

Abstract
The dramatic increase in healthcare cost has become a significant burden to the world. Many patients are denied the accessibility of medication because of the high price of drugs. Total biosynthesis of chiral drug intermediates is an environmentally friendly approach that helps provide more affordable pharmaceuticals. Here we have expanded the natural metabolic capability to biosynthesize a nonnatural amino acid L-homoalanine, which is a chiral precursor of levetiracetam, brivaracetam, and ethambutol. We developed a selection strategy and altered the substrate specificity of ammonium-assimilating enzyme glutamate dehydrogenase. The specificity constant kcat/Km of the best mutant towards 2-ketobutyrate is 50-fold higher than that towards the natural substrate 2-ketoglutarate. Compared to transaminase IlvE and NADH-dependent valine dehydrogenases, the evolved glutamate dehydrogenase increased the conversion yield of 2-ketobutyrate to L-homoalanine by over 300% in aerobic condition. As a result of overexpressing the mutant glutamate dehydrogenase and Bacillus subtilis threonine dehydratase in a modified threonine-hyperproducing Escherichia coli strain (ATCC98082, ΔrhtA), 5.4 g/L L-homoalanine was produced from 30 g/L glucose (0.18 g/g glucose yield, 26% of the theoretical maximum). This work opens the possibility of total biosynthesis of other nonnatural chiral compounds that could be useful pharmaceutical intermediates.
Keywords: chiral chemicals, directed evolution, metabolic engineering, synthetic biology
L-homoalanine is a nonnatural amino acid that can be a key chiral intermediate for the synthesis of several important drugs (Fig. 1A). It can be converted to S-2-aminobutyramide, which is the immediate precursor of the antiepileptic drugs levetiracetam (trade name Keppra) and brivaracetam. It can also be converted to S-2-aminobutanol for synthesizing the antituberculosis compound ethambutol. The optical purity of these drugs is critical for therapeutic safety and efficacy. The R-enantiomer of levetiracetam has no antiepileptic activity (1) and (R, R)-form of ethambutol can cause blindness (2). Even though ethambutol and levetiracetam are now generic drugs, in many countries the cost of just one month’s supply exceeds the entire annual per capita health expenditure (3). The prohibitive drug price has created global healthcare problems. For example, while epilepsy occurs to 50 million people worldwide, most of the patients cannot afford the levetiracetam treatment, and use cheaper but much less effective alternatives such as phenobarbital (4). This severe healthcare problem could be relieved by developing a cost-effective approach to L-homoalanine and thus reducing the manufacturing cost of levetiracetam.
Fig. 1.

Biosynthesis of L-homoalanine and its pharmaceutical applications. (A) Chemical synthetic routes of antiepileptic and antituberculosis drugs from the chiral intermediate L-homoalanine. (B) Constructing a nonnatural metabolic pathway for L-homoalanine fermentation. Engineered E. coli can overproduce the natural amino acid threonine from glucose. Threonine is converted to 2-ketobutyrate by threonine dehydratase. Then L-homoalanine is synthesized from 2-ketobutyrate by amination.
The conventional methods for commercial-scale preparation of nonnatural amino acids are complex and environmentally unfriendly. In one approach, chemically synthesized 2-ketoacids are asymmetrically converted to optically pure nonnatural amino acids by transaminases or dehydrognenases (5 and 6). Another approach is to use enzymes such as acylases or amidases to resolve racemic mixtures of nonnatural amino acids (5). In comparison, most of the natural L-amino acids can now be produced from glucose by microbial fermentation (7). Notably, L-glutamate, L-lysine, and L-threonine are produced more than 2 million tons annually (5). Unlike natural amino acids, total biosynthesis of nonnatural amino acids from simple sugars encounters significant technical challenges: First, artificial metabolic pathways have to be designed to expand the metabolic capability of cells (8); second, optimizing the designed unique metabolic pathways requires extensive protein evolution (9); third, metabolic flux should be driven to the production of the target compounds (10). We have successfully addressed these problems and achieved high-level production of the pharmaceutically relevant nonnatural amino acid L-homoalanine directly from glucose.
Results and Discussion
Expanding Metabolic Network for Biosynthesis of L-homoalanine.
