

Abstract

A variety of substituted benzisoxazoles has been prepared by the [3 + 2] cycloaddition of nitrile oxides and arynes. Both components, being highly reactive intermediates, have been generated in situ by fluoride anion from readily prepared aryne precursors and chlorooximes. The reaction scope is quite general, affording a novel, direct route to functionalized benzisoxazoles under mild reaction conditions.
Benzisoxazoles are present in a large number of pharmaceutically important products with antipsychotic,1 antitumor,2 anticonvulsant,3 antimicrobial,4 antithrombotic5 and cholinesterase-inhibiting (Alzheimer’s disease6) properties. The traditional approach to the synthesis of benzisoxazoles is a 3–4 step synthesis, which in some cases involves the strongly acidic conditions of a Friedel-Crafts reaction and the use of one of the substrates as a solvent, while in other cases it requires the use of strongly basic organometallics. Employing these methods, it is possible to synthesize particular benzisoxazoles on a large scale, but it is not as convenient to synthesize large numbers of diverse benzisoxazoles readily for biological activity screening.
The idea of forming two bonds at a time, by means of a [3 + 2] cycloaddition reaction of a benzyne with a nitrile oxide, has been reported previously,7 although the low yields (10 and 53%), limited possibilities for diversification, and the challenging experimental procedures employed were not very encouraging for the widespread utility of this approach.
Our recent interest8 in the chemistry of arynes generated from the corresponding o-(trimethylsilyl)aryl triflates under mild fluoride ion treatment has encouraged us to reexamine this approach to benzisoxazoles. Fluoride ion not only induces the formation of benzyne due to initial nucleophilic attack on the silicon of the trimethylsilyl group,9 but, being also a base, it could potentially10 induce formation of the nitrile oxide from the corresponding chlorooximes. Such an approach would eliminate potential problems arising from interaction of the reagents needed for generating the two highly reactive intermediates, as well as the requirement that the two species be generated separately from each other.
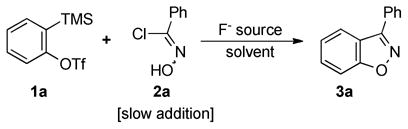
Indeed, our initial attempt to react o-(trimethylsilyl)-phenyl triflate (1a) with N-hydroxybenzimidoyl chloride (2a) in the presence of 3 equiv of CsF in acetonitrile led to formation of the desired 3-phenylbenzisoxazole (3a) in a 46% yield (Table 1, entry 1). High resolution GC-MS of the crude mixture revealed the expected dimerization products11 of phenyl nitrile oxide as the major byproducts of the reaction.
Table 1.
Optimization Studies of the Reaction of o-(Trimethylsilyl)phenyl Triflate (1a) and N-Hydroxybenzimidoyl Chloride (2a)
 | ||||||
|---|---|---|---|---|---|---|
| entry | 1a equiv | fluoride source (equiv) | solvent | temp (°C) | add. time of 2a (h) | % yieldb |
| 1 | 1.2 | CsF (3) | CH3CN | rt | - | 46 |
| 2 | 1.2 | TBAT (3) | CH3CN | rt | - | <5c |
| 3 | 1.2 | CsF (3) | THF | rt | - | <5c |
| 4 | 1.2 | CsF (3) | THF | 65 | - | <5c |
| 5 | 1.2 | CsF (3) | CH3CN | 65 | - | 9 |
| 6 | 1.2 | CsF (2.5) | CH3CN | rt | - | 23 |
| 7 | 2.0 | CsF (6) | CH3CN | rt | - | 61 |
| 8 | 3.0 | CsF (6) | CH3CN | rt | - | 58 |
| 9 | 2.0 | CsF (6) | CH3CN | rt | 5.0 | 70 |
| 10 | 2.0 | CsF (6) | CH3CN | rt | 2.5 | 90 |
| 11 | 2.0 | CsF (6) | CH3CN | rt | 1.0 | 73 |
All reactions were carried out on a 0.25 mmol scale.
Isolated yields, unless stated otherwise.
1H NMR spectroscopic yields.
