

Abstract
Neurons in sensory ganglia are surrounded by satellite glial cells (SGCs) that perform similar functions to the glia found in the CNS. When primary sensory neurons are injured, the surrounding SGCs undergo characteristic changes. There is good evidence that the SGCs are not just bystanders to the injury but play an active role in the initiation and maintenance of neuronal changes that underlie neuropathic pain. In this article the authors review the literature on the relationship between SGCs and nociception and present evidence that changes in SGC potassium ion buffering capacity and glutamate recycling can lead to neuropathic pain-like behavior in animal models. The role that SGCs play in the immune responses to injury is also considered. We propose the term gliopathic pain to describe those conditions in which central or peripheral glia are thought to be the principal generators of principal pain generators.
Keywords: trigeminal ganglion, gap junctions, potassium channel, ATP, glutamate recycling
Our understanding of the role and importance of glial cells in nervous system function has changed a great deal since the original nineteenth century descriptions. The dogma that glial cells are passive players only providing structural support to neurons has long since been replaced by the understanding that there are many different types of glial cell and that they are involved in every function of the brain in both normal and pathological states.
Much of the new understanding of glia and pain comes from the study of CNS glia and less is known about glial cells in the peripheral nervous system (PNS). In the PNS, the Schwann cell might be the first, and for some the only, glial cell that comes to mind, and Schwann cell biology has been extensively studied, particularly in relation to degeneration and regeneration of axons. There are, however, other classes of glial cell in the PNS, most notably the satellite glial cells (SGCs) that surround neuronal somata in sensory and autonomic ganglia. Other glial cells are present in the PNS but are less well characterized and include microglia-like cells and possibly a less differentiated cell type that might be related to Schwann cells and/or SGCs. Although glial cells in the PNS have similar functions to their CNS counterparts, there are enough differences in the morphology, biochemistry, and function between PNS and CNS glia that they appear to be more analogs rather than homologs, and we need to be careful about making too many generalizations.
In addition to playing a role in the function of the normal nervous system, there is now increasing evidence that glial cells play a key role in dysfunction of the nervous system such as in the generation and/or maintenance of pain. In the CNS several key findings have been established. In many, if not all chronic pain models, astrocytes and microglial cells become reactive, as shown by up-regulation in the spinal cord of the glia fibrillary acidic protein (GFAP) and the cell-surface complement receptor 3 (or CD11b or OX-42), respectively Cao and Zhang 2008). Spinal glia activation has been shown in pain models, such as peripheral nerve injury (Raghavendra and others 2003; Sweitzer and others 2001), peripheral inflammation (Watkins and others 1997), and bone cancer (Vit and others 2006b).
In the PNS, Schwann cells have long been known to play an important role in degeneration and regeneration processes following nerve injury. More recently evidence has been accumulating showing that Schwann cells also have a direct role in the generation and maintenance of neuropathic pain (Campana 2007), directly by the secretion of pro-inflammatory cytokines, and indirectly by recruiting immune cells, such as macrophages, to the site of injury through the secretion of chemoattractants. On the other hand, there is good evidence that Schwann cells can, in addition to producing pro-inflammatory mediators, also release factors that facilitate the recovery from chronic pain states (Campana 2007).
In light of the role that CNS glia and PNS Schwann cells play in, or are altered by, pain states we will examine some of the recent findings examining the relationship between pain and PNS glia with emphasis on the least studied of the PNS glia, the SGC.
Overview
The basic cellular organization of sensory ganglia is well known and described in detail in several excellent studies (Hanani and others 2002; Pannese 1981; Pannese and others 1972). In this review we will highlight the major points with regard to sensory ganglia that are pertinent to understanding this review. Similar to the CNS, sensory ganglia contain three general classes of cells, neurons, glia, and peripheral support cells. The two most numerous, and best defined, glial cells in the ganglia are the equivalent of the CNS macroglia and are Schwann cells and the SGCs. The nature of other glial cell types such as microglia is less clear.
Schwann Cells
A distinct, and readily identifiable, glial cell in the primary sensory ganglia is the Schwann cell (Fig 1B). There is a large literature on the structure and function of Schwann cells (Bhatheja and Field 2006; Griffin and Thompson 2008; Jessen and Mirsky 2005; Simons and Trotter 2007). Schwann cells have received a great deal of attention because of their role in nerve pathology (Kuwabara 2004; Nave and others 2007; Ooi and Srinivasan 2004) and as possible repair cells for CNS demyelinating conditions (Jasmin and others 2000; Lavdas and others 2008; Stangel and Hartung 2002; Woodhoo and others 2007). In the PNS, the Schwann cell is a dynamic player and insulates nerve fibers in myelinating and nonmyelinating forms and, following injury, de-differentiates to take part in repair processes including phagocytosis. Similar to the astrocyte/SCG comparison, Schwann cells are compared with the CNS oligodendrocytes in terms of function, but are very different in terms of their morphology and myelination properties. Like the SGCs, Schwann cells are derived from the neural crest. As already noted, Schwann cells are also known to contribute to the inflammatory response that can change the nociceptive thresholds following nerve injury (Martini and others 2008).