To expand metabolism and include L-homoalanine in the metabolite family, we have introduced a unique metabolic pathway into Escherichia coli (Fig. 1B). In the native metabolic network, glucose is converted to threonine after “glycolysis” and “aspartate biosynthesis” (11). Threonine can then be converted to 2-ketobutyrate by threonine dehydratase TdcB or IlvA as a precursor for isoleucine biosynthesis. Here we divert 2-ketobutyrate to biosynthesize the nonnatural amino acid L-homoalanine.
There are two major challenges in this expanded biosynthetic pathway: the first challenge we face is to find the right amination enzyme to convert 2-ketobutyrate into L-homoalanine and existing enzymes may not work since L-homoalanine cannot be detected in normal cells (12) even though 2-ketobutyrate is a natural metabolite; the second is evolving metabolism to drive the carbon flux towards 2-ketobutyrate. In most organisms, 2-ketobutyrate is synthesized via threonine. Alternative routes include “the pyruvate pathway” starting with the condensation of acetyl-CoA and pyruvate to form citramalate (13), “the glutamate pathway” via ß-methylaspartate and ß-methyloxaloacetate (14), or “γ elimination” of activated substrates such as O-phospho-homoserine and O-acetyl-homoserine (15). We choose “the threonine pathway” because there are existing technologies in fermentation industry to develop bacteria strains that could produce more than 100 g/L threonine (11).
Limitation of Natural Enzymes on Amination of 2-ketobutyrate.
In order to check if any endogenous transaminase of E. coli can work on 2-ketobutyrate, we fed 10 g/L 2-ketobutyrate to wild-type E. coli strain BW25113 growing in M9 medium. After 24 h, HPLC analysis showed that only 182 mg/L L-homoalanine was produced (Fig. 2A). Since the branched-chain amino acid aminotransferase IlvE (16) is likely to be a functional candidate, we cloned and overexpressed IlvE in BW25113, which increased the production of L-homoalanine to 1.2 g/L (Fig. 2B). Additional feeding of 10 g/L amino donor glutamate further increased the L-homoalanine titer to 3.2 g/L. These experiments demonstrated that transaminases such as IlvE could aminate 2-ketobutyrate into L-homoalanine. However, high concentration of glutamate is needed to drive the reaction equilibrium because transamination is a reversible reaction process. Even under such extreme conditions, the conversion rate of transaminating 2-ketobutyrate to L-homoalanine is only 32%.
Fig. 2.

Comparison of different amination enzymes on the production of L-homoalanine. E. coli cultures were inoculated in M9 medium with addition of 10 g/L 2-ketobutyrate and incubated at 37 °C for 24 h. (A) Wild-type E. coli BW25113. Overexpression of (B) transaminase IlvE (C) valine dehydrogenase from Streptomyces avermitilis (D) valine dehydrogenase from Streptomyces coelicolor (E) valine dehydrogenase from Streptomyces fradiae (F) evolved glutamate dehydrogenase GDH1 (G) evolved glutamate dehydrogenase GDH2. Error bars: standard deviations from three independent experiments.
Direct reductive amination of ketoacids with ammonia is a preferable choice to produce amino acid because it avoids the usage of glutamate as the amino donor (17). Compared to transamination, reductive amination could potentially simplify the metabolic manipulation and reduce the production cost. It has been shown previously that valine dehydrogenases from various Streptomyces species are active on reductive amination of 2-ketobutyrate in vitro (18, 19). We thus cloned and overexpressed in BW25113 the valine dehydrogenases from Streptomyces avermitilis, Streptomyces coelicolor, and Streptomyces fradiae. Unfortunately, these dehydrogenases only slightly increased the L-homoalanine titer (around 240 ∼ 260 mg/L, Fig. 2C–E) as compared to the BW25113 background (182 mg/L). The possible reason is that valine dehydrogenase is a catabolic enzyme that favors the reaction direction towards degradation of L-homoalanine instead of biosynthesis in aerobic condition.
Evolving Glutamate Dehydrogenase to Aminate 2-ketobutyrate.
Glutamate is formed by reductive amination of 2-ketoglutarate with ammonia. The reaction is catalyzed by glutamate dehydrogenase (GDH) or glutamate synthase in the presence of cofactor NADPH (Fig. 3A). This ammonia utilization process is very efficient since glutamate is the universal nitrogen source of other amino acids. The catalytic activity of GDH towards biosynthesis of glutamate is more than 10 times higher than that towards degradation of glutamate (20), which makes GDH an ideal biosynthetic enzyme for amino acid production. However, since native GDH is active only on 2-ketoglutarate, we need to engineer GDH to obtain new substrate specificity towards 2-ketobutyrate for L-homoalanine biosynthesis.