Our optimization studies of this process are summarized in Table 1. The use of tetrabutylammonium triphenyldifluorosilicate (TBAT), an alternative anhydrous fluoride source, failed to provide the desired product under analogous conditions (entry 2). The use of THF, a less polar solvent, did not lead to formation of the desired benzisoxazole either (entries 3 and 4). An attempt to run the reaction at an elevated temperature to increase the rate of [3 + 2] cycloaddition at the expense of nitrile oxide dimerization did not work well (entry 5). To decrease the rate of formation of the nitrile oxide, thus decreasing self-dimerization products, we attempted to use less of the base CsF and at the same time decrease the rate of benzyne formation, which resulted in a lower (23%) yield of benzisoxazole (entry 6). Since the rate of nitrile oxide dimerization seemed to be greater than the rate of [3 + 2] cycloaddition, we examined the use of an excess of the benzyne precursor (entries 7 and 8). Indeed, the use of 2 equiv of the benzyne precursor was found to increase the yield to 61%. To further increase the local concentration of benzyne relative to the nitrile oxide, we examined slow addition of the chloroxime to the reaction mixture via syringe pump (entries 9–11). The optimal time of addition, which best aligned the rates of formation of the benzyne and the nitrile oxide, was found to be 2.5 h, which allowed the benzisoxazole (3a) to be isolated in a 90% yield (entry 10).
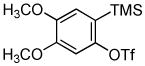
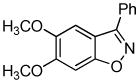
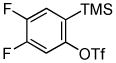
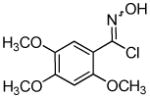
Employing the optimal conditions shown in Table 1, entry 10, we examined the scope of this reaction using various aryne precursors (Table 2, entries 1–3). Both electron-rich and electron-poor aryne precursors successfully participated in the [3 + 2] cycloaddition reaction, providing the desired benzisoxazoles. Symmetrical 3,4-dimethoxybenzyne provided the corresponding benzisoxazole 3b in a 65% yield (entry 1). The lowest yield (36%) was observed in the reaction of the highly reactive 3,4-difluorobenzyne (entry 2).
Table 2.
Synthesis of 3-Substituted Benzisoxazoles by the Reaction of Chlorooximes and Aryne Precursors in the Presence of CsFa
| entry | benzyne precursor | chlorooxime | product(s) | yield % isolated |
|---|---|---|---|---|
| 1 |
 1b 1b
|
2a |
 3b 3b
|
65 |
| 2 |
 1c 1c
|
2a |
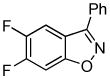 3c 3c
|
36 |
| 3 |
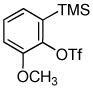 1d 1d
|
2a |
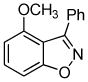 3d + 3d + 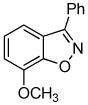 3e 3e
|
74 (1.8:1)b |
| 4 | 1a |
 2b 2b
|
 3f 3f
|
57 |
| 5 | 1a |
 2c 2c
|
 3g 3g
|
93 |
| 6 | 1a |
 2d 2d
|
 3h 3h
|
81 |
| 7 | 1a |
 2e 2e
|
 3i 3i
|
93 |
| 8 | 1a |
 2f 2f
|
 3j 3j
|
82 |
| 9 | 1a |
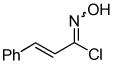 2g 2g
|
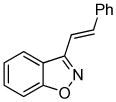 3k 3k
|
70 |
| 10 | 1a |
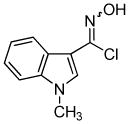 2h 2h
|
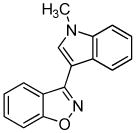 3l 3l
|
61 |
| 11 | 1a |
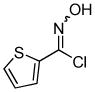 2i 2i
|
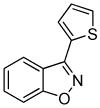 3n 3n
|
54 |
Reaction conditions: a solution of the appropriate chlorooxime (0.25 mmol) in 3 mL of acetonitrile was added via syringe pump to a stirring mixture of silylaryl triflate (0.50 mmol) and CsF (1.50 mmol) in 3 mL of acetonitrile over the course of 2.5 h.
The ratio was determined by 1H NMR spectroscopy.
Unsymmetrical 3-methoxybenzyne provided a mixture of regioisomers in a ~1.8:1 ratio (entry 3).12 Interestingly, electronic factors dominate over steric factors with isomer 3d being the major product in the mixture (Scheme 1).
Scheme 1.
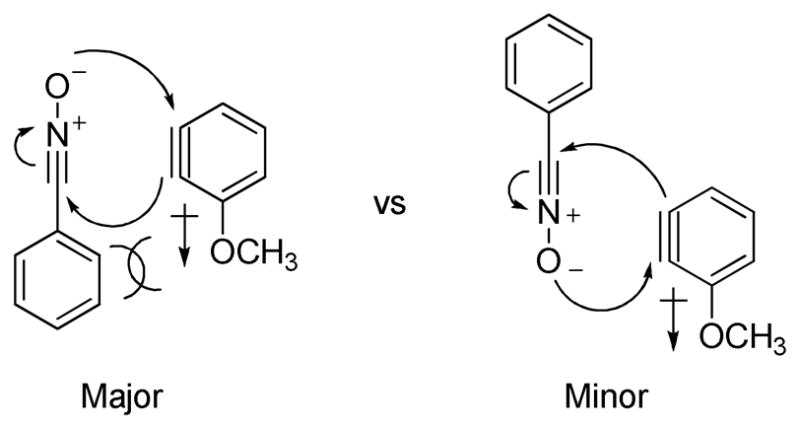
Electronic and Steric Factors in the Reaction of 3-Methoxybenzyne and Phenyl Nitrile Oxide
Various chlorooximes were prepared from the corresponding readily available aldehydes in 53–99% overall yield as follows. Reaction of the aldehyde with hydroxylamine hydrochloride in the presence of Na2CO3 afforded the corresponding oximes, which without isolation were chlorinated using NCS and catalytic amounts of Py at room temperature in chloroform (Scheme 2).13 An alternative protocol,14 which employs chlorination by oxone and HCl, failed to provide chlorooximes bearing alkyl moieties.
