

Abstract
Apoptosis of neurons in the maturing neocortex has been recorded in a wide variety of mammals, but very little is known about its effects on cortical differentiation. Recent research has implicated the RhoA GTPase subfamily in the control of apoptosis in the developing nervous system and in other tissue types. Rho GTPases are important components of the signaling pathways linking extracellular signals to the cytoskeleton. To investigate the role of the RhoA GTPase subfamily in neocortical apoptosis and differentiation, we have engineered a mouse line in which a dominant-negative RhoA mutant (N19–RhoA) is expressed from the Mapt locus, such that all neurons of the developing nervous system are expressing the N19–RhoA inhibitor. Postnatal expression of N19–RhoA led to no major changes in neocortical anatomy. Six layers of the neocortex developed and barrels (whisker-related neural modules) formed in layer IV. However, the density and absolute number of neurons in the somatosensory cortex increased by 12–26% compared with wild-type littermates. This was not explained by a change in the migration of neurons during the formation of cortical layers but rather by a large decrease in the amount of neuronal apoptosis at postnatal day 5, the developmental maximum of cortical apoptosis. In addition, overexpression of RhoA in cortical neurons was seen to cause high levels of apoptosis. These results demonstrate that RhoA-subfamily members play a major role in developmental apoptosis in postnatal neocortex of the mouse but that decreased apoptosis does not alter cortical cytoarchitecture and patterning.
Introduction
Programmed neuronal cell death plays a central role in the dynamic organization of developing neuronal networks (Purves, 1990). The classical neurotrophic model describes populations of peripheral neurons that compete for limiting amounts of survival factors (e.g., NGF, BDNF, and neurotrophins 3 and 4/5) (Bibel and Barde, 2000). Failure to bind neurotrophins results in an apoptotic response and neuronal death. The activation of apoptotic machinery in the periphery is well investigated, but corresponding mechanisms in developing brain are poorly understood. Apoptosis plays a crucial role in embryonic development of the cerebral cortex, cerebellum, and brainstem. The key apoptosis proteins caspase-3, caspase-9, Apaf1, Bax, Bcl-XL, and survivin have been demonstrated to regulate and execute apoptosis through mouse knock-out studies (for review, see Kuan et al., 2000). However, the proximal causes of apoptosis within the embryonic brain remain unknown.
Neuronal loss is known to occur in postnatal mammalian neocortex (Heumann et al., 1978; Finlay and Slattery, 1983; Heumann and Leuba, 1983; Price and Blakemore, 1985), marked by a wave of apoptosis that peaks at postnatal day 5 (P5) to P7 in rodents (Pearlman, 1985; Ferrer et al., 1990, 1992; Spreafico et al., 1995; Verney et al., 2000). Aside from IGF-1 (Chrysis et al., 2001; Hodge et al., 2007), the molecular players in postnatal cortical apoptosis are not known.
The Rho GTPases RhoA, RhoB, and RhoC belong to and define the RhoA-subfamily within the Ras superfamily of small GTP-binding proteins (Hall, 1998). They cycle between a GDP-bound, inactive and a GTP-bound, active state. These two states are primarily regulated by the guanine nucleotide exchange factors (GEFs) and the GTPase-activating proteins families (Etienne-Manneville and Hall, 2002), which promote the activation and inactivation, respectively, of GDP- and GTP-bound Rho. One of the most important targets of active RhoA is Rho-associated kinase (ROCK), a serine/threonine kinase with multiple substrates (Katoh et al., 1998). Although much is known about the modeling of axons and dendrites by Rho GTPases (Luo, 2000), the role that they play in the control of apoptosis is controversial. In vitro studies have indicated that Rho can induce apoptosis in hippocampal (Donovan et al., 1997) and cortical (Zhang et al., 2007) neurons. In vivo studies have delivered conflicting evidence, with one investigation showing that apoptosis of spinal cord motor neurons increased during inhibition of Rho activity (Kobayashi et al., 2004), whereas a rat model of spinal cord injury showed that Rho inhibition reduced injury-related apoptotic levels (Dubreuil et al., 2003).
To investigate Rho GTPases in the control of postnatal cortical apoptosis, we developed a mouse line in which a dominant-negative inhibitor of Rho GTPases is expressed specifically in neurons. We found that inhibition of Rho GTPases in postnatal cortical neurons in vivo resulted in a significant reduction in apoptosis of excitatory neurons and a corresponding increase in the absolute number and density of neurons in the cortex. Despite the increase in neuronal numbers, cortical lamination and pattern formation were unaltered. These findings suggest that postnatal apoptosis does not contribute to cytoarchitectonic differentiation and cellular patterning of the neocortex.
Materials and Methods
Generation of the N19–RhoA mouse line.
All animal experiments were in compliance with the regulations of Baden–Württemberg. To construct the targeting vector, a human RhoA cDNA containing the N19 mutation and an N-terminal hemagglutinin (HA) tag was inserted into a vector (pLSL; courtesy of Dr. Silvia Arber, University of Basel, Basel, Switzerland) downstream of a transcriptional stop cassette flanked by loxP sites. The resultant cassette was inserted into a targeting vector (courtesy of Dr. Silvia Arber) (Hippenmeyer et al., 2005) containing genomic Mapt sequence and a neomycin-selectable marker. The linearized targeting vector was electroporated into J1 embryonic stem (ES) cells as described previously (Tucker et al., 2001), and 28 neomycin-resistant colonies were analyzed by Southern blot, using external genomic probes as described (Tucker et al., 2001).
Targeted, euploid ES cells were injected into C57BL/6 blastocysts. Two high-contribution male chimeras derived from two different ES cell lines were bred with C57BL/6 wild-type mice to generate two independent N19–RhoA mouse lines. Germ-line transmission of the targeted Mapt allele was confirmed by Southern blots of mouse tail DNA. Subsequent generations were maintained on a C57BL/6 background. For genotyping the N19–RhoA mice, the primers 5′-TACGACGTGCCCGACTAC-3′ and 5′-GCTGTGTCCCACAAAGCC-3′ delivered a 220 bp amplicon. EIIa::CRE mice were genotyped using the primers 5′-GCCGAAATTGCCAGGATCAG-3′ and 5′-AGCCACCAGCTTGCATGATC-3′, giving a 486 bp amplicon. For the Southern blot analysis of the efficiency of the Cre-based excision of the stop cassette, a 600 bp N19–RhoA cDNA fragment was used as a probe.
Rhotekin beads.
Rhotekin-expressing bacteria (Ren et al., 1999) were cultured in 20 ml of Luria broth (LB) media with ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) on a shaker at 37°C overnight. The following day, 2–4 ml of overnight culture were added to 4× 500 ml LB media with ampicillin and chloramphenicol on a shaker at 37°C and induced with 500 μl of 0.5 m isopropyl-β-d-thiogalactopyranoside until the OD600 became between 0.7 and 0.8, followed by 3 h incubation at 30°C on a shaker. The bacterial cultures were placed in 500 ml tubes and centrifuged for 10 min at 4000 rpm, and the pellets were washed twice with 200 ml of cold 1× PBS. The pellets were resuspended and centrifuged for 10 min at 4000 rpm at 4°C. These pellets were resuspended in 4× 50 ml lysis buffer, the cells were sonicated for 15 s six times at 130 W, and 500 μl of 10% Triton X-100 was added per tube to make the final concentration of 0.1% Triton X-100 and incubated for 15 min on ice, rocking. Lysates were decanted in eight ultracentrifuge tubes on ice and centrifuged for 12 min at 27,000 rpm (Hitachi HimacCS 100fx). The beads (1.3 ml) (Glutathione Sepharose 4B; GE Healthcare) for 2 L of medium was washed in a 50 ml tube twice with 15 ml of 1× PBS at 4°C. Beads were centrifuged at 1000 rpm at 2°C for 1 min. Rhotekin lysates were added to the beads and incubated for 45 min, shaking horizontally to avoid making bubbles. The beads were washed with 20 ml of cold wash buffer [50 mm Tris-HCl, pH 7.5, 0.5% Triton X-100, 150 mm NaCl, 5 mm MgCl2, 10 μg/ml aprotinin (Roche), 10 μg/ml leupeptin (Roche), and 10 μg/ml PMSF] for 1 min at 1000 rpm at 2°C, and then the supernatant was removed. Beads were resolved in 20 ml of wash buffer/10% glycerol, and aliquoted beads were kept at −80°C.