Fig. 3.

Selection strategy to evolve glutamate dehydrogenase (GDH) for amination of 2-ketobutyrate. (A) Wild-type GDH assimilates ammonia directly into glutamate. (B) Knocking out transaminase genes avtA and ilvE from the chromosome makes wild-type E. coli valine auxotrophic, which can be complemented by a mutant GDH active on aminating 2-ketoisovalerate. (C) 2-ketobutyrate is chemically similar to the valine precursor, 2-ketoisovalerate. A mutant GDH active on 2-ketoisovalerate is likely to be active on 2-ketobutyrate.
To this end, we have developed a selection strategy to evolve GDH. Deletion of transaminase genes avtA and ilvE from the chromosome makes wild-type E. coli valine auxotrophic (21), whose growth in minimal medium can be rescued by a mutant GDH active on aminating the valine precursor 2-ketoisovalerate (Fig. 3B). Since the chemical structures of 2-ketobutyrate and 2-ketoisovalerate are similar, we reasoned that GDH variants active on 2-ketoisovalerate could be active on 2-ketobutyrate (Fig. 3C).
Based on the crystal structure of Clostridium symbiosum glutamate dehydrogenase (PDB: 1BGV) (22), residues K89, T193, V377, and S380 are within a radius of 6 Å of the γ-carbon of glutamate substrate (Fig. 4A). Sequence alignment of GDH between C. symbiosum and E. coli shows that the binding pocket is conserved, and the corresponding residues of E. coli GDH are K92, T195, V377, and S380 (Fig. 4B). These sites were subjected to site-saturation mutagenesis by PCR gene assembly using primers containing degenerate NNK (N = A, T, G, C; K = G, T) codons. The assembled fragments were ligated into plasmid pZE12 (23) and the resulting library was transformed into the valine auxotrophic E. coli (ΔavtA, ΔilvE). The library contained 2 million independent clones and the selection was performed in M9 minimal medium. Two mutants were isolated: GDH1 has K92L, T195A, V377A, and S380C mutations; GDH2 has K92V and T195S mutations.
Fig. 4.

Construction of GDH library for evolution. (A) Binding pocket of Clostridium symbiosum glutamate dehydrogenase (PDB: 1BGV) complexed with its natural substrate glutamate. Residues K89, T193, V377, and S380 are within a radius of 6 Å of the γ-carbon of glutamate. (B) Sequence alignment of C. symbiosum and E. coli GDH. The binding pocket is conserved, and the corresponding residues of E. coli GDH are K92, T195, V377, and S380. These residues were subjected to site-saturation mutagenesis with randomized NNK codon. A library size of 2 million members was transformed into valine auxotrophic E. coli and selected for mutants growing in M9 minimal medium. (C) Growth curve of E. coli (ΔavtA, ΔilvE) transformants in minimal medium. −Val means absence of valine. +Val means presence of valine. Or cells are transformed with GDH1 (K92L, T195A, V377A, and S380C mutations) or GDH2 (K92V and T195S mutations) mutant. Cells did not grow up in absence of valine in the minimal medium, while GDH mutants could rescue cell growth without valine addition.
Characterization of the Evolved Glutamate Dehydrogenase Mutants.
As can be seen in Fig. 4C, while E. coli (ΔavtA, ΔilvE) without plasmid could not grow in the absence of valine in minimal medium, GDH mutants could support cell growth without valine addition. In particular, the growth speed of GDH2 transformant in minimal medium was very close to that in valine-supplemented medium, which means that GDH2 successfully rescued the valine auxotrophic phenotype. The evolved GDH mutants were transformed and overexpressed in BW25113. Feeding of 10 g/L 2-ketobutyrate into such transformants produced 1.6 g/L (GDH1) and 5.1 g/L (GDH2) L-homoalanine (Fig. 2F, G). Interestingly, the production yield is in agreement with the growth speed of these two GDH mutants in minimal medium. These results suggest that our selection strategy works well and the selected GDH2 is more active on both 2-ketoisovalerate and 2-ketobutyrate than GDH1. GDH2 transformant has a conversion yield of 50% and a productivity of 5.1 g/L/day, much better than the IlvE transformant plus high concentration of glutamate.
