

Abstract
 α-Mannopyranosyl phosphosugars are obtained in 61 to 90 % yields from 4,6-O-benzylidene-protected mannosyl thioglycosides bearing ester functionality in the 3-O-position by coupling reactions with ammonium salts of phosphosugars on activation with 1-benzenesulfinyl piperidine, 2,4,6-tri-tert-butylpyrimidine, and trifluoromethanesulfonic anhydride. Due to the presence of the disarming ester group, only the formation of the α-isomer was observed.
α-Mannopyranosyl phosphosugars are obtained in 61 to 90 % yields from 4,6-O-benzylidene-protected mannosyl thioglycosides bearing ester functionality in the 3-O-position by coupling reactions with ammonium salts of phosphosugars on activation with 1-benzenesulfinyl piperidine, 2,4,6-tri-tert-butylpyrimidine, and trifluoromethanesulfonic anhydride. Due to the presence of the disarming ester group, only the formation of the α-isomer was observed.
Glycosyl phosphosugars are fragments of several glycoproteins. They are also structural blocks of poly(glycosyl phosphates) which are present in the cell walls and capsules of numerous bacteria. Such capsular polysaccharides are composed of mono- or polysaccharide units linked with phosphate diester bridges through hemiacetal and alcohol hydroxyl groups neighboring units.1 Recently, Vinogradov et al. determined the structure of the capsular polysaccharide from Campylobacter jejuni RM1221, the causative agent of human gastroenterits.2 This polysaccharide 1 has a regular structure made up of a linear main chain of trisaccharide repeating units, comprised of two α- and one β-6-deoxy-d-manno-hepto-pyranose residues punctuated by a phosphodiester linkage. Continuing our interest in the stereocontrolled synthesis of mannoheptoside containing oligosaccharides and their deoxy congenors,3 we have now directed our attention to the synthesis of this repeating unit and report here on the accomplishment of an essential first step – the highly stereocontrolled preparation of α-mannopyranosyl phosphosugars.
 Glycosyl phosphosugars have previously been synthesized by the phosphate diester and phosphite triester methods,4 as well as by the H-phosphonate approach.5 More recently, Boons et al employed glycosyl phosphoramidites in a approach to the synthesis of the proteophosphoglycans from Leishmania.6 In our laboratory we prepared β-mannopyrannosyl phosphoisoprenoids diastereoselectively by coupling of a 2,3-di-O-benzyl-4,6-O-benzylidene protected mannopyranosyl sulfoxide with tetrabutylammonium phosphates.7 The stereoselectivity observed in these reactions was highly solvent dependent, being highly β-selective in toluene, but giving α,β-mixtures in dichloromethane. In view of the importance of the acetals of the 4,6-O-benzylidene type in our general strategy toward the Campylobacter jejuni RM1221 repeating unit, it was necessary to adapt this chemistry so as to render it highly α-selective ideally, in the optimal solvent for this chemistry - dichloromethane. To this end we opted to investigate the influence of an ester in the 3-O-position, which we had previously shown to result in very high α-selectivity in the synthesis of a range of typical glycosides despite the presence of the 4,6-O-benzylidene group.8,9
Glycosyl phosphosugars have previously been synthesized by the phosphate diester and phosphite triester methods,4 as well as by the H-phosphonate approach.5 More recently, Boons et al employed glycosyl phosphoramidites in a approach to the synthesis of the proteophosphoglycans from Leishmania.6 In our laboratory we prepared β-mannopyrannosyl phosphoisoprenoids diastereoselectively by coupling of a 2,3-di-O-benzyl-4,6-O-benzylidene protected mannopyranosyl sulfoxide with tetrabutylammonium phosphates.7 The stereoselectivity observed in these reactions was highly solvent dependent, being highly β-selective in toluene, but giving α,β-mixtures in dichloromethane. In view of the importance of the acetals of the 4,6-O-benzylidene type in our general strategy toward the Campylobacter jejuni RM1221 repeating unit, it was necessary to adapt this chemistry so as to render it highly α-selective ideally, in the optimal solvent for this chemistry - dichloromethane. To this end we opted to investigate the influence of an ester in the 3-O-position, which we had previously shown to result in very high α-selectivity in the synthesis of a range of typical glycosides despite the presence of the 4,6-O-benzylidene group.8,9
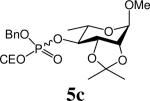
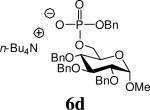
The ammonium salts of phosphosugars required as glycosyl acceptors were prepared using the phosphoramidite strategy.10 Thus, a range of phosphosugars 6 were obtained from known carbohydrates 2 (d-glucose, l-rhamnose or d-mannose derived) by coupling with benzyl 2-cyanoethyl (CE) N,N-diisopropylphosphoramidite 37 in the presence of benzimidazolium trifluoromethanesulfonate 411 as promoter followed in situ by oxidation with m-CPBA (Scheme 1). The diesters 5 were isolated in high yields after this two-step sequence as approximately 1/1 mixtures of diastereoisomers (Table 1). This mixture was confirmed by the 31P NMR which showed two signals. The 2-cyanoethyl protecting group was removed by treatment of 5 with tetrabutylammonium hydroxide in a CH2Cl2/water mixture (Scheme 1) to give the ammonium phosphosugars 6 quantitatively (Table 1), and these were employed in the subsequent glycosylation reactions without purification.12
Scheme 1.