Rhotekin pulldown assay.
Cerebral cortex from postnatal animals was carefully microdissected and flash frozen in liquid nitrogen. Tissue was lysed (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 5 mm MgCl2) with 10 μg/ml aprotinin (Roche), 10 μg/ml leupeptin (Roche), and 10 μg/ml PMSF (Carl Roth GmbH) by sonication three times for 15 s. Brain lysate (450 μg) was incubated with 200 μl Rhotekin beads for 19 h at 4°C. Lysates were centrifuged at 4°C for 15 min at 2900 rpm, and the pellet was resuspended and washed three times in 50 mm Tris-HCl, pH 7.5, 0.5% Triton X-100, 150 mm NaCl, 5 mm MgCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml PMSF for 5 min with rotation. The pellet was resuspended in 15 μl of sample buffer and electrophoresed in acrylamide gels, and RhoA levels were analyzed by Western blot. The antibodies (Abs) used included mouse anti-RhoA (sc-418, 1:500; Santa Cruz Biotechnology) and rabbit anti-actin (A-2066, 1:1000; Sigma-Aldrich).
Western blotting.
Western blotting was performed on homogenized brain as described previously (Willaredt et al., 2008), with either a mouse anti-RhoA Ab (sc-418, 1:1000; Santa Cruz Biotechnology) or a rabbit anti-HA Ab (sc-805, 1:1000; Santa Cruz Biotechnology). For a positive control, HN10 cells were transiently transfected with the HA-tagged N19–RhoA construct used above, and protein lysates were prepared 48 h after transfection.
In situ hybridization.
In situ hybridization on paraffin sections was performed (Shakèd et al., 2008) with a human RhoA cDNA (Deutsches Ressourcenzentrum für Genomforschung GmbH).
Barrel cortex analyses.
Four P9 and four P65 mice (two control and two transgenic each age) were used to analyze the barrel cortex. In these experiments, serial sections through flattened cortices and coronal brain sections were processed with routine histological methods for visualization of barrel patterns. The brains were split in half along the sagittal plane. The left cortex from each half was removed and flattened between two glass slides. Thirty-micrometer-thick coronal and tangential sections were kept in PBS, pH 7.4. Alternate sections were stained for either Nissl, cytochrome oxidase (CO) histochemistry or serotonin transporter (5-HTT) immunohistochemistry for P9 brains. For CO histochemistry, free-floating sections were incubated with phosphate buffer containing 0.5 mg/ml cytochrome C (type III; Sigma), 0.5 mg/ml diaminobenzidine (DAB) (Sigma), and 40 mg/ml sucrose for 2–4 h at 37°C in a shaker incubator. Alternate sections were stained with 2% cresyl violet for Nissl staining. For 5-HTT immunohistochemistry, sections were treated as below (immunohistochemical analysis) for peroxidase detection using an anti-5-HTT rabbit polyclonal Ab (1:10,000; Diasorin). Sections were mounted on slides, dehydrated, cleared in xylene, and coverslipped with Permount.
Immunohistochemical analysis and cell counting.
Free-floating 20 μm brain sections were rinsed in 2% H2O2 in 10% methanol for 20 min to block endogenous peroxidase activity, and biotinylated secondary antibody and peroxidase-labeled avidin–biotin complex were applied according to the instructions of the manufacturer (Vectastain Elite ABC; Vector Laboratories). Sections were washed in PBS for 30 min, developed in 0.05% DAB (Sigma) with the addition of 0.006% H2O2, washed in PBS for 10 min, and mounted (Aquatex medium). The following primary antibodies were used, with clone name, source, and dilution indicated: mouse anti-neuronal-specific nuclear protein (NeuN) (MAB377, 1:1000; Millipore Corporation), mouse anti-parvalbumin (clone PARV-19, 1:2000; Sigma), rabbit anti-calretinin (catalog #7699/4, 1:3000; Swant), mouse anti-calbindin (catalog #300, 1:3000; Swant), rabbit anti-cleaved-caspase-3 (clone 5A1, 1:200; Cell Signaling Technology), rabbit anti-phospho-histone H3 (Ser10, rabbit 06-570, 1:200; Millipore Corporation), and rabbit anti-ER81 (1:200; kind gift from Dr. Silvia Arber). For caspase-3, phospho-histone H3, and ER81 stains, secondary Abs were used as described previously (Brachmann et al., 2007).
Sections were photographed with a Leica DMLB microscope equipped with a Leica DFC320 CCD camera and two different air objectives: a 40× HCX PLAN APO objective (numerical aperture, 0.85) and a 5× HC PL FLUOTAR objective (numerical aperture, 0.15), using the program Firecam 3.1 (Leica). Somatosensory cortex and the anterior extent of the visual cortex was used for analyses. Ten slides were chosen at equidistant locations within this area, photographed on both the right and left sides, and subsequently quantitated. In NeuN-stained preparations, the darkly stained nuclei were easy to identify at both magnifications. In the photos taken with a 5× objective, the entire width of the cortex could be identified, nuclei in specific layers were counted by hand, and these data are presented in Figure 4. Photographs taken at 40× were assigned to specific layers, and a correction for the thickness of the section and the size of the nuclei was made according to Abercrombie (1946). Because the size of the nuclei did not vary significantly between wild-type (layer IV, 10.0 ± 0.9 μm; layer V, 11.8 ± 1.0 μm; layer VI, 10.8 ± 0.8 μm; mean ± SEM; n > 150 for all layers) and N19–RhoA (layer IV, 10.2 ± 0.9 μm; layer V, 12.8 ± 0.9 μm; layer VI, 10.8 ± 0.7 μm; mean ± SEM; n > 150 for all layers) mice, this correction did not affect the relative increase in neuron number when comparing wild-type with N19–RhoA mice, and the results agreed with the counts made using sections photographed with the 5× objective. With the 40× objective, we recorded an increase in layer IV of 23.4 ± 1.9%, in layer V of 18.0 ± 2.7%, and in layer VI of 21.0 ± 5.8% (n = 4; mean ± SEM, Student's t test). All counting was performed in a blind manner.
Figure 4.
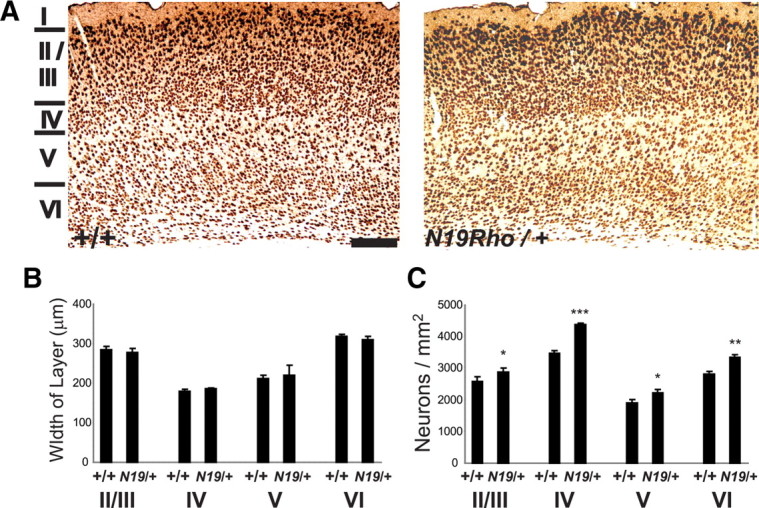
N19–RhoA mice show an increased density of neurons in somatosensory cortex. A, Immunohistochemical staining of somatosensory cortex in P65 wild-type (+/+) and N19–RhoA/+ mice, using an antibody recognizing NeuN. For each coronal section, dorsal is to the top, and lateral is to the right. Layers I–VI are indicated to the left. B, Quantification of the dorsoventral width of layers II/III, IV, V, and VI. C, Quantification of NeuN-positive neurons in layers II/III, IV, V, and VI. *p < 0.5, **p < 0.01, ***p < 0.001, Student's t test (n = 4). Scale bar: A, 200 μm.