To characterize the enzymes, both the wild-type glutamate dehydrogenase GDHwt and mutant GDH2 were added an N-terminal 6xHis-tag, overexpressed, and purified through Ni-NTA columns. The kinetic parameters for activation of 2-ketoglutarate (cognate substrate) and 2-ketoisovalerate or 2-ketobutyrate (nonnatural substrates) were determined by monitoring the consumption of NADPH at 340 nm (Table 1). The enzymatic assay indicates that towards 2-ketoglutarate the specificity constant kcat/Km of GDH2 is nearly 3,000-fold smaller than that of GDHwt. While the two enzymes have a similar Km towards 2-ketoisovalerate, GDH2 has a five times higher kcat than GDHwt and can produce valine for cell growth in physiological condition. Compared to GDHwt, GDH2 has a significant enhanced catalytic activity on 2-ketobutyrate with a 4-fold decrease in Km (8.4 mM vs. 35.4 mM) and a 2-fold increase in kcat (90.2 s-1 vs. 47.2 s-1). The increase in binding affinity is very helpful since high concentration of 2-ketobutyrate interferes with cellular metabolic activities (24). On the basis of the crystallographic model for the active site of GDH, amino group of K92 hydrogen-bonds to the γ-carboxyl group of 2-ketoglutarate and determines the substrate specificity (22). Mutation of lysine to valine increases the hydrophobic character of the binding pocket, which explains why the specificity constant kcat/Km of GDH2 towards 2-ketobutyrate is 50-fold higher than that towards the natural substrate 2-ketoglutarate.
Table 1.
Kinetic parameters of wild type and mutant glutamate dehydrogenases
| GDHwt | GDH2 | ||||||
| Substrate | Structure | Km (mM) | kcat (s-1) | kcat/Km (mM-1 s-1) | Km (mM) | kcat (s-1) | kcat/Km (mM-1 s-1) |
| 2-ketoglutarate | 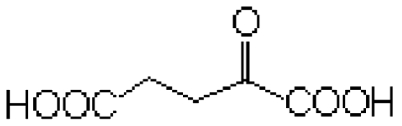 |
0.30 ± 0.02 | 187.2 ± 8.4 | 624 | 69.2 ± 4.9 | 14.0 ± 0.6 | 0.2 |
| 2-Ketoisovalerate | 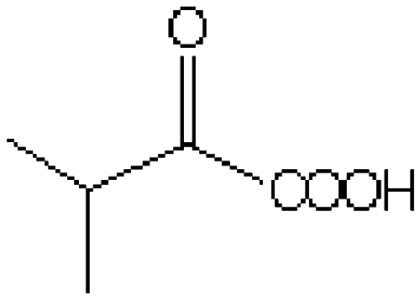 |
152.7 ± 9.5 | 10.4 ± 0.7 | 0.07 | 145.6 ± 6.5 | 47.1 ± 4.5 | 0.3 |
| 2-ketobutyrate | 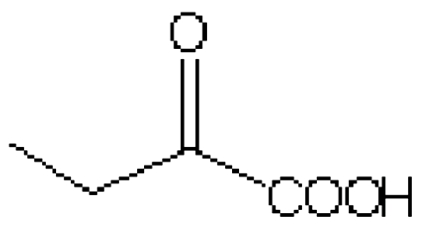 |
35.4 ± 7.4 | 47.2 ± 5.0 | 1.3 | 8.4 ± 1.7 | 90.2 ± 5.4 | 10.7 |
Optimizing the Full Biosynthetic Pathway of L-homoalanine.
Through directed evolution, we obtained a mutant glutamate dehydrogenase GDH2 that is highly active on amination of 2-ketobutyrate. When we overexpressed GDH2 in BW25113, 0.1 g/L L-homoalanine was produced from 30 g/L glucose (column #2 in Fig. 5A). Without overexpression of GDH2, no homoalanine was detected (column #1 in Fig. 5A). Now the remaining challenge is to engineer metabolism to divert the carbon flux towards 2-ketobutyrate.
Fig. 5.

Production of L-homoalanine with different combinations of E. coli strains and threonine dehydratases. (A) (1) BW25113 (2) BW25113 with overexpression of GDH2, (3) ATCC98082 with overexpression of TdcB & GDH2, (4) ATCC98082 with overexpression of IlvAEC & GDH2, (5) ATCC98082 with overexpression of IlvABS & GDH2, (6) ATCC98082 (ΔrhtA) with overexpression of IlvABS and GDH2. (B) The yield of L-homolalanine biosynthesis from glucose. Error bars: standard deviations from three independent experiments.