Preparation of tetrabutylammonium phosphosugars 6
Table 1.
Preparation of Acceptors 6
| Entry | SugarOH 2 | Phosphosugars 5 | % Yield | Ammonium Salt 6 | % Yield |
|---|---|---|---|---|---|
| 1 |

|

|
98 |

|
100 |
| 2 |

|

|
84 |

|
94 |
| 3 |

|
|
81 |

|
99 |
| 4 |

|

|
82 |
|
99 |
| 5 |

|

|
86 |

|
95 |
| 6 |

|

|
82 |

|
100 |
| 7 |

|

|
81 |

|
100 |
The α-thiomannoside 7 was prepared from the phenyl 2-O-benzyl-4,6-O-benzylidene-1-thia-α-d-mannopyranoside13 by acetylation in 98% yield (Scheme 2).
Scheme 2.

Preparation of α-Mannopyranosyl phosphosugars 8
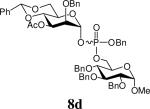
Glycosylation the acceptors 6 with donor 7, carried out by means of the 1-benzenesulfinyl piperidine (BSP)/Tf2O preactivation protocol14 in the presence of the hindered base 2,4,6-tri-tert-butylpyrimidine (TTBP),15 was achieved in dichloromethane at - 78 °C (Scheme 2). These reactions were rapid and, in line with our expectations, gave the α-anomers 8 with very high anomeric selectivity and good to high yield following silica gel chromatography16 (Table 2). No significant selectivity was observed at the level of the stereogenic phosphorus center and compounds 8 were therefore characterized as approximately 1/1 mixtures of diastereoisomers.
Table 2.
Glycosylation reactions of 7
| Entry | Ammonium salt 6 | α-Mannopyranosyl Phosphosugars 8 | % Yield |
|---|---|---|---|
| 1 | 6a |

|
73 |
| 2 | 6b |

|
74 |
| 3 | 6c |

|
90 |
| 4 | 6d |
|
61 |
| 5 | 6e |

|
66 |
| 6 | 6f |

|
90 |
| 7 | 6g |

|
67 |
Perhaps not surprisingly in view of the occasional use of glycosyl phosphates as glycosyl donors17 we observed that compounds 8 were generally not stable in methanolic solution when they gave the corresponding methyl glycosides.18
Finally, we turned our attention to deprotection. After several dead ends,19 we had recourse to treatment of compounds 8 with sodium in liquid ammonia, when cleavage of all benzyl esters and ethers, benzylidene acetals and acetate esters was achieved cleanly in excellent yields (Scheme 3).
Scheme 3.