Scheme 2.
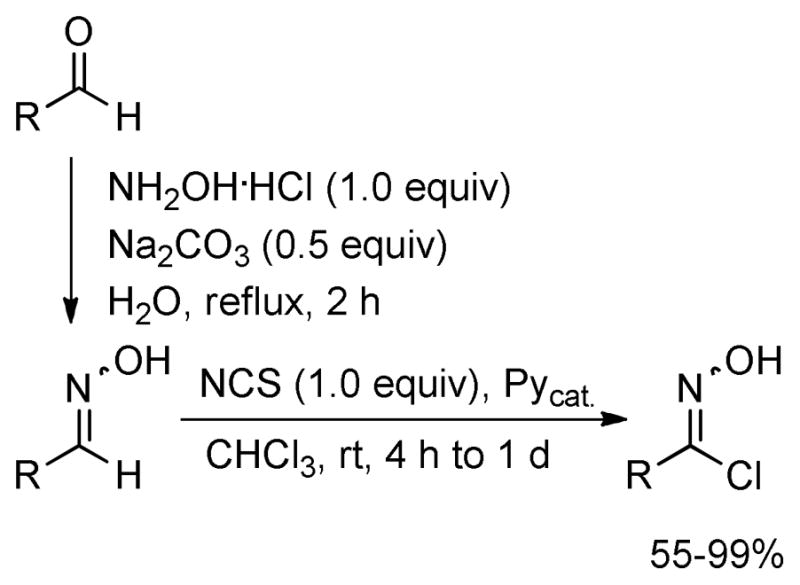
Preparation of the Chlorooximes
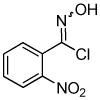
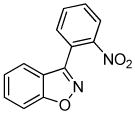
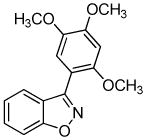
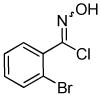
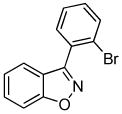
We next investigated the scope of the reaction between o-(trimethylsilyl)phenyl triflate (1a) and various chlorooximes (Table 2, entries 4–11). Aromatic chlorooximes provided the corresponding benzisoxazoles in good to excellent yields. Lower yields were observed when electron-withdrawing substituents reside on the benzene ring. For example, 3-(2-nitrophenyl)benzisoxazole (3f) was isolated in a 57% yield (entry 4).15 Excellent yields were observed using chlorooximes bearing electron-donating (through resonance) substituents (entries 5–7), and even chlorooxime 2e bearing a bulky o-bromo substituent afforded benzisoxazole 3i in a 93% isolated yield.
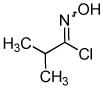
Despite the reported instability of chlorooximes bearing alkyl moieties,16 3-isopropylbenzisoxazole was isolated in an 83% yield. Surprisingly, an alkene was also readily tolerated in the reaction. Thus, (E)-3-styrylbenzisoxazole (3k) was isolated in a 70% yield (entry 9).
Next, we studied the cycloaddition reaction of benzyne with chlorooximes derived from heterocyclic aldehydes. 3-(N-Methylindolyl)chlorooxime (2h) furnished the desired product 3l in a 61% yield (entry 10). Unfortunately, we could not obtain the desired product from the 2-furyl chlorooxime. The reaction produced many inseparable products, presumably due to Diels-Alder reactions9 of the furan ring with the benzyne. On the other hand, the 3-thiophenyl chlorooxime 2i successfully provided the desired benzisoxazole 3n in a 54% yield (entry 11).
In summary, a simple, convenient and efficient protocol has been developed for the synthesis of benzisoxazoles. The reaction tolerates a variety of functional groups and provides an alternative route to potentially important benzisoxazoles bearing aryl, alkyl, alkenyl and heterocyclic substituents at the 3 position of the benzisoxazole moiety. Further applications of this process and the synthesis of biologically active products are currently underway.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (GM079593 and GM070620) and the Kansas University NIH Center of Excellence in Chemical Methodology and Library Development (P50 GM069663) for their generous financial support. We also thank Dr. Feng Shi at Iowa State University for the preparation of non-commercially available benzyne precursors.
