X mutants are shown. (B) Analysis of positions and bandwidths of the E46X mutants. Absorbance (closed circles) and fluorescence emission (open circles) maxima are indicated. The vertical bars depict the width of the spectra at 3/4 height. The horizontal axis represents the residue substituted at position 46. The residues were sorted from most red-shifted to most blue-shifted absorbance maximum.
To examine the effects of the substitutions on the shape of the absorbance and fluorescence emission bands, we extracted their widths. The pKa of the pCA chromophore is strongly increased in some of the mutants, such that even at pH 9 a shoulder is present near 350 nm (Fig. 3A) (21). The shoulder at 350 nm in some of the absorbance spectra can be fully attributed to the presence of a small amount of acid denatured PYP (21). Thus, we determined the width of all spectra at 3/4 height, avoiding contributions to the extracted width of the ∼450 nm absorbance bands by the peak at ∼350 nm. This yielded two observations.
First, the average width of the absorbance spectra of the E46X mutants (1,973 cm-1) is significantly larger than that of the fluorescence emission spectra (1,139 cm-1). We attribute the larger width of the PYP absorbance spectra to the increased width of the S1 energy surface compared to that of the S0 state (see SI Text). Second, the width of the absorbance spectra of the E46X mutants is affected much more (spanning 1,428 cm-1) than the width of the fluorescence emission spectra (spanning 342 cm-1) (Fig. 4B). The widths of the red flank and blue flank of the absorbance and fluorescence emission bands were determined independently, and the ratio of these values was used as a measure for asymmetry in the shape of the bands. The absorbance spectra were found to have a significant asymmetry (0.54 on average for the E46X mutants) towards the blue flank of the absorbance band (also see Fig. 3B), while the fluorescence emission spectra are nearly symmetrical (average of 1.03). The asymmetry of the absorbance bands in the E46X mutants varies significantly (by a factor 2.35), in contrast to the fairly constant asymmetry of the fluorescence emission spectra (variation by a factor 1.35) (Fig. 4C).
These results can be used to determine the dominant spectral tuning effect of residue 46 in PYP based on three considerations. (i) The correlation between 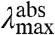 and
and 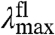 expected for the three distinct spectral tuning effects considered here are depicted in Fig. 5, together with the experimentally determined correlation. Since the
expected for the three distinct spectral tuning effects considered here are depicted in Fig. 5, together with the experimentally determined correlation. Since the 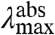 values of the mutants are significantly altered while their
values of the mutants are significantly altered while their 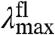 values are affected much less, the data for the E46X mutants closely match the W-tuning case. (ii) The widths of the absorbance spectra of the mutants are significantly affected, while those for the fluorescence emission spectra are not. (iii) The asymmetry in the shape of the absorbance spectra changes significantly in the mutants, in contrast to the similar shape of the fluorescence emission spectra. These three results agree with the predictions made for a change in W of the S1 state (case iii above), while they are in striking conflict with the predictions for a change in ΔE or ΔRe (case i and ii). Thus, we conclude that the main factor causing the changes in the spectra of the E46X proteins is a change in W of the S1 energy surface. In future work changes in the W of the S1 state in the E46X mutants could be experimentally verified based on resonance Raman intensity profile measurements. The experimental results strongly argue against a change in ΔE as the main spectral tuning effect of substitutions at position 46.
values are affected much less, the data for the E46X mutants closely match the W-tuning case. (ii) The widths of the absorbance spectra of the mutants are significantly affected, while those for the fluorescence emission spectra are not. (iii) The asymmetry in the shape of the absorbance spectra changes significantly in the mutants, in contrast to the similar shape of the fluorescence emission spectra. These three results agree with the predictions made for a change in W of the S1 state (case iii above), while they are in striking conflict with the predictions for a change in ΔE or ΔRe (case i and ii). Thus, we conclude that the main factor causing the changes in the spectra of the E46X proteins is a change in W of the S1 energy surface. In future work changes in the W of the S1 state in the E46X mutants could be experimentally verified based on resonance Raman intensity profile measurements. The experimental results strongly argue against a change in ΔE as the main spectral tuning effect of substitutions at position 46.