Figure 1.
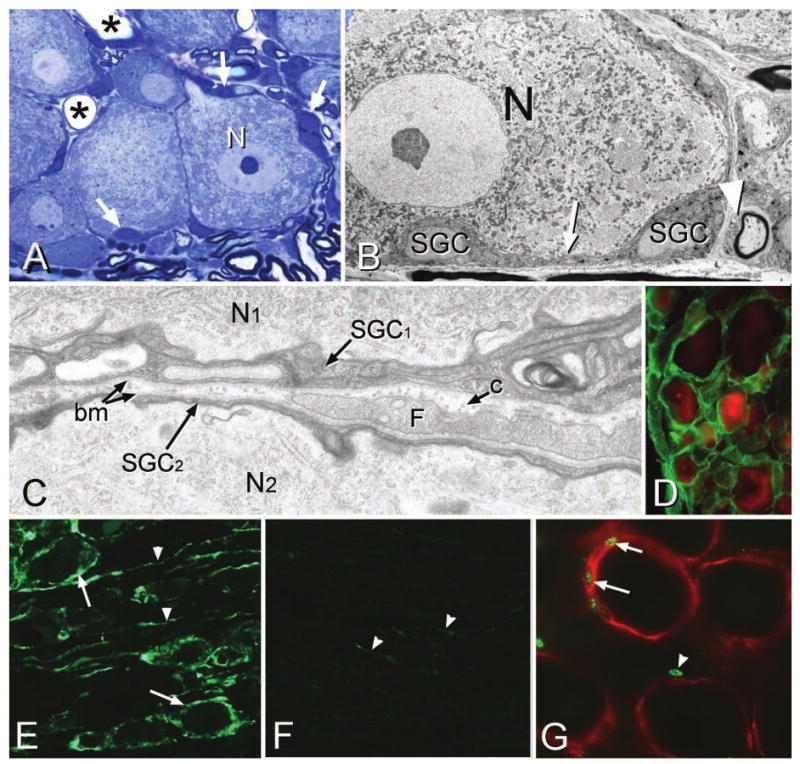
A, Methylene blue semi-thin Epon section showing a large dorsal root ganglion neuron (N) surrounded by small, intensely stained satellite ganglion cells (SGCs, arrows). Only the soma of the SGCs are easily visible at this magnification; the attenuated cytoplasm appears as a thin, dark blue line surrounding the neurons. Asterisks indicate the lumen of capillaries. B, Electron micrograph (EM) showing a neuron (N) and surrounding SGCs. A region of attenuated SGC cytoplasm between the soma is indicated by the arrow. A Schwann cell is present (arrowhead). C, At high magnification the relationship between two neurons (N1, N2) and their surrounding SGCs (SGC1 and SGC2) is seen. Each SGC presents a basement membrane (bm) on its non-neuronal face, and a fibroblast process (F) and collagen fibers (c) occupy the space between the SGCs. D, The basement membrane is present around all SGCs. Green = laminin immunostaining; red = neurons (NeuN). E, Glia fibrillary acidic protein (GFAP) is up-regulated in both SGCs (arrows) and unidentified elongated processes (arrowheads) in the ganglion following nerve injury. F, Only a few faintly stained GFAP elements (arrowheads) are visible in control ganglia. G, Following nerve injury, BrdU-positive nuclei (arrows) are present in SGCs (red, immunolabeled for SK3) surrounding neurons but are also present in non-SGC nuclei (arrowhead) located in regions between SGCs.
Satellite Glial Cells
The neuronal component of sensory ganglia are the cell bodies of the large, spherical, unipolar, dorsal root ganglion neurons and these are surrounded by a layer of SGCs (Fig. 1A–D). The SGCs that surround the sensory neuron somata usually consist of a single layer and are connected to each other by gap junctions. Each SGC is separated from its parent neuron by a gap of about 20 nm and the non-neuronal face of the SGC has a basement membrane (Fig. 1C, D). The SGC is often thought of as the PNS equivalent of the CNS astrocyte, but although astrocytes and SGCs share common functions such as insulation, ion sink and neurotransmitter recycling, the morphology of the two types of cell is quite different. The SGCs have a circular, flattened “tortilla” morphology, whereas the astrocytes are commonly multipolar with very attenuated cytoplasmic extensions. The SGCs share some immunocytochemical similarities to astrocytes but, interestingly, seem to differ in the expression of GFAP, the signature marker of astrocytes. GFAP immunoreactivity is readily detectible in the resting state in astrocytes but is not readily detectable in SGCs, at least by immunohistochemistry. Similar to astrocytes, however, GFAP expression does increase following nerve injury and become detectable by immunocytochemistry (Fig. 1E, F). Both SGCs and astrocytes express many other components in common including ion channels such as the inwardly rectifying K+ channel, Kir4.1, and glutamate recycling components such as the glutamate-aspartate transporter (GLAST).