Since 2-ketobutyrate is derived from threonine, we switched the production host from wild-type E. coli BW25113 to a threonine overproducer ATCC98082 (11). E. coli strain ATCC98082 can produce 8 g/L threonine from 30 g/L glucose. Overexpression of GDH2 and threonine dehydratase TdcB in ATCC98082 resulted in production of 0.18 g/L L-homoalanine (column #3 in Fig. 5A) and 3.7 g/L threonine. The high concentration of remaining threonine means that the catabolic enzyme TdcB is not active enough to fully convert threonine into 2-ketobutyrate. Another consequence is the accumulation of 4 g/L glutamate because low concentration of 2-ketobutyrate cannot compete with 2-ketoglutarate for amination by GDH2. We then cloned the biosynthetic threonine dehydratases IlvA from E. coli (IlvAEC) and Bacillus subtilis (25) (IlvABS). In combination with GDH2, these dehydratases significantly increased the production of L-homoalanine (column #4 and 5 in Fig. 5A). IlvABS was the best enzyme identified and its transformant produced 3.8 g/L L-homoalanine. However, there was still 2.5 g/L threonine remained in the fermentation medium. It is known that ATCC98082 has an rhtA23 mutation which enhances about 10-fold the expression of the rhtA gene (11), whose protein product is a strong threonine exporter. We reasoned that the active efflux of threonine might account for the extracellular accumulation of threonine. After deletion of rhtA from the ATCC98082 chromosome, threonine no longer accumulated and the concentration of L-homoalanine increased to 5.4 g/L (column #6 in Fig. 5A). This final strain has a productivity of 2.7 g/L/day and a yield of 0.18 g/g glucose (Fig. 5B), which is 26% of the theoretical maximum (the calculation detail is described in SI Text). Interestingly, even though GDH used significant amounts of cellular reducing power, improving NADPH availability by phosphoglucose isomerase (pgi) knockout (26) decreased the L-homoalanine production titer to 3.0 g/L, which indicated that threonine biosynthesis was affected by the intraceulluar redox status.
Conclusions
This work expanded the E. coli metabolism to biosynthesize a nonnatural amino acid L-homoalanine directly from glucose. The success here demonstrates that metabolic manipulation not only allows the production of natural metabolites, but also enables the microbial synthesis of nonnatural metabolites. While traditional metabolic engineering deals with flux engineering, to achieve industry-level biosynthesis of unique chemicals, three steps (pathway expansion, protein evolution, and flux enhancing) should be taken. Protein evolution is a key step since unique enzymes need to be developed to perform nonnatural activities. Here we have evolved the glutamate dehydrogenase to fix ammonia directly onto 2-ketobutyrate, which avoids the usage of glutamate as nitrogen donor and significantly improves the yield of L-homoalanine.
Developing a fermentation process for L-homoalanine (5.4 g/L titer and 0.18 g/g glucose yield in shake flasks) provides a renewable supply of this chiral chemical and enables the synthesis of levetiracetam without expensive chiral chromatography. The greener manufacturing process of levetiracetam could potentially reduce the drug cost (27, 28), which may help 50 million epilepsy patients worldwide considering 90% of these people are in developing countries and do not receive the appropriate treatment (4).
Materials and Methods
Vector Construction.
All cloning procedures were carried out in the E. coli strain XL10-gold (Stratagene). PCR reactions were performed with KOD polymerase (Novagen). Oligos were synthesized by Operon Biotechnologies (see SI Text for sequence details). A gene fragment encoding lac repressor LacI (8) was inserted into the SacI site of plasmid pZE12 (23) to yield plasmid pZElac. The ilvE gene was amplified from the genomic DNA of E. coli K12 using the primers IlvEaccfwd and IlvExbarev. The valine dehyrogenase genes were amplified from the genomic DNA of Streptomyces avermitilis, Streptomyces coelicolor, and Streptomyces fradiae using the primer pairs VDHsaaccfwd/VDHsaxbarev, VDHscaccfwd/VDHscxbarev, and VDHsfaccfwd/VDHsfxbarev. The glutamate dehydrogenase gene gdhA was amplified from the genomic DNA of E. coli K12 using the primers GDHecaccfwd and GDHecxbarev. All the PCR products were digested with Acc65I and XbaI and ligated into pZElac to yield plasmids pZElac_IlvE, pZElac_VDHsa, pZElac_VDHsc, pZElac_VDHsf and pZElac_GDH. The E. coli threonine dehydratase genes tdcB and ilvA were amplified from the genomic DNA of E. coli K12 using the primer pairs TdcBaccfwd/TdcBsalrev and IlvAecaccfwd/IlvAecsalrev. The Bacillus threonine dehydratase gene ilvA were amplified from the genomic DNA of Bacillus subtilis using the primer pair IlvAbsaccfwd/IlvAbssalrev. These fragments were digested with Acc65I and SalI. Then they were ligated with mutant GDH gene fragment digested with SalI and XbaI (amplified with primer pair GDHecsalfwd/GDHecxabrev). The ligated fragments were inserted into pZElac to create plasmids pZElac_tdcB_GDH, pZElac_ilvAEC_GDH and pZElac_ilvABS_GDH.