Correlating Fluorescence Quantum Yield and Excited State Lifetime.
The above assignment of the main effect of the E46X mutations on spectral tuning in PYP to a change in W of the S1 state is based on the assumption that the fluorescence emission spectra of the mutants provide reliable information on the electronic transitions in these proteins. However, in view of the very low Φfl of wt PYP (0.19%) (32), it is possible that in (some of) these mutants a small fraction of the protein is misfolded and becomes highly fluorescent. If this misfolding involves a slight perturbation of the active site structure, the absorbance spectrum of the mutant could be almost unchanged and difficult to detect experimentally. In that case the fluorescence emission spectrum would not be representative of the electronic transitions in the bulk of the protein molecules detected in the absorbance spectra, and the above reasoning would not hold.
To examine this possibility, we performed subpicosecond time-resolved pump-probe measurements to determine the lifetime of the S1 electronically excited state τexc in the E46X mutants. Previously, we reported that the fluorescence quantum yield Φfl in these mutants varies by a factor 7 (21). We selected wt PYP and the E46H, E46K, and E46Y mutants for this analysis because they span the entire range of Φfl values. The pump-probe measurements were performed with detection in the region 400–515 nm, since the electronically excited state has an absorbance band in this region (33, 34). The resulting time traces (Fig. 6A) were fitted as exponential decays. Data at longer time delays are shown in Fig. S2. In line with literature data (34), wt PYP exhibited biphasic kinetics with time constants of 0.6 and 2.7 ps. The PYP mutants followed monoexponential behavior. The extracted τexc of the proteins was plotted against their Φfl (Fig. 6B), revealing a linear correlation between τexc and Φfl.
Fig. 6.

Correlation of excited state lifetime and fluorescence quantum yield Φfl in wt PYP and the E46H, E46K, and E46Y mutants. (A) Determination of excited state lifetime by sub-ps transient absorbance pump-probe spectroscopy of wt PYP (black line) and its E46H (blue line), E46K (green line), and E46Y (red line) mutants. (B) Correlation between excited state lifetime and Φfl in wt PYP and the three mutants. The error bars depict the standard deviations.
The good correlation between τexc and Φfl provides strong evidence that the fluorescence spectra for the mutants are representative of the entire population of PYP molecules. In the case that a small percentage of the population in a mutant is misfolded and responsible for the increased Φfl, no correlation with τexc would be expected. Thus, the conclusion drawn above that the W of the S1 state is altered in the E46X mutants remains valid.
Dissecting Spectral Tuning Effects of Residue 46 on the S0 and S1 Free Energy Surfaces.
The data reported here allow a dissection of the three possible spectral tuning effects (changes in ΔE, ΔRe, and W) of substitutions at position 46 on the absorbance spectrum of PYP. The observed spectral properties of the mutants indicate that the substitutions do not cause significant changes in the difference between the Re values for the S0 and S1 states of the pCA in PYP (see SI Text). If all substitutions would only alter the W of the S1 energy surface, the fluorescence emission spectra would not be expected to change. However, the data reveal a small variation in the mutant 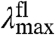 (over 421 cm-1). In addition,
(over 421 cm-1). In addition, 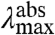 and
and 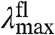 are linearly correlated with a slope of 0.16 (Fig. 5). In the case of pure ΔE-tuning a slope of 1 would be observed. Thus, we propose that of the 1,755 cm-1 in shifts for the absorbance spectra of the mutants, maximally ∼1/6th is caused by effects of the substitutions on the ΔE between the S0 and S1 states. We conclude that changes in W of S1 are the major factor in causing the shifts in
are linearly correlated with a slope of 0.16 (Fig. 5). In the case of pure ΔE-tuning a slope of 1 would be observed. Thus, we propose that of the 1,755 cm-1 in shifts for the absorbance spectra of the mutants, maximally ∼1/6th is caused by effects of the substitutions on the ΔE between the S0 and S1 states. We conclude that changes in W of S1 are the major factor in causing the shifts in 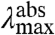 of the E46X mutants.
of the E46X mutants.