A key difference between the astrocytes and SGCs is their embryonic origin. SGCs are derived from multipotent neural crest cells (Jessen and Mirsky 2005) whereas astrocytes originate in the ventricular and subventricular zone of the neural plate. The different embryonic origin of the SGCs and astrocytes warns against drawing too many inferences about the other's function based on studies of either cell type alone. Schwann cells (see below) are also derived from the neural crest, a fact that underlies the close relationship between SGCs and Schwann cells (Le Douarin and others 1991). Studies have shown that in several different types of dissociated ganglion culture, SGCs can up-regulate Schwann cell markers such as Schwann cell myelin proteins and the zinc finger transcription factor Krox-20 (Murphy and others 1996) and down-regulate SGC specific markers such as Erm (Hagedorn and others 2000). In the adult, but not in the embryonic stage, the SGC phenotype is probably maintained by the repression of genes such as Krox-20 (Murphy and others 1996).
SGC Response to Injury
Proliferation
Unlike neurons, but like CNS astrocytes, SGCs divide following insult to the peripheral nerve including nerve damage (Fig. 1G; Lu and Richardson 1991, 1993), neuronal viral infection (Elson and others 2003), and skin scarification (Elson and others 2004).
SGC Reactivity and GFAP Up-Regulation
Following injury to the proximal or distal part of a peripheral nerve, there are many changes that occur in ganglion neurons including injury responses, up- and down-regulation of neurotransmitters, changes in neuron phenotype, and sometimes cell death (Devor 1991; Zimmermann 2001).
Along with the neuronal changes following injury, there are also changes in the SGCs. One noticeable change is an increase in GFAP expression in SGCs (Fig. 1E, F). This immediately reminds one of activation of astrocytes in the CNS following injury. This indicates that SGCs do express GFAP, but it is normally only present at levels in nonpathological conditions and below the detectability of immunochemistry. Whether the increase in GFAP in SGCs is associated with the same changes that trigger GFAP increases in astrocytes is not known. One significant finding regarding GFAP expressions comes from studies investigating the role of the neurotransmitter glutamate in neuron-to-glia signaling. In vitro studies show that an increase in extracellular glutamate causes the activation of astrocytes, as measured by a marked increase in GFAP. The glutamate activation of astrocytes is mediated by the type II metabotropic glutamate receptor mGluR 2/3, which is expressed on astrocytes (Romao and others 2008). Because GFAP acts to anchor GLAST at the plasma membrane of glial cells (Sullivan and others 2007), an increase in extracellular glutamate triggers an increase in GLAST, which necessitates the increase in GFAP.
If glutamate also activates SGCs, it is probably via the mGluR8 receptor because this receptor has the highest expression in L5 DRG and is abundant on SGCs whereas the other classes of receptor are less common (Carlton and Hargett 2007). Based on the data shown in astrocytes (Romao and others 2008) the increase in GFAP seen in SGCs after nerve injury may be triggered by increased glutamate released in the sensory ganglion resulting from increased neuronal firing Amir and Devor 2003a, 2003b).
Gap Junctions
The number of gap junctions between SGCs decreases with age (Procacci and others 2008) and increases following nerve injury (Huang and others 2005; Ohara and others 2008; Vit and others 2006a). Because gap junctions are a means for moving molecules between SGCs, both for buffering and signaling, it is reasonable to suppose that changes in gap junctions could cause alterations in the extracellular environment (Fig. 2A, B, D–F). One consequence of changing the extracellular environment would be a change in neuronal excitability and nociception. Because the number of gap junctions increases after nerve injury, it has been speculated that changes in SGC gap junctions are a factor in generating or maintaining neuropathic pain (Cherkas and others 2004; Hanani and others 2002). Recently we were able to show this is indeed the case using RNA interference (RNAi; Ohara and others 2008) to specifically target connexin 43 (Cx43), one of the major structural components of gap junctions expressed by glial cells in the CNS (Nagy and others 1997; Ochalski and others 1997; Yamamoto and others 1990), and in sensory ganglia (Procacci and others 2008; Vit and others 2006a).
Figure 2.