Developmental analysis of cortical layer formation.
Brains from newborn (P1) or P3 N19–RhoA and control littermate pups were perfused transcardially and removed for processing to paraffin blocks. For paraffin sectioning, tissue was dehydrated in an ascending ethanol row, isopropanol, and xylene. Brains from N19–RhoA and control littermates were embedded in one common paraffin block and were sectioned at 10 μm on a Leica sliding microtome. Sections were deparaffinated using xylene and isopropanol and were rehydrated in a descending ethanol row. Antigen unmasking was performed by boiling for 10 min in either citric buffer (pH 6) or Tris EDTA (pH 9) in a microwave oven at 600 W. Sections were washed with Tris buffer containing 2% milk powder (Tris+), blocked in 20% goat serum in PBS for 1 h, and incubated with primary antibodies in 5% goat serum in PBS for 4 h. Sections were washed three times with Tris+, incubated for 20 min with appropriate biotinylated secondary antibodies (Vector Laboratories), washed three times with Tris+, incubated for 10 min with streptavidin/biotin complex (Vector Laboratories), washed three times in Tris (without milk powder), incubated for 10 min with DAB substrate (Vector Laboratories), washed, and counterstained with hematoxylin. Sections were dehydrated in an ascending ethanol row, followed by isopropanol and xylene. Coverslips were applied using Eukitt. Primary antibodies were directed against Reelin (mouse IgG, 1:500; Calbiochem), Satb2 (mouse IgG, 1:1000; from V. Tarabykin), Ctip2 (rat IgG, 1:500; Abcam), Brn2 (polyclonal goat, 1:200; Santa Cruz Biotechnology), Tbr2 (polyclonal rabbit, 1:500; Abcam), and Pax6 (polyclonal rabbit, 1:500; Millipore Bioscience Research Reagents).
In vitro apoptosis assays.
Five to seven embryonic day 16.5 (E16.5) embryos were removed from timed pregnant CD-1 mice, the cerebral hemispheres cut apart with a scalpel, the meninges were peeled away, and ∼3 mm of the cortex lying above the hippocampus was cut out and digested in 0.25% trypsin (Invitrogen) at 37°C for 15 min. After three washes in HBSS/10 mm HEPES, pH 7.3, cells were triturated with a fire-polished Pasteur pipette, and 700,000 cells were centrifuged 7 min at 100 × g and electroporated in 100 μl nucleofection solution (mouse neuron nucleofector kit, Program O5; Amaxa Biosystems), containing either 5 μg of an expression vector expressing enhanced green fluorescent protein (pEGFP–N1), a RhoA expression vector [wild-type human RhoA cDNA with a 5′-localized HA tag cloned into the EcoRI/NotI site of the pcDNA3.1(+) vector], or myc-tagged wild-type human Rac1 or cdc42 expression plasmids (kind gift from Dr. Robert Grosse, Philipps University of Marburg, Marburg, Germany). Transfected cells were plated in 24-well plates at a concentration of 30,000 cells per 13 mm coverslip (pretreated with 1 mg/ml poly-l-lysine and 1 μg/ml laminin) in MEF medium (DMEM/10% fetal calf serum/2 mm glutamine), incubated at 37°C/5% CO2, with an exchange of MEF medium to Neurobasal medium/1× B27 supplement/2 mm glutamine (Invitrogen) after 24 h, and fixed after 48 h in culture for 10 min with 4% paraformaldehyde, pH 7.4, at room temperature. Immunocytofluorescence was performed as described previously (Shakèd et al., 2008), with the following changes: cells were permeabilized for 5 min with 0.2% Triton X-100 only at the beginning of the staining, followed by a quenching of the cells for 5 min with 0.1% sodium borohydride. The following primary antibodies were used: FITC-coupled goat anti-GFP (G8965×12A, 1:2000; US Biologicals), mouse anti-HA tag (clone 6E2, 1:200; Cell Signaling Technology), mouse anti-c-Myc tag (clone 9E10, sc-40, 1:100; Santa Cruz Biotechnology), rabbit anti-cleaved-caspase-3 (Asp175, clone 5A1, 1:200; Cell Signaling Technology). Secondary Abs were used as described previously (Brachmann et al., 2007). 4′,6′-Diamidino-2-phenylindole (DAPI) (2 μg/ml) was used to stain nuclei. The stained cortical neurons were analyzed with epifluorescent microscopy (Microscope BX61WI; Olympus) using identical exposure conditions and a digital CCD camera (F-View II; Soft Imaging GmbH) in combination with imaging software (analysis, Soft Imaging GmbH). One hundred cells positive for either pEGFP–N1 or Rho–HA were assayed for their cellular and nuclear morphology and for cleaved caspase-3 activity. Statistical analysis was performed using the Mann–Whitney U test.
Results
A mouse line expressing a dominant-negative inhibitor of Rho specifically in newborn neurons of the nervous system
To inhibit the activity of proteins in the RhoA GTPase subfamily, we used the N19–RhoA construct, a dominant-negative inhibitor in which the threonine at position 19 has been mutated to asparagine (N) (Qiu et al., 1995). N19–RhoA is predicted to block the GTPase activity of endogenous RhoA-subfamily GTPases through competitive binding to GEFs (Feig, 1994), and it has been suggested that this substitution also disturbs Rho GTPase activity by interfering with an essential Mg2+ ion required for guanine nucleotide binding in all Ras superfamily GTP-binding proteins (Farnsworth and Feig, 1991). It has been shown to specifically inhibit members of the RhoA-subfamily GTPases and to demonstrate no direct effect on other Rho GTPase family members, such as cdc42 or Rac, either biochemically (Ren et al., 1999) or in cell-based assays (Hall, 1998; Wojciak-Stothard et al., 1999; Bouzahzah et al., 2001). However, it cannot be definitively excluded that N19–RhoA may also affect cdc42 or Rac-based pathways by binding to promiscuous GEFs that interact with both RhoA- and cdc42/Rac-subfamily members (Rossman et al., 2005). To distinguish the mutant RhoA protein from the endogenous murine RhoA, a human N19–RhoA cDNA was modified to express an HA tag at its N terminus.
The N19–RhoA cDNA was engineered to allow for its inducible expression after exposure to the Cre recombinase. A cassette was constructed in which the N19–RhoA cDNA lies downstream of a transcriptional stop cassette flanked by two loxP sites (Fig. 1B). This entire cassette was placed into the first exon of the gene (Mapt) encoding tau, which had been used previously to express EGFP specifically in newborn neurons (Tucker et al., 2001). We chose an inducible approach because we were concerned that the long-term expression of N19–RhoA in all neurons of the mouse could cause potentially lethal side effects if N19- RhoA were constitutively expressed from the Mapt locus. Because of the floxed stop cassette, transcription from the endogenous tau promoter at the altered locus should produce a truncated, non-protein-coding transcript, and the introduced cDNA should not be transcribed (Fig. 1A). Exposure to the Cre recombinase should cause recombination between the two loxP sites, excision of the stop cassette, and subsequent expression of the N19–RhoA protein (Fig. 1A). The targeting vector was electroporated into J1 ES cells, and 49 G418-resistant colonies were picked, of which 28 were analyzed by Southern blot. Using 5′ (Fig. 1B,D) and 3′ (Fig. 1B,E) external probes, five clones were found to be targeted using both probes, indicating a targeting efficiency of 18%. Two independent euploid clones were used to generate chimeras, both of which transmitted the targeted allele through the germ line. With respect to all data presented in this paper, similar phenotypes were observed for both lines.