In the case of ΔE-tuning it is difficult to distinguish between mutations that destabilize the S0 state by a certain amount and mutations that stabilize the S1 state to the same extent; in both cases the absorbance and fluorescence emission spectra would be red-shifted by exactly the same amount. A similar degeneracy applies to shifts in the Re of the S0 and S1 states. However, in the case of W-tuning a change in W of S1 can be distinguished from a change in W of the S0 state. In the case that the W of the S0 state is significantly changed with respect to that of the S1 state, this would result in changes in the position and shape of the fluorescence emission spectra, while leaving the absorbance spectra largely unchanged. Conversely, a change in the W of the S1 state would only have significant effects on the absorbance spectra. Since the variation in the position and shape of the emission spectra is much smaller than that of the absorbance spectra (Fig. 4), we conclude that substitutions at position 46 mainly affect the W of the S1 energy surface, while leaving the W of the S0 energy surface largely unaffected.
Above, we derived the prediction that an increase in the W of S1 will result a positive correlation between the 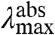 and width of the absorbance band. This correlation is indeed observed in the data (Fig. S1). Interestingly, a similar observation was reported for the visual rhodopsins in 1975 (35), indicating its more general relevance. For PYP we find that the same correlation holds for the fluorescence emission spectra, although over a much more narrow spectral range (Fig. S1). This may be explained by proposing that mutations have similar effects on the W of the S0 and S1 states, but that the W of the S1 state is altered more strongly.
and width of the absorbance band. This correlation is indeed observed in the data (Fig. S1). Interestingly, a similar observation was reported for the visual rhodopsins in 1975 (35), indicating its more general relevance. For PYP we find that the same correlation holds for the fluorescence emission spectra, although over a much more narrow spectral range (Fig. S1). This may be explained by proposing that mutations have similar effects on the W of the S0 and S1 states, but that the W of the S1 state is altered more strongly.
How can substitutions at position 46 alter the W of the S1 energy surface? We propose that changes in bond order of the S1 state resulting from charge redistribution caused by altered interactions between the pCA and residue 46 (see SI Text) are responsible for the changes in the W of the S1 state. More generally, altered electron delocalization is usually discussed in terms of a change in ΔE, but can also modify W.
A New Approach to Study Spectral Tuning.
The approach to investigating spectral tuning presented here is based on the effect of a mutation on both the absorbance and fluorescence emission spectrum of the protein. Since such data can be obtained for a wide range of chromophoric proteins, the approach should be broadly applicable.
By investigating multiple substitutions at a single position in the protein, the effect of this position on the S0 and S1 energy surfaces can be probed. In general, it is possible that different substitutions have different effects on the ΔE, Re, and W of these surfaces. This would be apparent in the absorbance and fluorescence data, leading to a complex view of the changes in the energy surfaces underlying the tuning effects of this position. Experimentally, this situation would be detected as strong deviations from a linear dependence of 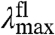 on
on 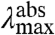 (see Fig. 5). For residue 46, one of the residues with the largest spectral tuning effects in PYP (see ref. 18), we found that a single factor—changes in the W of the S1 energy surface—dominates spectral tuning by this position.
(see Fig. 5). For residue 46, one of the residues with the largest spectral tuning effects in PYP (see ref. 18), we found that a single factor—changes in the W of the S1 energy surface—dominates spectral tuning by this position.
Published studies on spectral tuning have largely focused on (i) the residues responsible for spectral tuning in a specific protein, and (ii) the types of interactions (such as hydrogen bonding) causing spectral tuning. Experimental and computational work on PYP has confirmed that the chromophore absorbance band at 446 nm is a π-π∗ transition, has identified Glu46 and Tyr42 as the main contributors to the effect of the protein on the pCA absorbance spectrum, and has revealed that the hydrogen bonding interactions between these two side chains and the pCA are critical for spectral tuning in PYP (18–21, 24–27). These studies have all focused on the energy gap between the S0 and S1 states, i.e., ΔE-tuning.
The approach described here allows a third type of information to be obtained: What changes in the ΔE, Re, and W of the S0 and S1 energy surfaces result in spectral tuning? We have shown here that these three cases can be experimentally distinguished by examining the effect of mutations on both the absorbance and fluorescence emission spectra of the chromophore. A general assumption in the field is that changes in ΔE are responsible for biological spectral tuning. For residue 46 in H. halophila PYP we found that in fact changes in W for the S1 state are the dominant factor, revealing a unique mechanism for spectral tuning.