Knocking out Chromosomal Genes.
Gene deletion was performed using P1 transduction and the strains used for the P1 transduction were obtained from the Keio collection (29). Colonies containing the correct deletions were transformed with plasmid pCP20 to remove the kanamycin resistance marker. Valine transaminase genes avtA and ilvE were deleted from the E. coli strain BW25113 chromosome to make a valine auxotroph designated as ValK. The threonine exporter gene rhtA was inactivated from the chromosome of threonine-hyperproduction E. coli strain ATCC98082 to improve the production of L-homolalanine.
Construction and Selection of GDH Library.
Oligonucleotides encoding degenerate NNK (N is A,T,G,C; K is G,T) codons at the sites corresponding to Lys-92, Thr-195, Val377, and Ser380 in E. coli GdhA were used for library construction. Four separate PCRs were performed by using pZElac_GDH as the template and the following pairs of primers: GDHecaccfwd and GDH_k92lib_rev, GDH_k92lib and GDH_T195lib_rev, GDH_T195lib and GDH_VSlib_rev, GDH_VSlib and GDHecxabrev. The DNA fragments obtained from these PCRs were electrophoresed and purified by using Zymo-spin columns (Zymo Research). Equimolar quantities of the fragments were mixed and subjected to 10 rounds of PCR. The primers GDHecaccfwd and GDHecxabrev were subsequently added, and the reaction mixture was subjected to 25 more rounds of PCR. The resulting 1.4-kb PCR product was digested Acc65I and XbaI and ligated into PZElac digested with the same enzymes. The ligation mixture was transformed into electrocompetent ElectroMAX DH10B cells (Invitrogen), yielding 2 million independent transformants. The plasmid DNA from the pooled transformants was isolated and used to transform into valine auxotroph ValK through electroporation, yielding 10 million independent clones.
Pooled transformants (500 μL, ∼109 cells) were incubated in 30 mL of M9 medium containing 20 g/L glucose and 50 mg/L ampicillin with shaking at 37 °C for 2 d. 50 μL of culture was subcultured into two new culture tubes (100x dilution). After another five rounds of successive subculturing the enrichment (to ensure the best mutant dominated in the culture), two mutants were isolated: GDH1 has K92L, T195A, V377A and S380C mutations; and GDH2 has K92V and T195S mutations.
Protein Purification and Enzymatic Assay.
Both gene fragments encoding wild-type glutamate dehydrogenase and GDH2 were amplified using primers GDHbamfwd and GDHbamrev. After digestion with BamHI, the gene fragments were inserted into expression plasmid pQE9 (Qiagen). The resulting expression plasmids were transformed into E. coli strain BL21(DE3) harboring pREP4 (Qiagen). Cells were inoculated from an overnight preculture at 1/100 dilution and grown in 200 mL 2XYT rich medium containing 50 mg/L ampicillin and 25 mg/L kanamycin. At an OD600 of 0.6, recombinant proteins were expressed by induction of the cell cultures with 0.1 mM IPTG, followed by incubation at 30 °C overnight. Overexpressed proteins were then purified with with Ni-nitrilotriacetic acid columns. Protein concentration was determined by measuring UV absorbance at 280 nm.
Enzymatic assay was performed in assay buffer (100 mM Tris buffer, pH 8.0) containing 0.2M NH4Cl, 0.2 mM NADPH, and various concentrations of 2-ketoacids. The reactions were started by adding the purified enzymes, and the consumption of NADPH was monitored at 340 nm (extinction coefficient, 6.22 mM-1 cm-1). Kinetic parameters (kcat and Km) were determined by fitting initial velocity data to the Michaelis-Menten equation using Origin.