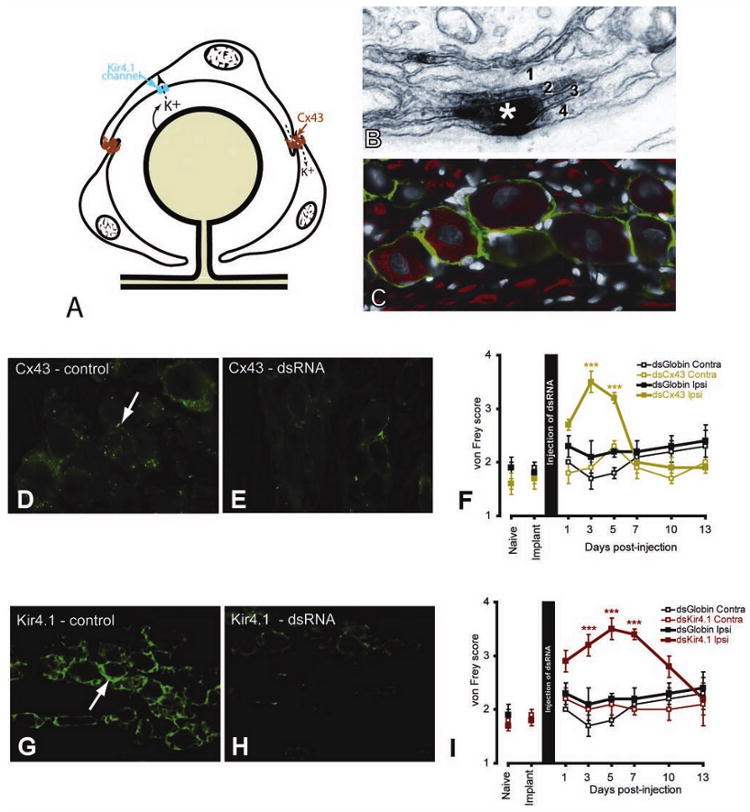
A, Diagram illustrating the role played by connexin 43 (Cx43) in intercellular transport and Kir 4.1 in the uptake of K+ ions. B, Cx43 postembedding electron micrograph staining (asterisk) showing the location of a gap junction between two satellite ganglion cells (SGCs). Where SGCs join there is commonly an interdigitation of processes from the SGCs (here labeled 1, 2, 3, 4). C, In the trigeminal ganglion, SGCs express the glia-specific K+ channel, Kir4.1 (green). Neurons (N) and axons are labeled with NF160 (red). D–I, Immunohistological and behavioral results of using dsRNA against Cx43 (D, E) and Kir4.1 (G–I). For each dsRNA, the control image (D, G) shows staining from an uninjected trigeminal ganglion and the second image (E, H) shows staining from a trigeminal ganglion 5 days after dsRNA injection. von Frey scores were measured for 13 days following injection of the dsRNAs (F, I). In each case there was a transient and reversible increase in von Frey scores for the active dsRNA but not for the control (globin) dsRNA nor on the side contralateral to the injection. *** P < .001, compared with contralateral side. (Additional information on protocols, statistical analysis, and results can be found in Ohara and others 2008; Vit and others 2008)
The effects of reducing Cx43 expression in the ganglion are more complex than we anticipated (Fig. 2D–F). As noted, nerve injury is associated with an increase in Cx43 expression in SGCs (Ohara and others 2008). This is believed to increase the coupling between SGCs (Hanani and others 2002; Huang and others 2005) and the intercellular passage of neuromediators associated with pain and/or glial activation, such as ATP, cAMP, Ca2+, and IP3 (Dina and others 2005). When we injected Cx43 dsRNA into the trigeminal ganglion in a model of facial neuropathic pain, we reduced the number of gap junctions and produced analgesia whereas the number of gap junctions remained unchanged in controls (Ohara and others 2008). These results were in agreement with pharmacological approaches using carbenoloxone, a broad gap junction inhibitor, that has also been shown to produce analgesia in different pain models by inhibiting the coupling between CNS astrocytes (Lan and others 2007; Qin and others 2006; Spataro and others 2004) and glial cells in the sensory ganglion (Lamotte and Ma 2008). A confounding element in these experiments is that it is likely that these gap junction inhibitors also had some peripheral effects (Dublin and Hanani 2007). Unexpectedly, knockdown of Cx43 in uninjured rats resulted in the expression of both spontaneous (increased eye closure) and evoked pain (facial allodynia to von Frey hairs; Fig. 2A-F) and decreased tolerance to innocuous stimulation in an operant conflict paradigm on the injected side. When the expression of Cx43 returned to baseline values, the nociceptive threshold also returned to normal values. The fact that both a decrease or an increase in Cx43 expression in SGCs leads to hyperalgesia highlights the functional complexity of SGCs in the modulation of neuronal excitability.