Materials and Methods
Mutagenesis, Expression, and Purification of PYP.
Mutagenesis of PYP was performed using Stratagene’s QuikChange Site-Directed Mutagenesis Kit, and mutants were overproduced using the pQE-80A plasmid (QIAGEN). PYP was purified in 96-well format as described in (21) using Ni-nitrilotriacetate agarose (QIAGEN) chromatography or batch purified as described in (16) with minor modifications (see SI Text). Pump-probe measurements and Φfl determinations were performed using batch purified PYP.
Absorbance and Fluorescence Spectroscopy.
All spectroscopic measurements were performed using PYP in 20 mM Tris HCl, 100 mM NaCl, 50 mM EDTA, pH 8.0. Absorbance spectra were measured in either a Synergy HT plate reader (Bio-Tek) or individually with a microcuvet in a Hewlett-Packard 8453 UV/Vis spectrophotometer. Fluorescence spectra were measured in a Fluoromax-3 fluorometer (Jobin-Yvon) using a 100 μL fluorescence microcuvet. Emission spectra were obtained using an excitation wavelength of 445 nm.
Ultrafast Pump-Probe Spectroscopy.
Two-color pump-probe measurements (see SI Text) were performed using the laser system described by Moran et al. (36). The pump beam was obtained by second-harmonic generation in beta barium borate (BBA) using pulses (2 kHz, 50 fs, 50 μJ pulses, 800 nm) from an amplified Ti:sapphire laser system. The tunable probe pulses (10–50 nJ, 80–90 fs pulses, 450–520 nm) were produced using an optical parametric amplifier. For all pump-probe experiments, the 0.2 mm path length sample was contained in a spinning cell rotated at 10 Hz, using focused pump (100 nJ) and probe (∼5 nJ) pulses.
Supplementary Material
ACKNOWLEDGMENTS.
The authors thank Iben B. Nielsen and Andrew M. Moran for help with the pump-probe experiments. W.D.H. gratefully acknowledges support from the National Institutes of Health Grant GM063805 and Oklahoma Center for the Advancement of Science and Technology (OCAST) Grant HR07-135S, and from startup funds provided by Oklahoma State University.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0903092107/DCSupplemental.
References
- 1.Scheer H. The pigments. In: Green BR, Parson WW, editors. Advances in Photosynthesis and Respiration. Vol 13. Dordrecht, The Netherlands: Kluwer; 2003. pp. 29–81. [Google Scholar]
- 2.Honig B, et al. External point-charge model for wavelength regulation in visual pigments. J Am Chem Soc. 1979;101:7084–7086. [Google Scholar]
- 3.Kochendoerfer GG, Lin SW, Sakmar TP, Mathies RA. How color visual pigments are tuned. Trends Biochem Sci. 1999;24:300–305. doi: 10.1016/s0968-0004(99)01432-2. [DOI] [PubMed] [Google Scholar]
- 4.Nakanishi K, Baloghnair V, Arnaboldi M, Tsujimoto K, Honig B. An external point-charge model for bacteriorhodopsin to account for its purple color. J Am Chem Soc. 1980;102:7945–7947. [Google Scholar]
- 5.Yan B, et al. Spectral tuning in bacteriorhodopsin in the absence of counterion and coplanarization effects. J Biol Chem. 1995;270:29668–29670. doi: 10.1074/jbc.270.50.29668. [DOI] [PubMed] [Google Scholar]
- 6.Ren L, et al. Molecular mechanism of spectral tuning in sensory rhodopsin II. Biochemistry. 2001;40:13906–13914. doi: 10.1021/bi0116487. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi S, et al. Structural determinants of spectral tuning in retinal proteins-bacteriorhodopsin vs sensory rhodopsin II. J Phys Chem B. 2001;105:10124–10131. [Google Scholar]
- 8.Meyer TE. Isolation and characterization of soluble cytochromes, ferredoxins and other chromophoric proteins from the halophilic phototrophic bacterium Ectothiorhodospira halophila. Biochim Biophys Acta. 1985;806:175–183. doi: 10.1016/0005-2728(85)90094-5. [DOI] [PubMed] [Google Scholar]