Production of L-homoalanine.
To test the conversion of 2-ketobutyrate to L-homoalanine, plasmids pZElac_IlvE, pZElac_VDHsa, pZElac_VDHsc, pZElac_VDHsf, pZElac_GDH and pZElac_GDH2 were transformed into BW25113. The transformants were inoculated in M9 medium with 5 g/L yeast extract, 10 g/L ammonium hydrochloride and 20 g/L glucose. Once OD reached ∼1.0, 10 g/L 2-ketobutyrate plus 0.1 mM IPTG were added and incubated at 37 °C for 24 h. Amino acids were quantified as o-phthaldialdehyde (OPA) derivatives by HPLC analysis.
To test the production of L-homoalanine from glucose, E. coli strain ATCC98082 harboring plasmid pZS_thrO (8) was transformed with pZElac_tdcB_GDH, pZElac_ilvAEC_GDH and pZElac_ilvABS_GDH. These transformants were subjected to fermentation using the following production medium: 30 g glucose, 17 g (NH4)2SO4, 2 g KH2PO4, 1 g MgSO4·7H2O, 2 g yeast extract, 0.1 g L-valine, 0.01 g FeSO4.7H2O and 0.01 g MnSO4.7H2O per liter. Antibiotics were added appropriately (ampicillin 50 mg/L, spectinomycin 25 mg/L). Overnight 2XYT culture were diluted 25x into fermentation medium and 0.1 mM isopropyl-b-D-thiogalactoside (IPTG) was added to induce protein expression. In shake flask experiments, the culture medium was buffered by addition of 30 g/L CaCO3. Cultures were incubated in 33 °C shaker (250 rpm) for 40–50 h until glucose was consumed.
Supplementary Material
Acknowledgments.
This work was supported in part by the University of California, Los Angeles and Department of Energy Institute for Genomics and Proteomics.
Footnotes
The authors declare a conflict of interest. UCLA will apply for a patent based on the technology and license the technology for commercialization.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0912903107/DCSupplemental.
References
- 1.Shorvon SD, Van Rijckevorsel K. A new antiepileptic drug. Journal of Neurology, Neurosurgery, and Psychiatry. 2002;72(4):426–429. doi: 10.1136/jnnp.72.4.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breuer M, et al. Industrial methods for the production of optically active intermediates. Angewandte Chemie International Edition. 2004;43(7):788–824. doi: 10.1002/anie.200300599. [DOI] [PubMed] [Google Scholar]
- 3.Moore-Gillon J. Multidrug-resistant tuberculosis: This is the cost. Ann N Y Acad Sci. 2001;953:233–240. doi: 10.1111/j.1749-6632.2001.tb11382.x. [DOI] [PubMed] [Google Scholar]
- 4.Scott RA, Lhatoo SD, Sander J. The treatment of epilepsy in developing countries: Where do we go from here? B World Health Organ. 2001;79:344–351. [PMC free article] [PubMed] [Google Scholar]
- 5.Leuchtenberger W, Huthmacher K, Drauz K. Biotechnological production of amino acids and derivatives: Current status and prospects. Appl Microbiol Biotechnol. 2005;69(1):1–8. doi: 10.1007/s00253-005-0155-y. [DOI] [PubMed] [Google Scholar]
- 6.Taylor PP, Pantaleone DP, Senkpeil RF, Fotheringham IG. Novel biosynthetic approaches to the production of unnatural amino acids using transaminases. Trends Biotechnol. 1998;16(10):412–418. doi: 10.1016/s0167-7799(98)01240-2. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda M. Amino acid production processes. Adv Biochem Eng Biot. 2002;79:1–35. doi: 10.1007/3-540-45989-8_1. [DOI] [PubMed] [Google Scholar]
- 8.Zhang K, Sawaya MR, Eisenberg DS, Liao JC. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc Natl Acad Sci USA. 2008;105(52):20653–20658. doi: 10.1073/pnas.0807157106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold FH. Combinatorial and computational challenges for biocatalyst design. Nature. 2001;409(6817):253–257. doi: 10.1038/35051731. [DOI] [PubMed] [Google Scholar]
- 10.Causey TB, Zhou S, Shanmugam KT, Ingram LO. Engineering the metabolism of Escherichia coli W3110 for the conversion of sugar to redox-neutral and oxidized products: Homoacetate production. Proc Natl Acad Sci USA. 2003;100(3):825–832. doi: 10.1073/pnas.0337684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debabov VG. The threonine story. Adv Biochem Eng Biot. 2002;79:113–136. doi: 10.1007/3-540-45989-8_4. [DOI] [PubMed] [Google Scholar]