Figure 1.

Inducible expression of dominant-negative RhoA (N19–RhoA) at the Mapt locus. A–C, Schematic indicating targeting and expression schemes: loxP, loxP sites; Tx Stop, transcriptional stop sequence; N19RhoA, HA-tagged N19–RhoA cDNA; pA, Pgk1 polyadenylation sequence; neo, G418-resistance cassette. Scheme is not to scale. A, Scheme indicating Cre-mediated induction of N19–RhoA expression. For details, see Results. B, Map of endogenous Mapt allele (top) and the targeting construct (bottom) that is inserted into exon 1 during homologous recombination. Endogenous Mapt genomic sequences are revealed by BamHI or KpnI digestion, followed by Southern blotting with 5′ or 3′external probes, respectively. C, Map of targeted Mapt allele, before (top) and after (bottom) Cre-mediated recombination. Genomic alterations caused by the knock-in construct are detected by Southern blotting with external 5′ or 3′ probes after BamHI or KpnI digestion, respectively. Cre-mediated recombination eliminates the floxed transcriptional stop sequence, as revealed by KpnI digestion and Southern blotting with a RhoA probe. D, E, Southern blot of ES cell clones using the 5′ (D) and 3′ (E) external probes. Targeted clones (t) show a wild-type band and a band corresponding to the targeted allele at 2.9 kb (D) and 8.1 kb (E). DNA markers are indicated (right) with size in kilobase pairs.
Inducible expression of N19–RhoA in postnatal brain
To remove the floxed stop cassette from the Mapt locus, the N19–RhoA mice were crossed with the EIIa::Cre mouse line, which expresses Cre recombinase under the control of the adenovirus EIIa promoter (Lakso et al., 1996). In this line, Cre recombinase is expressed already in the zygote, and all cells of the resulting embryo should show recombination at the loxP sites and removal of the floxed stop cassette. After Cre-based recombination, the N19–RhoA cassette can then be expressed specifically in neurons. Compound heterozygotes for both the N19–RhoA and the EIIa::Cre alleles (Cre+/N19–RhoA+) were generated, and cultures of cerebellar granule neurons (CGNs) were prepared from P5 Cre−/N19–RhoA+ and Cre+/N19–RhoA+ mice. The ability of the loxP sites to undergo recombination by Cre recombinase was examined using Southern blot analysis. KpnI-digested genomic DNA from CGN cultures was probed with a human RhoA cDNA. The cassette before recombination was seen to be 3.4 kb in the Cre−/N19–RhoA+ cultures (Fig. 1D, 2A). Cre-mediated recombination would remove the 1.4 kb floxed transcriptional stop sequence, with a corresponding decrease in the size of the RhoA-hybridizing band on Southern blotting (Fig. 1D). Indeed, in the Cre+/N19–RhoA+ cultures, no signal could be detected at 3.4 kb, whereas a strong band was seen at 2.0 kb, indicating a 100% efficiency in the removal of the floxed stop cassette in CGN during exposure to Cre recombinase (Fig. 2A). Protein lysates were prepared from embryonic and postnatal brain of Cre+/N19–RhoA+ mice to examine expression of the N19–RhoA construct and were examined with Western blot analysis using an antibody recognizing the HA-tagged N19–RhoA. Surprisingly, no embryonic expression could be detected (Fig. 2B), despite the fact that EGFP is very strongly expressed from the tau locus as early as 9.5 d postcoitum (Tucker et al., 2001). Expression could first be detected at P3 in the brain, with expression continuing at P20 (Fig. 2C). The expression of the N19–RhoA protein was shown to act as an inhibitor of endogenous Rho function by performing a Rhotekin assay on lysates from the cerebral cortex of N19–RhoA-expressing or control mice. A large reduction in active GTP-bound Rho was observed (Fig. 2D). To examine the distribution of RhoA expression, in situ hybridization was performed on brain sections from postnatal N19–RhoA transgenic mice. At P5 and P9, expression could be seen throughout the neocortex and hippocampus, with particularly high levels in layers IV/V of the cortex and the pyramidal layers of CA1, CA3, and the dentate gyrus of the hippocampus (Fig. 2E,F).
Figure 2.

Postnatal expression of N19–RhoA inhibits endogenous Rho activity. A, Complete recombination of the targeted Mapt allele during in vivo exposure to EIIa::Cre recombinase was detected in acute cultures of cerebellar granule neurons, indicated by Southern blots on KpnI-digested genomic DNA with a RhoA cDNA probe. Band sizes are explained in Figure 1C. DNA markers are indicated (right) with size in kilobase pairs. B, C, Western blot analysis indicates HA-tagged N19–RhoA expression (arrow) in the brain of E13.5 (B) and newborn (C) animals after exposure to Cre recombinase, using an anti-HA antibody. B, HN10, Positive control in which HA-tagged N19–RhoA expression construct was transfected into HN10 cells. D, N19–RhoA acts as a dominant-negative inhibitor of RhoA function in vivo. Brain lysates from N19–RhoA or control mice were lysed, incubated with Sepharose beads coupled to the Rho-binding domain of Rhotekin, and protein bound to Rho-binding domain subsequently analyzed by Western blotting, using an anti-RhoA antibody to detect wild-type RhoA (22 kDa). Loading levels were analyzed using an anti-actin antibody (42 kDa). B–D, Protein markers (in kilodaltons) are indicated (right). E, F, In situ hybridization analysis of N19–RhoA expression in the telencephalon of P5 (E) and P9 (F) N19–RhoA mice. Dorsal is to the top. Scale bars: 200 μm.
Cortical anatomy is not disrupted in N19–RhoA transgenic mice
We examined the barrel cortex of P9 and P65 N19–RhoA transgenic mice to determine whether laminar differentiation and whisker-related barrel formation were affected. Six laminae of the parietal cortex were visible with Nissl (Fig. 3B,C,E) stains in transgenic mice, and there were no noticeable differences from the controls at both ages. CO histochemistry, a routine method for visualization of whisker-related patterns (i.e., cortical barrels) also revealed distinct patterning in the transgenic mice at both ages (Fig. 3D). In addition, we examined thalamocortical patterning with 5-HTT immunohistochemistry in P9 mice. Monoamine transporters are transiently expressed in the primary sensory thalamic nuclei in mice (Lebrand et al., 1998), and 5-HTT immunohistochemistry has been established as a reliable marker for thalamocortical afferent terminal patterns in the barrel cortex (Iwasato et al., 2000; Rebsam et al., 2002). The thalamocortical afferent patterning in the barrel cortex was similar between the N19–RhoA transgenic and control mice, in both the tangential and coronal planes (Fig. 3A).
Figure 3.
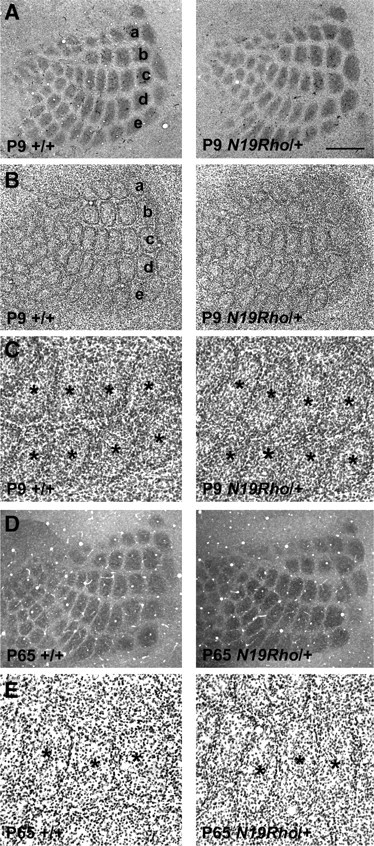
Normal barrel patterns in N19–RhoA mice. A, B, At P9, thalamocortical afferent terminal patterns, as assessed with 5-HTT immunohistochemistry (A), are similar between the control and N19–RhoA mice as are the barrels as cytoarchitectonic entities, revealed by Nissl stain (B). Whisker-related barrel rows (a–e) are indicated. C, High-magnification views of Nissl-stained barrels in rows d and e. Asterisks mark barrel centers. D, Cytochrome oxidase staining in P65 mice also shows clear barrel patterns in both the control and N19–RhoA mice. These micrographs were montaged from serial sections. E, High-magnification views of Nissl-stained barrels in P65 mice also revealed clear barrels. Barrel centers are marked with asterisks. Scale bars: A, B, 500 μm; C, E, 200 μm; D, 700 μm.