- 9.Meyer TE, Yakali E, Cusanovich MA, Tollin G. Properties of a water soluble, yellow protein isolated from a halophilic phototrophic bacterium that has photochemical activity analogous to sensory rhodopsin. Biochemistry. 1987;26:418–423. doi: 10.1021/bi00376a012. [DOI] [PubMed] [Google Scholar]
- 10.Hellingwerf KJ, Hendriks J, Gensch T. Photoactive yellow protein, a new type of photoreceptor protein: Will this “yellow lab” bring us where we want to go? J Phys Chem A. 2003;107:1082–1094. [Google Scholar]
- 11.Sprenger WW, Hoff WD, Armitage JP, Hellingwerf KJ. The Eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorbance spectrum of the photoactive yellow protein. J Bacteriol. 1993;175:3096–3104. doi: 10.1128/jb.175.10.3096-3104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoff WD, et al. Measurement and global analysis of the absorbance changes in the photocycle of the photoactive yellow protein from Ectothiorhodospira halophila. Biophys J. 1994;67:1691–1705. doi: 10.1016/S0006-3495(94)80643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kort R, et al. Evidence for the trans-cis isomerization of the p-coumaric acid chromophore as the photochemical basis of the photocycle of photoactive yellow protein. FEBS Lett. 1996;382:73–78. doi: 10.1016/0014-5793(96)00149-4. [DOI] [PubMed] [Google Scholar]
- 14.Hoff WD, et al. Thiol ester-linked p-coumaric acid as a new photoactive prosthetic group in a protein with rhodopsin-like photochemistry. Biochemistry. 1994;33:13959–13962. doi: 10.1021/bi00251a001. [DOI] [PubMed] [Google Scholar]
- 15.Baca M, et al. Complete chemical structure of photoactive yellow protein: Novel thioester-linked 4-hydroxycinnamyl chromophore and photocycle chemistry. Biochemistry. 1994;33:14369–14377. doi: 10.1021/bi00252a001. [DOI] [PubMed] [Google Scholar]
- 16.Xie A, Hoff WD, Kroon AR, Hellingwerf KJ. Glu46 donates a proton to the 4-hydroxycinnamate anion chromophore during the photocycle of photoactive yellow protein. Biochemistry. 1996;35:14671–14678. doi: 10.1021/bi9623035. [DOI] [PubMed] [Google Scholar]
- 17.Borgstahl GEO, Williams DR, Getzoff ED. 1.4 Å structure of photoactive yellow protein, a cytosolic photoreceptor: Unusual fold, active site and chromophore. Biochemistry. 1995;34:6278–6287. doi: 10.1021/bi00019a004. [DOI] [PubMed] [Google Scholar]
- 18.Kumauchi M, Hara M, Stalcup P, Xie A, Hoff WD. Identification of six new photoactive yellow proteins: Diversity and structure-function relationships in a bacterial blue light photoreceptor. Photochem Photobiol. 2008;84:956–969. doi: 10.1111/j.1751-1097.2008.00335.x. [DOI] [PubMed] [Google Scholar]
- 19.Genick UK, et al. Active site mutants implicate key residues for control of color and light cycle kinetics of photoactive yellow protein. Biochemistry. 1997;36:8–14. doi: 10.1021/bi9622884. [DOI] [PubMed] [Google Scholar]
- 20.Mihara K, Hisatomo O, Imamoto Y, Kataoka M, Tokunaga F. Functional expression and site-directed mutagenesis of photoactive yellow protein. J Biochem. 1997;121:876–880. doi: 10.1093/oxfordjournals.jbchem.a021668. [DOI] [PubMed] [Google Scholar]
- 21.Philip AF, Eisenman KT, Papadantonakis GA, Hoff WD. Functional tuning of photoactive yellow protein by active site residue 46. Biochemistry. 2008;47:13800–13810. doi: 10.1021/bi801730y. [DOI] [PubMed] [Google Scholar]