- 12.Epelbaum S, et al. Branched-chain amino acid biosynthesis in Salmonella typhimurium: A quantitative analysis. J Bacteriol. 1998;180(16):4056–4067. doi: 10.1128/jb.180.16.4056-4067.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charon NW, Johnson RC, Peterson D. Amino acid biosynthesis in the spirochete Leptospira: Evidence for a novel pathway of isoleucine biosynthesis. J Bacteriol. 1974;117(1):203–211. doi: 10.1128/jb.117.1.203-211.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phillips AT, Nuss JI, Moosic J, Foshay C. Alternate pathway for isoleucine biosynthesis in Escherichia coli. J Bacteriol. 1972;109(2):714–719. doi: 10.1128/jb.109.2.714-719.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donini S, et al. A threonine synthase homolog from a mammalian genome. Biochem Biophys Res Commun. 2006;350(4):922–928. doi: 10.1016/j.bbrc.2006.09.112. [DOI] [PubMed] [Google Scholar]
- 16.Inoue K, et al. Branched-chain amino acid aminotransferase of Escherichia coli: Overproduction and properties. J Biochem. 1988;104(5):777–784. doi: 10.1093/oxfordjournals.jbchem.a122549. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Jantama K, Moore JC, Shanmugam KT, Ingram LO. Production of L-alanine by metabolically engineered Escherichia coli. Appl Microbiol Biotechnol. 2007;77(2):355–366. doi: 10.1007/s00253-007-1170-y. [DOI] [PubMed] [Google Scholar]
- 18.Priestley ND, Robinson JA. Purification and catalytic properties of L-valine dehydrogenase from Streptomyces cinnamonensis. Biochem J. 1989;261(3):853–861. doi: 10.1042/bj2610853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnbull AP, Baker PJ, Rice DW. Analysis of the quaternary structure, substrate specificity, and catalytic mechanism of valine dehydrogenase. J Biol Chem. 1997;272(40):25105–25111. doi: 10.1074/jbc.272.40.25105. [DOI] [PubMed] [Google Scholar]
- 20.Sharkey MA, Engel PC. Modular coenzyme specificity: A domain-swopped chimera of glutamate dehydrogenase. Proteins. 2009;77(2):266–278. doi: 10.1002/prot.22433. [DOI] [PubMed] [Google Scholar]
- 21.Wang MD, Liu L, Wang BM, Berg CM. Cloning and characterization of the Escherichia coli K-12 alanine-valine transaminase (avtA) gene. J Bacteriol. 1987;169(9):4228–4234. doi: 10.1128/jb.169.9.4228-4234.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stillman TJ, Baker PJ, Britton KL, Rice DW. Conformational flexibility in glutamate dehydrogenase. Role of water in substrate recognition and catalysis. J Mol Biol. 1993;234(4):1131–1139. doi: 10.1006/jmbi.1993.1665. [DOI] [PubMed] [Google Scholar]
- 23.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O, and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25(6):1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniel J, Dondon L, Danchin A. 2-Ketobutyrate: A putative alarmone of Escherichia coli. Mol Gen Genet. 1983;190(3):452–458. doi: 10.1007/BF00331076. [DOI] [PubMed] [Google Scholar]
- 25.Shulman A, et al. Allosteric regulation of Bacillus subtilis threonine deaminase, a biosynthetic threonine deaminase with a single regulatory domain? Biochemistry. 2008;47(45):11783–11792. doi: 10.1021/bi800901n. [DOI] [PubMed] [Google Scholar]
- 26.Chemler JA, Fowler ZL, McHugh KP, Koffas MAG. Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab Eng. 2009 doi: 10.1016/j.ymben.2009.07.003. 10.1016/j.ymben.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Gobert J, Geerts JP, Bodson G. ((S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide. 4,696,943. US Patent. 1987
- 28.Futagawa T, et al. Process for the preparation of levetiracetam. 6,107,492. US Patent. 2000
- 29.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2:1–11. doi: 10.1038/msb4100050. (2006.0008) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.