Ion Channels and K+ Buffering
Recently, it has emerged that severe and characteristic changes in nociceptive processing can result from altered expression of a single neuronal ion channel (Cox and others 2006). We recently showed that silencing a single ion channel in SGC can also result in pain states. One ion channel present on SGCs, but not found on ganglionic neuron, is the inwardly rectifying potassium channel, Kir4.1 (Fig. 2A, C). We produced reversible silencing of Kir4.1 in SGCs by directly injecting double-stranded RNA against Kir4.1 (Fig. 2H) into the trigeminal ganglion (Vit and others 2008). Silencing Kir4.1 expression resulted in the appearance of both spontaneous (increased eye closure) and evoked pain (facial allodynia to von Frey hairs, Fig. 2I) and decreased tolerance to innocuous stimulation in an operant conflict paradigm on the side of the injection. When Kir4.1 expression returned to baseline levels, normal pain behavior was restored. The relevance of this result to chronic pain is highlighted by the finding that after nerve injury, Kir4.1 expression in SGCs is reduced. Similarly, Kir4.1 expression is decreased centrally after severe crush injury to the spinal cord (Olsen and Sontheimer 2008), a condition also associated with chronic pain (Siddall and Loeser 2001).
Based on our previous observation, we hypothesize that when neuronal excitation occurs, K+ is released in the perineuronal environment and the increased extracellular K+ leads to increased neuronal excitability. Such hyperexcitability is associated with increased or spontaneous pain sensation. Currently we are measuring changes in neuronal excitability in the ganglia that lack Kir4.1 following dsRNA treatment, to validate this hypothesis. Kir4.1 is not only involved in K+ homeostasis but also in the uptake of glutamate by glial cells. Using RNAi, the suppression of Kir4.1 expression was shown to reduce glutamate uptake in cultured astrocytes in vitro (Kucheryavykh and others 2007). Moreover, reduction of glutamate uptake has been shown in astrocyte-specific Kir4.1 knockout mice in vivo (Djukic and others 2007). Although we did not measure extracellular glutamate levels after transient inhibition of Kir4.1 dsRNA treatment, it is possible that glutamate levels were increased, which is also expected to cause increased neuronal excitability. Thus the loss of Kir4.1 probably affects the clearance of both K+ and glutamate, which are two of the main functions of astrocytes and SGCs in the maintenance of neuronal excitability. Finally, it is worth mentioning that a recent genetic study identified Kir4.1 as an essential component for the full effect of analgesic drugs (Smith and others 2008).
Glutamate Recycling
Another important function of astrocytes is to recycle extracellular glutamate (Fig. 3A) (Bak and others 2006). SGCs in peripheral ganglia appear to have similar capabilities. In the CNS, astrocytes express at least two glial specific glutamate transporters, GLAST and glutamate transporter-1 (GLT-1), are involved in the uptake of glutamate released by neurons at synapses. Once in the astrocyte, glutamate is amidated to glutamine by the enzyme glutamine synthetase. Glutamine is returned to neurons where phosphate-activated glutaminase converts glutamine to glutamate. Thus the astrocytes play the main role in the clearance of glutamate from the extracellular space and as a consequence modulate neuronal activity. In peripheral sensory ganglia, SGCs have been shown to express both GLAST and glutamine synthetase, suggesting that these cells might have a role in the clearance of perineuronal glutamate.
Figure 3.
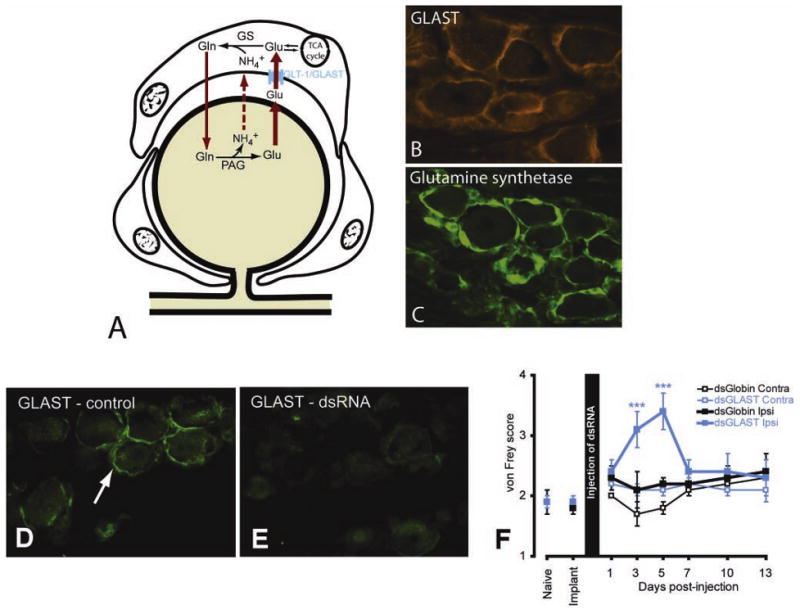
A, Diagram illustrating the role of satellite ganglion cells (SGCs) in glutamate recycling. The glial glutamate transporter (GLAST) transports extracellular glutamate (Glu) into the SGC where glutamine synthetase (GS) converts glutamate to glutamine (Gln), which is eventually recycled to the neuron for conversion into glutamate. In the trigeminal ganglion, SGCs express GLAST (B, red) and glutamine synthetase (C, green). D, GLAST immunostaining of SGC from a control ganglion (green, arrow). E, Five days following injection of GLAST dsRNA into the trigeminal ganglion there is an obvious reduction of GLAST immunostaining. F, von Frey scores for 13 days following injection of the dsRNAs into the trigeminal ganglion. There was an increase in von Frey scores for the GLAST dsRNA but no increase in von Frey score was observed for the control (globin) dsRNA, nor on the side contralateral to the injection. *** P < .001, compared with contralateral side.