The absolute number and density of neurons is increased in the somatosensory cortex in N19–RhoA transgenic mice
To assess the number of neurons within the barrel cortex, an antibody recognizing NeuN, which is expressed by all cortical neurons except for Cajal-Retzius cells (Mullen et al., 1992; Lyck et al., 2007), was used. Careful counting of the neurons in specific layers revealed a large and significant increase in the density of NeuN-positive neurons in layers II/III, IV, V, and VI of the somatosensory cortex of the N19–RhoA transgenic mice when counted at maturity (P65) such that, in layer II/III, neuronal density increased by 11.6 ± 1.3%, in layer IV by 26.4 ± 1.9%, in layer V by 17.2 ± 1.5%, and in layer VI by 18.7 ± 1.2% (n = 4; mean ± SEM, Student's t test) (Fig. 4A,C). A similar increase was also observed in juvenile mice at P24 (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), indicating that the increase in neuronal number is already established at an early age. Because the dorsoventral width of the layers did not change significantly in the N19–RhoA transgenic mice (Fig. 4B), it can be concluded that the absolute number of neurons had increased by 12–26% in these cortical layers.
The density of interneuron populations is not affected in somatosensory cortex of N19–RhoA mice
NeuN is expressed by both excitatory projection neurons, which constitute the vast majority of the neurons in the cortex, and inhibitory interneurons, which make up 20–30% of the neuronal population (Markram et al., 2004). To distinguish which population was demonstrating an increase in the number in the cortex of the N19–RhoA mice, we performed stainings for interneuron subpopulations characterized by the expression of parvalbumin (Fig. 5A,B), calbindin (Fig. 5C), and calretinin (Fig. 5D). In all three cases, quantification of positive neurons revealed no difference in number between wild-type and N19–RhoA cortex (Fig. 5E). Because these interneurons do not show any change in number in the N19–RhoA cortex, we conclude that the change in neuronal density in the N19–RhoA cortex is attributable to an increase in the number of excitatory projection neurons and not in that of the interneuron population.
Figure 5.

The density of interneurons is not affected in somatosensory cortex of N19–RhoA mice. A, B, Immunohistochemical staining of somatosensory cortex in P65 wild-type (+/+) and N19–RhoA/+ mice, using antibodies recognizing parvalbumin (A, B), calbindin (C), and calretinin (D). For each coronal section, one telencephalic half is shown, with dorsal to the top and lateral to the right. Arrows (white) indicate positive neurons. B, Magnification of A. E, Quantification of interneurons. Scale bars: A, 500 μm; B–D, 200 μm.
Neuronal migration and cell type specification of cortical neurons is not affected in N19–RhoA mice
RhoA activity has been implicated in the control of radial migration of cortical neurons (Hand et al., 2005), and it has been shown to be involved in a large number of migratory processes throughout the developing nervous system (Liu and Jessell, 1998; Rupp and Kulesa, 2007; Carmona-Fontaine et al., 2008; Groysman et al., 2008). To investigate the possibility that the increased number of neurons in deeper cortex layers of N19–RhoA mice results from abnormal radial migration of upper layer neurons, we performed immunohistochemistry for typical layer-specific proteins in brain sections of newborn (P1) (Fig. 6) and P3 mice. We found the expression patterns of Reelin, Satb2, Ctip2, Brn2, Tbr2, and Pax6 (Fig. 6A–F) to be unchanged in N19–RhoA mice. In addition, in E16.5 cbs/cbs mutants, the number of mitotic cells at the cortical ventricular zone was not significantly altered in the cortex (n = 4; p = 0.69, Student's t test) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Finally, we examined the expression of the layer V-specific transcription factor ER81 at P24 in juvenile mice, when layer formation is complete. Comparison of the distribution of ER81-positive neurons between wild-type and N19–RhoA/+ cortex revealed that, in both cases, expression of ER81 was restricted to layer V (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material), which confirmed that migration of cortical neurons is normal. Together, these results demonstrate that the specification and migration of cortical neurons was normal in the N19–RhoA-expressing mice.
Figure 6.

Migration and specification of cortical neurons is not affected in N19–RhoA mice. Characterization of radial migration and specification in coronal sections of somatosensory cortex from newborn (P1, day of birth) N19–RhoA-transgene-positive mice with or without EIIa::Cre recombinase. A–F, Immunohistochemistry using primary antibodies raised against the characteristic layer-specific proteins: A, Reelin; B, Satb2; C, Ctip2; D, Brn2; E, Tbr2; F, Pax6. For each coronal section, dorsal is to the top and lateral is to the right. II, III, IV, V, VI, Layers II–VI, respectively; MZ, IZ, VZ, SVZ, marginal, intermediate, ventricular, and subventricular zones, respectively. Scale bars, 50 μm.
Apoptosis is severely reduced in somatosensory cortex of N19–RhoA mice
To test whether reduced apoptosis plays a role in the increase of neurons in cortical layers IV–VI of the N19–RhoA transgenic mice, the numbers of apoptotic cells were examined at P5. Postnatal days 5–8 have been reported to be the developmental peak of apoptosis in both the mouse and rat cortex (Pearlman, 1985; Ferrer et al., 1990, 1992; Spreafico et al., 1995; Verney et al., 2000). Apoptotic cells were detected using an antibody recognizing cleaved, activated caspase-3, a processed protein that is not only produced early during the apoptotic process but is also responsible for executing the proteolytic cascade common to both intrinsic and extrinsic apoptotic pathways (Porter and Jänicke, 1999) (Fig. 7A–C). A dramatic reduction of 67.5% was observed in the cortex of the N19–RhoA transgenic mice at this time point (wild-type cortex, 94.7 ± 6.4 caspase-3-positive cells per section; N19–RhoA/+ cortex, 28.0 ± 4.7 caspase-3-positive cells per section; n = 4; p < 0.001, Student's t test). Caspase-3-positive cells in both wild-type and N19–RhoA transgenic mice showed layer-appropriate morphology of neurons from upper (Fig. 7B) and lower (Fig. 7C) layers, and morphological analysis indicated that >90% of the caspase-3-positive cells were neurons (n = 314) (Fig. 7C).
Figure 7.