Footnotes
Supporting Information Available: Detailed experimental procedures and characterization data for all new products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Davis L, Effland RC, Klein JT, Dunn RW, Geyer HM, III, Petko WM. Drug Design and Discovery. 1992;8:225. [PubMed] [Google Scholar]; (b) Strupczewski JT, Allen RC, Gardner BA, Schmid BL, Stache U, Glamkowski EJ, Jones MC, Ellis DB, Huger FP, Dunn RW. J Med Chem. 1985;28:761. doi: 10.1021/jm00383a012. [DOI] [PubMed] [Google Scholar]; (c) Janssen PAJ, Niemegeers CJE, Awouters F, Schellekens KHL, Megens AAHP, Meert TF. J Pharmacol Exp Ther. 1988;244:685. [PubMed] [Google Scholar]; (d) Strupczewski JT, Bordeau KJ, Chiang Y, Glamkowski EJ, Conway PG, Corbett R, Hartman HB, Szewczak MR, Wilmot CA, Helsley GC. J Med Chem. 1995;38:1119. doi: 10.1021/jm00007a009. [DOI] [PubMed] [Google Scholar]
- 2.(a) Gopalsamy A, Shi M, Golas J, Vogan E, Jacob J, Johnson M, Lee F, Nilakantan R, Petersen R, Svenson K, Chopra R, Tam MS, Wen Y, Ellingboe J, Arndt K, Boschelli F. J Med Chem. 2008;51:373. doi: 10.1021/jm701385c. [DOI] [PubMed] [Google Scholar]; (b) Jain M, Kwon CH. J Med Chem. 2003;46:5428. doi: 10.1021/jm020581y. [DOI] [PubMed] [Google Scholar]
- 3.(a) Stiff DD, Zemaitis MA. Drug Metab Dispos. 1990;18:888. [PubMed] [Google Scholar]; (b) Uno H, Kurokawa M, Masuda Y, Nishimura H. J Med Chem. 1979;22:180. doi: 10.1021/jm00188a011. [DOI] [PubMed] [Google Scholar]
- 4.Priya BS, Basappa, Swamy SN, Rangappa KS. Bioorg Med Chem. 2005;13:2623. doi: 10.1016/j.bmc.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 5.Nuhrich A, Varache-Lembege M, Renard P, Devaux G. Eur J Med Chem. 1994;29:75. [Google Scholar]
- 6.(a) Villalobos A, Blake JF, Biggers CK, Butler TW, Chapin DS, Chen YL, Ives JL, Jones SB, Liston DR, Nagel AA, Nason DM, Nielsen JA, Shalaby IA, White WF. J Med Chem. 1994;37:2721. doi: 10.1021/jm00043a012. [DOI] [PubMed] [Google Scholar]; (b) Rangappa KS, Basappa J Phys Chem. 2005;18:773. [Google Scholar]
- 7.(a) Minisci F, Quilico A. Gazz Chim Ital. 1964;46:428. [Google Scholar]; (b) Sasaki T, Yoshioka T. Bull Chem Soc Japan. 1969;42:826. doi: 10.1246/bcsj.42.1308. [DOI] [PubMed] [Google Scholar]
- 8.Shi F, Waldo JP, Chen Y, Larock RC. Org Lett. 2008;10:2409. doi: 10.1021/ol800675u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Himeshima Y, Sonoda T, Kobayashi H. Chem Lett. 1983:1211. [Google Scholar]
- 10.In the literature Na2CO3 or aliphatic amines are most commonly used, see: Lam PYS, Adams JJ, Clark CG, Calhoun WJ, Luettgen JM, Knabb RM, Wexler RR. Bioorg Med Chem Lett. 2003;13:1795. doi: 10.1016/s0960-894x(03)00130-6.
- 11.Kelly DR, Baker SC, King DS, de Silva DS, Lord G, Taylor JP. Org Biomol Chem. 2008;6:787. doi: 10.1039/b716915a. [DOI] [PubMed] [Google Scholar]
- 12.The ratios were determined by 1H NMR spectroscopy.
- 13.Larsen KE, Torssell KBG. Tetrahedron. 1984;40:2985. [Google Scholar]
- 14.Vela MA, Kohn H. J Org Chem. 1992;57:6650. [Google Scholar]
- 15.Electron-withdrawing groups are known to stabilize nitrile oxides thus making them more reluctant to cyclize, see: Wakefield BJ, Wright DJ. J Chem Soc C. 1970:1165.
- 16.Kumar V, Kaushik MP. Tetrahedron Lett. 2006;47:1457. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.