Although we and others have shown that SGCs express GLAST and glutamine synthetase (Fig. 3B, C), it is not known if glutamate is released by neurons within the ganglia. Other neuromediators such as ATP and substance P have been shown to be released by ganglion cell bodies (Burnstock 2007; Takeda and others 2005; Zarei and others 2004), and this together with the presence of the entire machinery for the glutamate-glutamine cycle in SGCs suggests that glutamate is released in the ganglion and that SGCs may play a key role in modulating glutamate homeostasis. In support of this idea, we found that reducing the expression of GLAST in the trigeminal ganglia using RNAi (Fig. 3D, E) correlated with increased evoked pain, in the form of facial allodynia to mechanical stimulation (Fig. 3F). The mechanism underlying this sensory change is currently under investigation, but we believe that a lack of glutamate clearance from the perineuronal environment by SGCs is involved. An increase in extracellular glutamate brought about by reduced uptake by SGCs would be predicted to increase neuronal excitability through the activation of glutamate receptors.
Our results are in agreement with several studies reporting a role for glutamate transporters in different pain models. Nerve injury has been shown to lead to a decrease in expression of astrocytic GLAST and GLT-1 in the spinal cord (Sung and others 2003). This decrease in glutamate transporter expression is accompanied by reduced glutamate uptake and an increase in extracellular glutamate, which in turn is associated with neuropathic pain behavior (Sung and others 2003). Another study examined the expression of glial glutamate transporters in the spinal cord of rats with chemotherapy-induced hyperalgesia (Weng and others 2005). Taxol-treated rats showed a significant decrease of both GLAST and GLT-1 expression, suggesting that glutamate transporters play a role in the development of hyperalgesia. Further studies are needed to understand the underlying mechanism by which excess glutamate leads to chronic pain, given that glutamate receptors are also present on glial cells and not just on neurons (Porter and McCarthy 1997).
ATP Signaling
An important, but not obvious, underlying concept when discussing SGCs is that these cells are rarely subject to direct injury. Rather, it is the neurons that are injured by damage to the central or peripheral part of the axon. Thus, any change we see in SGCs must be a secondary change driven by alterations to the neuron, and this implies some active signaling mechanism between neurons and SGCs. There are, in fact, a large number of molecules that could be involved in such signaling and many substances are released from injured (and noninjured) neurons, such as nitric oxide, tumor necrosis factor-α (TNF-α), and ATP.
ATP appears to be involved in pain-related processing through the activation of metabotropic (P2Y family) or ionotropic (P2X) purinergic receptors. SGCs express several different purinergic receptors, and there is evidence that these receptors are involved in neuron-to-SGC communication and more specifically are involved in nociceptive related processes. The P2Y4 receptor has been shown to be exclusively expressed by SGCs in sensory ganglia (Vit and others 2006a; Weick and others 2003). ATP released following nerve injury may activate P2Y4 receptors on SGCs resulting in an increase in intracellular Ca2+ (Fig. 4), which, in turn, can trigger activation of K+ channels. The resulting change in extracellular K+ may result in increased nociceptive neuron excitability (Filippov and others 2004; Weick and others 2003; see section on K+ buffering above).
Figure 4.