Deprotection with sodium in liquid ammonia
In summary, we have shown that α-mannopyranosyl phosphosugars can be obtained in good yield and excellent selectivity through the use of a donor carrying a stereodirecting, but non-participating 3-O-carboxylate ester. This chemistry is currently being applied to the syntheses of capsular polysaccharide repeating units from Campylobacter jejuni RM1221, on which we will report in due course.
Experimental Section
General procedure for the formation of phosphosugars 5
To a solution of sugars 2 (1.0 equiv) and benzimidazolium trifluoromethanesulfonate 4 (1.5 equiv) in dry CH2Cl2 (10 mL/mmol of 2) was added at room temperature 3 (1.2 equiv). The reaction mixture was stirred 2 h at room temperature, before it was cooled to – 78 °C and m-CPBA (2 equiv) was added. The reaction mixture was stirred 1 h at this temperature, warmed to room temperature and stirred overnight at room temperature before it was quenched with saturated aqueous NaHCO3 and then extracted several times with CH2Cl2. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under reduced pressure. The crude product was purified by chromatography on silica gel (EtOAc:Heptane, 1:1) to afford phosphosugars 5.
General procedure for cyanoethyl ester cleavage
To a solution of phosphosugars 5 (1.0 equiv) in a mixture CH2Cl2:H2O (2:1, 9mL/mmol of 5) was added at room temperature tetrabutylammonium hydroxide (1 M in water, 1.2 equiv). The reaction mixture was stirred 2 h at room temperature before it was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was washed with brine, dried (MgSO4) and concentrated under reduced pressure. The obtained ammonium salt was used for the next step without purification.
General procedure for glycosylation
To a cooled (- 78 °C) solution of 7 (1.0 equiv), BSP (1.2 equiv) and TTBP (1.5 equiv) in dry CH2Cl2 (20 mL/mmol of 7) was added Tf2O (1.2 equiv). The resulting orange reaction mixture was stirred 30 min at – 78 °C and then a solution of ammonium phosphosugars 6 (1.5 equiv) in dry CH2Cl2 (1 mL/mmol) was added. The reaction mixture was stirred 20 min at – 78 °C, warmed slowly to room temperature and stirred 1 h at room temperature. The reaction was quenched with saturated aqueous NaHCO3 and then extracted twice with CH2Cl2. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under reduced pressure. The crude product was purified by chromatography on silica gel (30% EtOAc in Heptane) to afford the α-d-mannopyranosyl phosphosugars 8.
General procedure for deprotection with sodium and liquid ammonia
The protected α-d-mannopyranosyl phosphosugars 8 was dissolved in dry THF (0.1 mol/L) and placed in a three-necked flask equipped with a bubbler, an ammonia inlet and a cold finger condenser. The system was cooled to – 78 °C, flushed with argon and the condenser was filled with dry ice/acetone before ammonia gas was passed through the system. To the ensuing stirred solution was added Na as small spheres until the solution retained a dark blue color, after which stirring was continued for 0.5 h before the reaction was quenched with saturated aqueous NH4Cl, warmed to room temperature and concentrated under reduced pressure. The residue was purified on Sephadex LH20 (eluent absolute EtOH) to afford the α-d-mannopyranosyl phosphosugars 9.
Benzyl 2-cyanoethyl (methyl 2,4,6-tri-O-benzyl-α-d-mannopyranosid-3-yl) phosphate (5g)