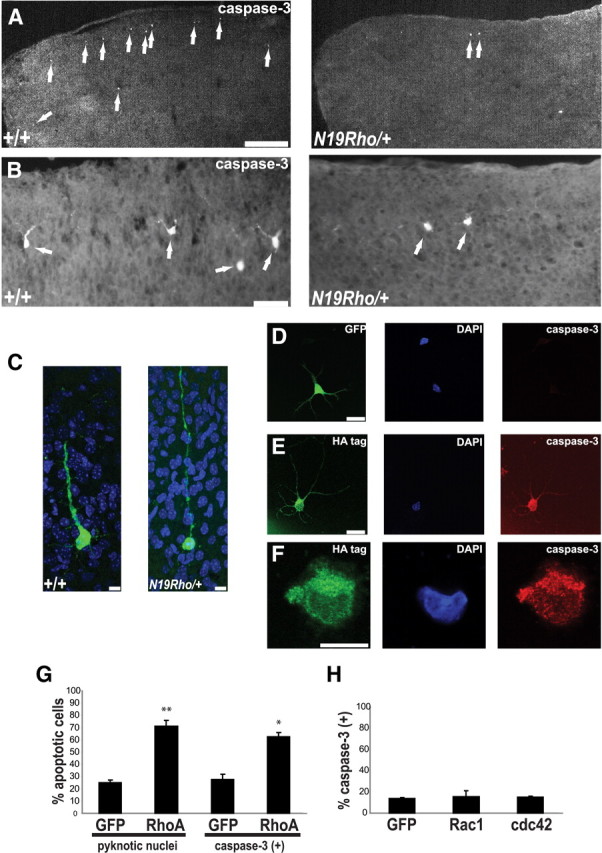
Apoptosis is severely reduced in somatosensory cortex of N19–RhoA mice. A–C, Immunofluorescence analysis of apoptosis in cerebral cortex of P5 wild-type (+/+) and N19–RhoA/+ mice, using an antibody recognizing cleaved, activated caspase-3. For each coronal section, one telencephalic half is shown, with dorsal to the top and lateral to the right. Arrows (white) indicate positive neurons. C, Caspase-3-positive cells (green) in layers IV–VI display the typical morphology of cortical projection neurons in both wild-type (+/+) and N19–RhoA/+ mice. D–H, Expression of wild-type RhoA (D–G), Rac1 (H), or cdc42 (H) in isolated cortical neurons leads to apoptosis only during expression of RhoA. Cortical neurons isolated from E16.5 embryonic cortex 48 h after electroporation with a GFP-tagged (D, green stain), an HA-tagged wild-type RhoA (E, F, green stain), or a myc-tagged Rac1 (H) or cdc42 (H) expression construct. D–F, Red indicates staining for activated caspase-3. G, H, Quantitation of GFP-, HA-, or myc-tagged positive cells for their fraction of pyknotic nuclei (G) or the fraction positive for an antibody recognizing activated-caspase-3 (G, H). In each case, >100 neurons were counted. *p < 0.05, **p < 0.01, Mann–Whitney U test; n = 3. C–F, Blue indicates DAPI-labeled nuclei. Scale bars: A, 300 μm; B, 50 μm; C, F, 10 μm; D, E, 20 μm.
Because inhibition of Rho activity resulted in a decrease in the levels of apoptosis in vivo in postnatal cortex, we tested whether apoptosis could be promoted by the overexpression of wild-type RhoA in cortical neurons. Acutely isolated E16.5 cortical neurons were electroporated with expression constructs expressing either EGFP alone or HA-tagged wild-type RhoA and cultivated on poly-lysine-coated coverslips. RhoA-expressing neurons demonstrated high levels of apoptosis 48 h after transfection, as assayed by either expression of activated caspase-3 (Fig. 7D–G) or the presence of pyknotic nuclei (Fig. 7D–G). Indeed, the levels of apoptosis seen in both the control (EGFP-expressing) and RhoA-expressing neurons (Fig. 7G) were very similar to those reported for cortical cultures prepared from wild-type mice and mice lacking the antiapoptotic protein Bcl-XL (Shindler et al., 1997). To test whether other members of the Rho GTPase subfamily could also induce apoptosis, constructs expressing myc-tagged wild-type Rac1 or cdc42 cDNAs were transfected into acutely isolated E16.5 cortical neurons. In distinct contrast to RhoA, neither of these constructs showed any influence on apoptotic levels (Fig. 7H).
Discussion
In this paper, we demonstrate that the RhoA-subfamily signaling pathway regulates the postnatal apoptosis of cortical neurons. Inhibition of Rho activity in vivo greatly reduced the amount of apoptosis occurring in postnatal cortex and resulted in a concomitant increase in the density and absolute number of neurons in the adult cortex. Overexpression of wild-type RhoA in vitro led to the apoptosis of cortical neurons, but this was not the case for other members of the Rho GTPase family, such as cdc42 or Rac1. A significant increase in neuronal numbers in the somatosensory cortex did not interfere with lamination or barrel formation in layer IV. In layer IV of the rodent somatosensory cortex, thalamocortical afferent terminals from the ventroposteromedial thalamic nucleus form distinct patches reflecting the distribution of whiskers on the contralateral snout, and cortical neurons organize around these patches forming barrels (Woolsey and Van der Loos, 1970; Rebsam et al., 2002). The mechanisms underlying the formation of cell-dense barrel walls and cell-sparse barrel hollows in the cytoarchitectonic organization in the mouse somatosensory cortex are not clear. At the time of arrival and terminal arborization of thalamocortical axons, the developing layer IV is uniformly populated with neurons. Thalamocortical afferents are the first elements to show whisker-specific patterning, followed by layer IV neuronal patterning (Erzurumlu and Jhaveri, 1990; Senft and Woolsey, 1991). A potential mechanism contributing to barrel formation could be apoptotic events in postnatal barrel cortex. Our results, however, indicate that a significant reduction in layer IV neuronal apoptosis, caused by an inhibition of Rho activity, and a concomitant increase in neuronal numbers do not interfere with barrel formation. Barrel patterning most likely involves active distribution of layer IV neuronal somata and dendritic arbors with respect to thalamocortical axon arbor patches.
Apoptosis has been demonstrated to be a critical process during embryonic development in which to control the neuronal populations in both the CNS and PNS. This is the first report to show that Rho signaling is important for the survival of neurons in vivo in cerebral cortex during postnatal development. Many investigations have outlined the critical roles that components of the apoptotic machinery play in developmentally regulated cell death. For example, gene targeting of the proapoptotic protein Bax (Shindler et al., 1997), the downstream effector caspases caspase-3 (Kuida et al., 1996; Roth et al., 2000) and caspase-9 (Hakem et al., 1998; Kuida et al., 1998), and the tumor suppressor gene Pten (Groszer et al., 2001) have all shown reductions in levels of apoptosis and a subsequent expansion of neuronal populations. In contrast, knock-out mice for the antiapoptotic protein Bcl-XL die at E13 with very high levels of apoptosis in the spinal cord, brainstem, and dorsal root ganglia (Motoyama et al., 1995), and deficiency in Bax has been shown to prevent the cell death in Bcl-XL knock-out mice (Shindler et al., 1997). An epistatic pathway has been thereby delineated in which caspase-3 and caspase-9 activation lie downstream of the apoptosis-activating and -inhibiting activities of Bax and Bcl-XL, respectively (Shindler et al., 1997; Roth et al., 2000; Zaidi et al., 2001), although several other apoptosis proteins have also been shown to play a role in this process, such as Apaf1 (Cecconi et al., 1998; Yoshida et al., 1998), survivin (Jiang et al., 2005), and Pten (Groszer et al., 2001). Our results showed that inhibition of RhoA decreased the number of cleaved caspase-3-expressing neurons in cortex and that overexpression of wild-type RhoA can activate caspase-3, indicating that RhoA is involved upstream of this key executor of the apoptotic pathway. However, our results show a role of RhoA in the postnatal apoptosis in the cortex, not the embryonic apoptosis examined in the aforementioned studies. Intriguingly, RhoA has been reported to be necessary for motor neuron survival in embryonic spinal cord, using an approach very similar to the one used here (Kobayashi et al., 2004). This suggests that the role of RhoA-subfamily proteins in developmental apoptosis is very complex, because it can produce opposite effects in various neural tissues and at various developmental stages. We could not address the role of RhoA in apoptosis in embryonic cortex, because the transgene was not expressed until postnatal stages.