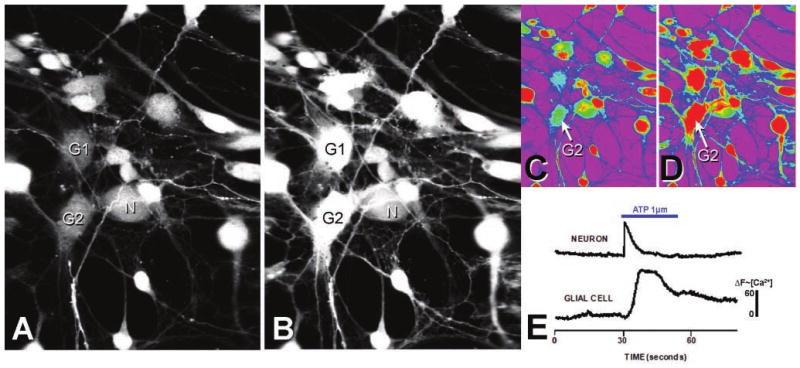
Calcium imaging of dissociated neurons and glia from the trigeminal ganglion. Dissociated trigeminal neurons (N) and glial cells (G1, G2) loaded with the calcium indicator Fluo-4 are shown before (A) and after (B) addition of 1 μM of ATP. C, D, Pseudocolor representation of the calcium intensity of A and B, respectively. Green indicated low and red indicated high calcium concentration. E, Shows the differences in the time course of calcium influx between a representative neuron and a glial cell after ATP application.
More recent studies have focused on the role of the P2X7 receptors in SGCs and in astrocytes. P2X7 receptors are expressed in the spinal cord and sensory ganglia (Kobayashi and others 2005; North 2002) and have been linked to excitotoxicity and nociception (Dublin and Hanami 2007; Sperlagh and others 2006). It has been shown recently that P2X7 receptors are expressed only by SGCs in DRGs and that activation of P2X7 receptors reduced nocifensive behavior and allodynia after complete Freund's adjuvant–induced inflammation (Chen and others 2008). P2X7 receptor–triggered analgesia is dependent on the down-regulation of P2X3 receptors in nociceptive neurons (Chen and others 2008).
An interesting finding relating to the glutamate studies detailed above is from CNS studies showing that P2X7 receptors are involved in intercellular Ca2+ signaling between astrocytes and neurons not only in response to ATP but also in response to glutamate (Hamilton and others 2008; Suadicani and others 2006). In addition, activation of P2X7 receptors decreases glutamate uptake and glutamine synthetase activity in astrocytes (Lo and others 2008).
In addition to the pathways discussed above, there are other routes by which ATP may be involved in nociceptive processing such as by stimulating the secretion of pro-inflammatory cytokines by SGCs (Inoue 2006). It also must be remembered that purinergic receptors are not confined to SGCs. Other purinergic receptors such as the P2X3 receptor are expressed by nociceptive neurons (Xiang and others 1998) and have been shown to play a role in pain processing (Cook and others 1997; Souslova and others 2000), as they become sensitized in response to inflammation or nerve injury (Chen and others 2005; Xu and Huang 2002).
Altogether, these studies reveal the importance of ATP as a mediator of pain acting on both neurons and glial cells and the understanding that the effect of ATP depends on the intricate relationship between both P2Y and P2X receptor families that is expressed on neuronal and glial cells.
Immune System in the Sensory Ganglion
The sensory ganglion is in a unique position to interface with the immune system inasmuch as there is no blood-brain barrier as found in the CNS. Similar to blood vessels outside the CNS, the endothelial cells of blood vessels within ganglia have no tight junctions (Jacobs and others 1976), with the consequence that sensory ganglion cells are able to detect distant events such as immune phenomena signaled by circulating peptides such as cytokines. Immune cells, in general, and macrophages, in particular, are able to move more freely in and out of the ganglion compared with almost any other part of the nervous system (Hu and McLachlan 2002). This rapid and unrestricted movement of immune cell into ganglia may represent an adaptive mechanism for rapid response to viral infection of sensory neurons. Although in the PNS there is no blood-brain barrier as defined in the CNS, sensory ganglion neurons are separated and protected from circulating molecules by blood vessel endothelial cell basement membrane, SGC basement membrane, and the SGCs themselves (Fig. 5A–C). The SGC basement membrane is recognized as an important part of the blood/ganglion-barrier, and anomalies of the extracellular matrix are associated with abnormal neuronal function (Kaksonen and others 2002) and pain (Previtali and others 2008).
Figure 5.