Mixture of diastereoisomers. Colourless oil, 81%. 1H NMR (500 MHz, CDCl3) δ: 2.26-2.50 (m, 4H), 3.36 (s, 6H), 3.71-3.86 (m, 6H), 3.88-4.14 (m, 8H), 4.51-4.60 (m, 4H), 4.68-4.80 (m, 12H), 4.98-5.12 (m, 4H), 7.20-7.40 (m, 40H); 13C NMR (75 MHz, CDCl3) δ: 19.1, 19.2, 19.3, 54.9, 61.6, 61.7, 61.8, 68.9, 69.6, 69.7, 69.8, 71.5, 73.1, 73.5, 74.0, 74.4, 74.7, 77.2, 78.8, 78.9, 79.0, 98.8, 166.2, 166.3, 127.5, 127.6, 127.7, 127.9, 128.3, 128.6, 135.4, 135.5, 137.9, 138.0, 138.2; 31P NMR (202 MHz, CDCl3) δ: −2.40, −2.16. ESIHRMS Calcd for C38H42NO9PNa [M+Na]+: 710.2495, found 710.2495.
Tetrabutylammonium benzyl (methyl 2,4,6-tri-O-benzyl-α-d-mannopyranosid-3-yl) phosphate (6g)
Colourless oil, 100%. [α]26D + 26.8 (c, 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 0.94 (t, J = 7.2 Hz, 12H), 1.31-1.44 (m, 8H), 1.48-1.62 (m, 8H), 3.21-3.28 (m, 8H), 3.31 (s, 3H), 3.66-3.84 (m, 3H), 3.94 (t, J = 9.5 Hz, 1H), 4.34 (s, 1H), 4.56-4.69 (m, 5H), 4.80 (d, J = 12.6 Hz, 1H), 4.97 (dd, J = 5.1 Hz, J = 12.7 Hz, 1H), 5.02 (d, J = 12.6 Hz, 1H), 5.03 (d, J = 12.4 Hz, 1H), 5.19 (d, J = 11.7 Hz, 1H), 7.16-7.35 (m, 18H), 7.42 (d, J = 6.7 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 13.7, 19.8, 24.0, 54.6, 58.7, 68.6, 70.0, 71.9, 73.2, 73.5, 74.4, 75.0, 75.6, 77.2, 100.1, 126.5, 126.9, 127.2, 127.6, 127.8, 127.9, 128.2, 138.7, 139.4, 139.7, 140.2; 31P NMR (202 MHz, CDCl3) δ: - 0.73. ESIHRMS Calcd for C35H38O9P [M-n-Bu4N+]−: 633.2253, found 633.2263.
Benzyl (3-O-acetyl-2-O-benzyl-4,6-O-benzylidene-α-d-mannopyranos-1-yl) (methyl 2,4,6-tri-O-benzyl-α-d-mannopyranosid-3-yl) phosphate (8g)
Mixture of diastereoisomers. Colourless oil, 67%. 1H NMR (500 MHz, CDCl3) δ: 2.03 (s, 3H), 2.04 (s, 3H), 3.36 (s, 6H), 3.70-3.87 (m, 10H), 4.41 (d, J = 11.7 Hz, 1H), 4.46-4.62 (m, 7H), 4.68-4.88 (m, 12H), 4.97-5.15 (m, 4H), 5.26-5.31 (m, 2H), 5.54 (s, 1H), 5.55 (s, 1H), 5.79-5.84 (m, 2H), 7.21-7.48 (m, 60H); 13C NMR (125 MHz, CDCl3) δ: 21.3, 55.3, 55.4, 66.1, 66.3, 68.6, 69.4, 69.8, 69.9, 70.0, 70.3, 72.0, 72.1, 73.4, 73.7, 73.8, 74.2, 74.3, 74.3, 74.4, 74.5, 74.6, 75.1, 75.4, 75.8, 75.9, 76.5, 76.6, 76.7, 79.6, 79.8, 96.7 (1JCH = 178 Hz), 96.8 (1JCH = 176 Hz), 99.3 (1JCH = 171 Hz), 99.4 (1JCH = 171 Hz), 102.2, 102.3, 126.6, 126.7, 127.4, 127.5, 127.6, 127.7, 127.8, 127.9, 128.0, 128.1, 128.2, 128.4, 128.5, 128.6, 129.0, 135.4, 135.5, 137.1, 137.2, 137.9, 138.0, 138.1, 138.2, 169.7, 169.9; 31P NMR (202 MHz, CDCl3) δ: - 3.65, - 3.47. ESIHRMS (Solvent MeCN) Calcd for C57H61O15PNa [M + Na]+ : 1039.3646, found 1039.3638.
Ammonium (methyl α-d-mannopyranosid-3-yl) (α-d-mannopyranos-1-yl) phosphate (9b)
White powder, 89% from 8g. [α]26D + 55 (c, 0.15, CH3OH); mp: 180 °C; 1H NMR (500 MHz, CD3OD) δ: 3.38 (s, 3H), 3.50-3.56 (m, 1H), 3.61 (t, J = 9.7 Hz, 1H), 3.70 (dd, J = 6.3 Hz, J = 11.5 Hz, 1H), 3.74-3.85 (m, 5H), 3.86-3.93 (m, 2H), 4.03-4.05 (m, 1H), 4.32 (td, J = 2.0 Hz, J = 8.0 Hz, 1H), 4.68 (s, 1H), 5.51 (d, J = 7.5 Hz, 1H); 13C NMR (75 MHz, CD3OD) δ: 55.4, 62.4, 62.8, 67.4, 68.4, 71.2, 71.9, 72.4 (d, J = 9.2 Hz), 74.4, 75.8, 77.8 (d, J = 4.8 Hz), 98.0 (d, J = 5.5 Hz), 102.5; 31P NMR (202 MHz, CD3OD) δ: −1.34. ESIHRMS Calcd for C13H24O14P [M−]−: 435.0904, found 435.0918.
Supplementary Material
Acknowledgment
We thank the NIH (GM57335) for partial support of this work.
Footnotes