The exact molecular mechanism by which RhoA activity promotes apoptosis is not clear. RhoA activation was found to be necessary for both thrombin-induced (Donovan et al., 1997) and phenylalanine-induced (Zhang et al., 2007) neuronal apoptosis in vitro. The latter study identified a downstream target of activated RhoA, the Rho-activated serine/threonine kinase ROCK, as transducing the apoptotic signal from Rho, whereas p38 kinase has been shown to be activated by RhoA in excitotoxic neuronal death in the adult brain (Semenova et al., 2007). ROCK activity has clearly been shown to control the actin–myosin-based cell contractility, membrane blebbing (Coleman et al., 2001; Sebbagh et al., 2001), and nuclear disintegration (Croft et al., 2005) characteristic of apoptosis, but whether these processes are even RhoA dependent is unclear (Coleman and Olson, 2002). In any case, the in vitro studies in neurons (Donovan et al., 1997; Zhang et al., 2007) and non-neuronal cells (Jiménez et al., 1995; Lai et al., 2003; Minambres et al., 2006), together with our in vivo results, clearly place RhoA high in the pathway controlling the decision to undergo apoptosis. With respect to postnatal cortical apoptosis, very little is known of the mechanism. The overexpression of IGF-I has been shown to promote the survival of neurons in postnatal cerebral cortex (Hodge et al., 2007). It would be of great interest to see whether IGF-1 exerts its effect through modulation of Rho activity. However, knock-out studies to demonstrate a necessary role of IGF-I in this process are notably lacking. Moreover, it is entirely unclear whether the postnatal apoptotic process is ligand mediated, as in the classical case of neurotrophins and the PNS, or ligand independent. Additional studies are needed to answer this intriguing question.
Our studies show that Rho signaling is involved in apoptosis mechanism during early postnatal development. Inhibition of Rho signaling led to a cortex with a 12–26% increase in the number of neurons. Because cell death occurs over an extended prenatal and postnatal period in the neocortex, it is difficult to estimate from our counts at P5 in the cortex of the N19–RhoA expressing mice whether a 67.5% decrease in activated caspase-3-positive neurons would lead to the observed increase in excitatory neuron numbers. A previous report (Verney et al., 2000) has used terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assays to quantitatively estimate layer-specific loss of neurons during the first 2 postnatal weeks in the parietal cortex of the mouse, the same area under investigation in this study. The authors reported losses of 10–23% in this timeframe, which is in agreement with the increases in neuronal numbers that we see during inhibition of Rho-subfamily GTPases using the N19–RhoA transgene. Simple counting of total neuronal number has revealed slightly higher levels of cell death in entire postnatal mouse cortex (24–30%) (Heumann et al., 1978). However, without detailed quantitative analyses of caspase-positive cells across different developmental ages, we cannot definitively conclude that Rho-subfamily GTPases are the sole controllers of neuronal numbers in the neocortex.
The aberrant increase in neuronal numbers occurs at the early postnatal stage in cortex, but the additional neurons persist into adulthood, which suggests that they may functionally integrate into the cortex. The supernumerary neurons are presumed to be excitatory, because a quantitation of parvalbumin-, calbindin-, and calretinin-positive interneurons indicated no change in these inhibitory populations in the N19–RhoA brain. It would be useful to see whether this increase in neuronal density results in any effects on behavior or on learning and memory. Neuronal cell loss is seen in a host of pathological conditions, including epilepsy (Henshall and Murphy, 2008), ischemia (Rami et al., 2008), microcephaly (Chen et al., 2009), alcohol abuse (Young et al., 2003), and neurodegenerative diseases such as Alzheimer's (Jellinger, 2006) and Parkinson's (Burke, 2008) disease. The protection against apoptosis seen here in early postnatal brain may be used to study neuronal loss in other contexts and potentially offers a new target for neuroprotective therapy.
Footnotes
This work was supported by the German Research Foundation [Deutsche Forschungsgemeinschaft: Sonderforschungsbereich 488, Teilprojekt B7/B9 (K.L.T.) and Exzellenzcluster 257 (V.T.)], the University of Heidelberg [Excellence Cluster Cellular Networks (K.L.T.)], National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant NS039050 (R.S.E.), the Max Planck Society (V.T.), and the Heisenberg Program (V.T.). We thank Joachim Kirsch for generous scientific support, Karin Gorgas for enormous help with anatomical analysis, Yves-Alain Barde for thoughtful commentary on this manuscript, Silvia Arber for the floxed stop and Mapt targeting constructs, Frank Zimmermann for blastocyst injections, Stefan Offermanns for EIIa::CRE mice and rhotekin-expressing bacteria, Julia Hoffmann for preparation of the rhotekin beads, Robert Grosse for Rac1 and cdc42 expression plasmids, Antonio Caputi for the anti-parvalbumin antibody, Ulrich Müller for the N19–RhoA cDNA, Günter Giese and Annemarie Scherbarth for assistance with confocal microscopy, and Dmitry Rusanov and Jana Hechler for superb technical assistance.
References
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;7:382–389. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- Bouzahzah B, Albanese C, Ahmed F, Pixley F, Lisanti MP, Segall JD, Condeelis J, Joyce D, Minden A, Der CJ, Chan A, Symons M, Pestell RG. Rho family GTPases regulate mammary epithelium cell growth and metastasis through distinguishable pathways. Mol Med. 2001;7:816–830. [PMC free article] [PubMed] [Google Scholar]
- Brachmann I, Jakubick VC, Shakèd M, Unsicker K, Tucker KL. A simple slice culture system for the imaging of nerve development in embryonic mouse. Dev Dyn. 2007;236:3514–3523. doi: 10.1002/dvdy.21386. [DOI] [PubMed] [Google Scholar]
- Burke RE. Programmed cell death and new discoveries in the genetics of parkinsonism. J Neurochem. 2008;104:875–890. doi: 10.1111/j.1471-4159.2007.05106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, Mayor R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 2008;456:957–961. doi: 10.1038/nature07441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Chen L, Melendez J, Campbell K, Kuan CY, Zheng Y. Rac1 deficiency in the forebrain results in neural progenitor reduction and microcephaly. Dev Biol. 2009;325:162–170. doi: 10.1016/j.ydbio.2008.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrysis D, Calikoglu AS, Ye P, D'Ercole AJ. Insulin-like growth factor-I overexpression attenuates cerebellar apoptosis by altering the expression of Bcl family proteins in a developmentally specific manner. J Neurosci. 2001;21:1481–1489. doi: 10.1523/JNEUROSCI.21-05-01481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Olson MF. Rho GTPase signalling pathways in the morphological changes associated with apoptosis. Cell Death Differ. 2002;9:493–504. doi: 10.1038/sj.cdd.4400987. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- Croft DR, Coleman ML, Li S, Robertson D, Sullivan T, Stewart CL, Olson MF. Actin-myosin-based contraction is responsible for apoptotic nuclear disintegration. J Cell Biol. 2005;168:245–255. doi: 10.1083/jcb.200409049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan FM, Pike CJ, Cotman CW, Cunningham DD. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci. 1997;17:5316–5326. doi: 10.1523/JNEUROSCI.17-14-05316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzurumlu RS, Jhaveri S. Thalamic axons confer a blueprint of the sensory periphery onto the developing rat somatosensory cortex. Brain Res Dev Brain Res. 1990;56:229–234. doi: 10.1016/0165-3806(90)90087-f. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Farnsworth CL, Feig LA. Dominant inhibitory mutations in the Mg2+-binding site of RasH prevent its activation by GTP. Mol Cell Biol. 1991;11:4822–4829. doi: 10.1128/mcb.11.10.4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA. Guanine-nucleotide exchange factors: a family of positive regulators of Ras and related GTPases. Curr Opin Cell Biol. 1994;6:204–211. doi: 10.1016/0955-0674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Bernet E, Soriano E, del Rio T, Fonseca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Soriano E, del Rio JA, Alcántara S, Auladell C. Cell death and removal in the cerebral cortex during development. Prog Neurobiol. 1992;39:1–43. doi: 10.1016/0301-0082(92)90029-e. [DOI] [PubMed] [Google Scholar]