- 22.Kroon A, et al. Spectral tuning, fluorescence, and photoactivity in hybrids of photoactive yellow protein, reconstituted with native or modified chromophores. J Biol Chem. 1996;271:31949–31956. doi: 10.1074/jbc.271.50.31949. [DOI] [PubMed] [Google Scholar]
- 23.Hoff WD, Van Stokkum IHM, Gural J, Hellingwerf KJ. Comparison of acid denaturation and light activation in the eubacterial blue-light photoreceptor photoactive yellow protein. Biochim Biophys Acta. 1997;1322:151–162. [Google Scholar]
- 24.Yoda M, Houjou H, Inoue Y, Sakurai M. Spectral tuning of photoactive yellow protein. Theoretical and experimental analysis of medium effects on the absorption spectrum of the chromophore. J Phys Chem B. 2001;105:9887–9895. [Google Scholar]
- 25.Yamato T, Ishikura T, Kakitani T, Kawaguchi K, Watanabe H. Spectral tuning of photoactive yellow protein. Photochem Photobiol. 2007;83:323–327. doi: 10.1562/2006-06-16-RA-930. [DOI] [PubMed] [Google Scholar]
- 26.Gromov EV, Burghardt I, Köppel H, Cederbaum LS. Electronic structure of the PYP chromophore in its native protein environment. J Am Chem Soc. 2007;129:6798–6806. doi: 10.1021/ja069185l. [DOI] [PubMed] [Google Scholar]
- 27.Molina V, Merchan M. On the absorbance changes in the photocycle of the photoactive yellow protein: A quantum-chemical analysis. Proc Natl Acad Sci USA. 2001;98:4299–4304. doi: 10.1073/pnas.071058098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer TE, et al. Site-specific mutations provide new insights into the origin of pH effects and alternative spectral forms in the photoactive yellow protein from Halorhodospira halophila. Biochemistry. 2003;42:3319–3325. doi: 10.1021/bi020702w. [DOI] [PubMed] [Google Scholar]
- 29.Anderson S, Crossoon S, Moffat K. Short hydrogen bonds in photoactive yellow protein. Acta Crystallogr D. 2004;60:1008–1016. doi: 10.1107/S090744490400616X. [DOI] [PubMed] [Google Scholar]
- 30.Sugishima M, et al. Structure of photoactive yellow protein (PYP) E46Q mutant at 1.2 angstrom resolution suggests how Glu46 controls the spectroscopic and kinetic characteristics of PYP. Acta Crystallogr D. 2004;60:2305–2309. doi: 10.1107/S0907444904024084. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura R, Hamada N, Ichida H, Tokunaga F, Kanematsu Y. Coherent oscillations in ultrafast fluorescence of photoactive yellow protein. J Chem Phys. 2007;127:215102. doi: 10.1063/1.2802297. [DOI] [PubMed] [Google Scholar]
- 32.Meyer TE, Tollin G, Causgrove TP, Cheng P, Blankenship RE. Picosecond decay kinetics and quantum yield of fluorescence of the photoactive yellow protein from the halophilic purple phototrophic bacterium, Ectothiorhodospira halophila. Biophys J. 1991;59:988–991. doi: 10.1016/S0006-3495(91)82313-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horn MA, et al. Femtosecond studies of the initial events in the photocycle of photoactive yellow protein (PYP) In: de Schryver FC, de Feyter S, Schweitzer G, editors. Femtochemistry. Weinheim, NY: Wiley-VCH; 2001. pp. 381–390. [Google Scholar]
- 34.Gensch T, et al. The primary photoreaction of photoactive yellow protein (PYP): Anisotropy changes and excitation wavelength dependence. Chem Phys Lett. 2002;356:347–354. [Google Scholar]
- 35.Greenberg AD, Honig B, Ebrey TG. Wavelength dependence of bandwidths of visual pigment spectra. Nature. 1975;257:823–824. doi: 10.1038/257823a0. [DOI] [PubMed] [Google Scholar]
- 36.Moran AM, et al. Optical coherence and theoretical study of the excitation dynamics of a highly symmetric cyclophane-linked oligophenylenevinylene dimer. J Chem Phys. 2006;124:194904. doi: 10.1063/1.2196041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.