Supporting Information Available: Preparation of 7, characterisation of 5a-f, 6a-f, 7, 8a-f and 9a, 1H, 13C and 31P NMR spectra of 7, 5a-g, 6a-g, 8a-g and 9a-b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a von Figura K, Hasilik A. Annu. Rev., Biochem. 1986;55:167–194. doi: 10.1146/annurev.bi.55.070186.001123. [DOI] [PubMed] [Google Scholar]; b Raetz CRH, Whitfield C. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nikolaev AV, Botvinko IV, Ross AJ. Carbohydr. Res. 2007;342:297–334. doi: 10.1016/j.carres.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert M, Mandrell RE, Parker CT, Li J, Vinogradov E. ChemBioChem. 2007;8:625–631. doi: 10.1002/cbic.200600508. [DOI] [PubMed] [Google Scholar]
- 3.a Crich D, Banerjee A. Org. Lett. 2005;7:1395–1398. doi: 10.1021/ol050224s. [DOI] [PubMed] [Google Scholar]; b Crich D, Banerjee A. J. Am. Chem. Soc. 2006;128:8078–8086. doi: 10.1021/ja061594u. [DOI] [PMC free article] [PubMed] [Google Scholar]; Crich D, Li M. J. Org. Chem. 2008;73:7003–7010. doi: 10.1021/jo801414c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Cawley TN, Letters R. Carbohydr. Res. 1971;19:373–382. [Google Scholar]; b Thiem J, Franzkowiak M. J. Carbohydr. Chem. 1989;8:1–28. [Google Scholar]
- 5.Nikolaev AV, Ivanova IA, Shibaev VN, Kochetkov NK. Carbohydr. Res. 1990;204:65–78. [Google Scholar]
- 6.a Elsayed GA, Boons G-J. Synlett. 2003;9:1373–1375. [Google Scholar]; b Majumdar D, Elsayed GA, Buskas T, Boons G-J. J. Org. Chem. 2005;70:1691–1697. doi: 10.1021/jo048443z. [DOI] [PubMed] [Google Scholar]
- 7.a Crich D, Dudkin V. Org. Lett. 2000;2:3941–3943. doi: 10.1021/ol006725p. [DOI] [PubMed] [Google Scholar]; b Crich D, Dudkin V. J. Am. Chem. Soc. 2002;124:2263–2266. doi: 10.1021/ja0123958. [DOI] [PubMed] [Google Scholar]
- 8.a Crich D, Cai W, Dai Z. J. Org. Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]; Crich D, Yao Q. J. Am. Chem. Soc. 2004;126:8232–8236. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]
- 9.For a discussion of this effect: Crich D, Hu T, Cai F. J. Org. Chem. 2008;73:8942–8953. doi: 10.1021/jo801630m.
- 10.Matteucci MD, Caruthers MH. J. Am. Chem. Soc. 1981;103:3185–3191. For a review, see: Beaucage S, Iyer RP. Tetrahedron. 1992;48:2223–2311.
- 11.a Hayakawa Y, Kataoka M, Noyori R. J. Org. Chem. 1996;61:7996–7997. doi: 10.1021/jo961569e. [DOI] [PubMed] [Google Scholar]; b Sakakura A, Hayakawa Y. Tetrahedron. 2000;56:4427–4435. [Google Scholar]
- 12.The ammonium salts 6 were not stable to silica gel chromatography.
- 13.Crich D, Li W, Li H. J. Am. Chem. Soc. 2004;126:15081–15086. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 14.Crich D, Smith M. J. Am. Chem. Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 15.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 16.Bock K, Pedersen C. The selectivity was determined by analyses 1H and 31P NMR of crude reaction rmixtures and confirmed by the determination of the 1JCH coupling constants of pure diastereoisomers. J. Chem. Soc., Perkin Trans. 2. 1974:293–297. [Google Scholar]
- 17.Hashimoto SI, Honda T, Ikegami S. J. Chem. Soc. Chem Commun. 1989;11:685–687. Plante OJ, Palmacci ER, Andrade RB, Seeberger PH. J. Am. Chem. Soc. 2001;123:9545–9554. doi: 10.1021/ja016227r. For a review, see: Zhu X, Schmidt RR. Angew. Chem. Int. Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036.
- 18.For this reason ESIMS analyses of compounds 8 were conducted with solutions in acetonitrile.
- 19.With a view to stabilizing the glycosyl phosphate linkage before removal of the benzylidene acetal, compounds 8 were first treated with NaI in acetone to afford quantitatively the corresponding stable sodium phosphates Unfortunately, hydrogenolyses of these compounds with a variety of catalysts (Pd/C or Pd(OH)2/C) were either incomplete or resulted in complex mixtures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.