- Finlay BL, Slattery M. Local differences in the amount of early cell death in neocortex predict adult local specializations. Science. 1983;219:1349–1351. doi: 10.1126/science.6828866. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Groysman M, Shoval I, Kalcheim C. A negative modulatory role for rho and rho-associated kinase signaling in delamination of neural crest cells. Neural Dev. 2008;3:27. doi: 10.1186/1749-8104-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron. 2005;48:45–62. doi: 10.1016/j.neuron.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Murphy BM. Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol. 2008;8:75–81. doi: 10.1016/j.coph.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Heumann D, Leuba G. Neuronal death in the development and aging of the cerebral cortex of the mouse. Neuropathol Appl Neurobiol. 1983;9:297–311. doi: 10.1111/j.1365-2990.1983.tb00116.x. [DOI] [PubMed] [Google Scholar]
- Heumann D, Leuba G, Rabinowicz T. Postnatal development of the mouse cerebral neocortex. IV. Evolution of the total cortical volume, of the population of neurons and glial cells. J Hirnforsch. 1978;19:385–393. [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, D'Ercole AJ, O'Kusky JR. Insulin-like growth factor-I (IGF-I) inhibits neuronal apoptosis in the developing cerebral cortex in vivo. Int J Dev Neurosci. 2007;25:233–241. doi: 10.1016/j.ijdevneu.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knöpfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Challenges in neuronal apoptosis. Curr Alzheimer Res. 2006;3:377–391. doi: 10.2174/156720506778249434. [DOI] [PubMed] [Google Scholar]
- Jiang Y, de Bruin A, Caldas H, Fangusaro J, Hayes J, Conway EM, Robinson ML, Altura RA. Essential role for survivin in early brain development. J Neurosci. 2005;25:6962–6970. doi: 10.1523/JNEUROSCI.1446-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez B, Arends M, Esteve P, Perona R, Sánchez R, Ramón y Cajal S, Wyllie A, Lacal JC. Induction of apoptosis in NIH3T3 cells after serum deprivation by overexpression of rho-p21, a GTPase protein of the ras superfamily. Oncogene. 1995;10:811–816. [PubMed] [Google Scholar]
- Katoh H, Aoki J, Ichikawa A, Negishi M. p160 RhoA-binding kinase ROKalpha induces neurite retraction. J Biol Chem. 1998;273:2489–2492. doi: 10.1074/jbc.273.5.2489. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi M, Matsushita N, Miyazaki J, Koike M, Yaginuma H, Osumi N, Kaibuchi K, Kobayashi K. Survival of developing motor neurons mediated by Rho GTPase signaling pathway through Rho-kinase. J Neurosci. 2004;24:3480–3488. doi: 10.1523/JNEUROSCI.0295-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Lai JM, Hsieh CL, Chang ZF. Caspase activation during phorbol ester-induced apoptosis requires ROCK-dependent myosin-mediated contraction. J Cell Sci. 2003;116:3491–3501. doi: 10.1242/jcs.00660. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrand C, Cases O, Wehrlé R, Blakely RD, Edwards RH, Gaspar P. Transient developmental expression of monoamine transporters in the rodent forebrain. J Comp Neurol. 1998;401:506–524. [PubMed] [Google Scholar]
- Liu JP, Jessell TM. A role for rhoB in the delamination of neural crest cells from the dorsal neural tube. Development. 1998;125:5055–5067. doi: 10.1242/dev.125.24.5055. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Lyck L, Krøigård T, Finsen B. Unbiased cell quantification reveals a continued increase in the number of neocortical neurones during early post-natal development in mice. Eur J Neurosci. 2007;26:1749–1764. doi: 10.1111/j.1460-9568.2007.05763.x. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Miñambres R, Guasch RM, Perez-Aragó A, Guerri C. The RhoA/ROCK-I/MLC pathway is involved in the ethanol-induced apoptosis by anoikis in astrocytes. J Cell Sci. 2006;119:271–282. doi: 10.1242/jcs.02723. [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Pearlman AL. The visual cortex of the normal mouse and the reeler mutant. In: Peters A, Jones EG, editors. Cerebral cortex, Vol 3, Visual cortex. New York: Plenum; 1985. pp. 1–18. [Google Scholar]
- Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Price DJ, Blakemore C. Regressive events in the postnatal development of association projections in the visual cortex. Nature. 1985;316:721–724. doi: 10.1038/316721a0. [DOI] [PubMed] [Google Scholar]
- Purves D. Cambridge, MA: Harvard UP; 1990. Body and brain: a trophic theory of neural connections. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Chen J, McCormick F, Symons M. A role for Rho in Ras transformation. Proc Natl Acad Sci U S A. 1995;92:11781–11785. doi: 10.1073/pnas.92.25.11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami A, Bechmann I, Stehle JH. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog Neurobiol. 2008;85:273–296. doi: 10.1016/j.pneurobio.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P. Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: a study of normal and monoamine oxidase a knock-out mice. J Neurosci. 2002;22:8541–8552. doi: 10.1523/JNEUROSCI.22-19-08541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Roth KA, Kuan C, Haydar TF, D'Sa-Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P. Epistatic and independent functions of caspase-3 and Bcl-X(L) in developmental programmed cell death. Proc Natl Acad Sci U S A. 2000;97:466–471. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp PA, Kulesa PM. A role for RhoA in the two-phase migratory pattern of post-otic neural crest cells. Dev Biol. 2007;311:159–171. doi: 10.1016/j.ydbio.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Renvoizé C, Hamelin J, Riché N, Bertoglio J, Bréard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- Semenova MM, Mäki-Hokkonen AM, Cao J, Komarovski V, Forsberg KM, Koistinaho M, Coffey ET, Courtney MJ. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat Neurosci. 2007;10:436–443. doi: 10.1038/nn1869. [DOI] [PubMed] [Google Scholar]
- Senft SL, Woolsey TA. Growth of thalamic afferents into mouse barrel cortex. Cereb Cortex. 1991;1:308–335. doi: 10.1093/cercor/1.4.308. [DOI] [PubMed] [Google Scholar]
- Shakèd M, Weissmüller K, Svoboda H, Hortschansky P, Nishino N, Wölfl S, Tucker KL. Histone deacetylases control neurogenesis in embryonic brain by inhibition of BMP2/4 signaling. PLoS ONE. 2008;3:e2668. doi: 10.1371/journal.pone.0002668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindler KS, Latham CB, Roth KA. Bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J Neurosci. 1997;17:3112–3119. doi: 10.1523/JNEUROSCI.17-09-03112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreafico R, Frassoni C, Arcelli P, Selvaggio M, De Biasi S. In situ labeling of apoptotic cell death in the cerebral cortex and thalamus of rats during development. J Comp Neurol. 1995;363:281–295. doi: 10.1002/cne.903630209. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Verney C, Takahashi T, Bhide PG, Nowakowski RS, Caviness VS., Jr Independent controls for neocortical neuron production and histogenetic cell death. Dev Neurosci. 2000;22:125–138. doi: 10.1159/000017434. [DOI] [PubMed] [Google Scholar]
- Willaredt MA, Hasenpusch-Theil K, Gardner HA, Kitanovic I, Hirschfeld-Warneken VC, Gojak CP, Gorgas K, Bradford CL, Spatz J, Wölfl S, Theil T, Tucker KL. A crucial role for primary cilia in cortical morphogenesis. J Neurosci. 2008;28:12887–12900. doi: 10.1523/JNEUROSCI.2084-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol. 1999;145:1293–1307. doi: 10.1083/jcb.145.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 1970;17:205–242. doi: 10.1016/0006-8993(70)90079-x. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, Holtzman DM, Roth KA, Olney JW. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003;10:1148–1155. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]
- Zaidi AU, D'Sa-Eipper C, Brenner J, Kuida K, Zheng TS, Flavell RA, Rakic P, Roth KA. Bcl-X(L)-caspase-9 interactions in the developing nervous system: evidence for multiple death pathways. J Neurosci. 2001;21:169–175. doi: 10.1523/JNEUROSCI.21-01-00169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gu X, Yuan X. Phenylalanine activates the mitochondria-mediated apoptosis through the RhoA/Rho-associated kinase pathway in cortical neurons. Eur J Neurosci. 2007;25:1341–1348. doi: 10.1111/j.1460-9568.2007.05404.x. [DOI] [PubMed] [Google Scholar]