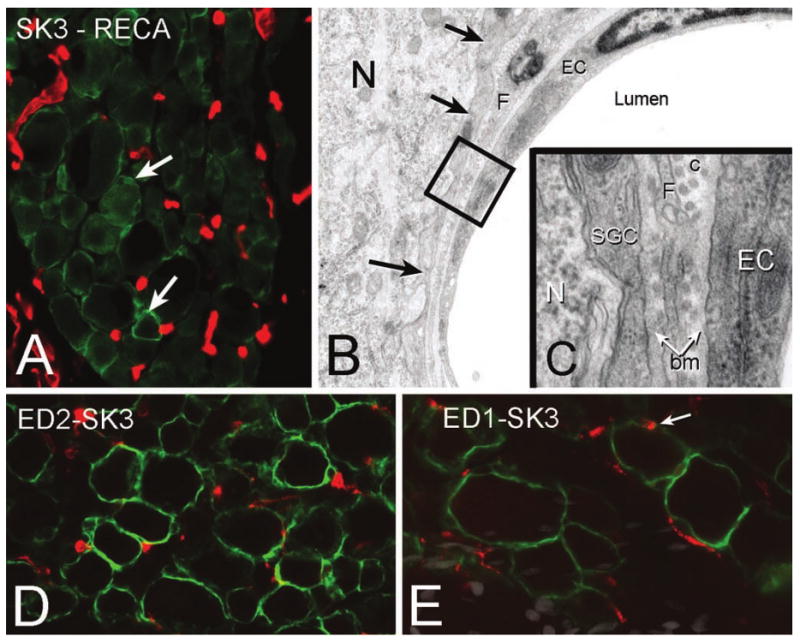
A, Section of the trigeminal ganglion immunostained for satellite ganglion cells (SGCs) (SK3, green; e.g., arrows) and endothelial cells (RECA, red) to show the plexus of capillaries. B, Electron micrograph showing the relationship between a capillary (EC = endothelial cell), SGC (arrows), and neuron (N). The area enclosed by the black square is shown at high magnification in C. bm = basement membrane; c = collagen; F = fibroblast. D, In the normal ganglion there is a population of resident macrophages as shown by ED2 immunolabeling (red) scattered throughout the ganglion between the SGCs (SK3, green). E, Following a chronic constriction injury to the infraorbital nerve, there is an increase in activated macrophages (ED-1, red). In some cases SGCs identified by SK3 immunolabeling (green) also appear to express ED-1 (arrow).
Each sensory ganglion harbors a large number of resident macrophages (Dijkstra and Damoiseaux 1993; Fig. 5D). Macrophages are considered the equivalent of CNS microglial cells (Mori and others 2003) and play a key role in regulating a number of pain conditions (Milligan and Watkins 2009). Resident macrophages are the first immune cells to respond to nerve injury, viral infection, and to foreign oligonucleotides, but within days, circulating macrophages, polymorphonuclear, and T lymphocytes invade the sensory ganglion (Hu and McLachlan 2002; Morin and others 2007). Following an immune system trigger, the number of macrophage in the sensory ganglion will peak at 3 to 7 days and can persist for weeks (Xie and others 2006). This persistence of macrophages in the ganglion can occur in the absence of any phagocytic event, for instance, in the presence of peripheral inflammation, and might favor regenerative processes but also ongoing pain (Milligan and Watkins 2009).
Activated macrophages are prevalent in all immune responses involving the sensory ganglion as well as for inflammation occurring in the vicinity of the ganglion (Dubovy and others 2007; Mori and others 2003; Xie and others 2006; Zhu and others 2005). Strikingly, signals from neurons can trigger invasion of the ganglion by macrophages, even when the injury or demyelination to the nerve is remote from the ganglion (Hu and McLachlan 2002). In the ganglion, activated macrophages divide and persist much longer than in peripheral tissue, contributing to the persistence of neuropathic pain (Hu and McLachlan 2002; Schreiber and others 2002). Activated macrophages respond to a mix of molecules including neurotransmitters, growth factors, and cytokines, most of which have been shown to cause increased pain behavior in animal models. Cytokines known to elicit this pain behavior include interlukin-1β (IL-1β), IL-6, IL-8, and MCP-1 (Xie and others 2006). MCP-1 is critical in attracting circulating macrophages by acting on the chemotactic cytokine receptor 2 (CCR2). Following nerve injury, the expression of MCP-1 and its receptor increases markedly in sensory ganglia. Reduction in the number of macrophages or antagonism of CCR2 blocks the appearance and continued expression of neuropathic pain (Abbadie and others 2003; Liu and others 2000), whereas stimulation of CCR2 is hyperalgesic (White and others 2005). These data suggest a pivotal role for macrophages in combating pain.
SGCs exhibit features of inflammatory cells. For instance, SGCs release pro-nociceptive peptides (Mori and others 2003). Similar to macrophages, SGCs are activated by MCP-1 through the CCR2 receptor (Jeon and others 2008; Lamotte and Ma 2008; Tanaka and others 2004). Intrathecal injection of MCP-1 induces tactile allodynia (Tanaka and others 2004), an effect that could be mediated by both the SGCs and resident macrophages. Once activated, SGCs, like macrophages, produce TNF-α (Dubovy and others 2006) and TNF-α activates SGCs causing an increase in phosphorylated extracellular signal-regulated protein kinase (Perk; Takahashi and others 2006). After nerve injury, the long-lasting increase of pERK in SGCs has been associated with chronic pain (Doya and others 2005). Another way in which SGCs behave similarly to immune cells is that activated SGCs also produce high levels of IL-1β (Takeda and others 2007), which in the trigeminal ganglion increases the firing rate of nociceptive neurons. Infusion of an IL-1β receptor antagonist blocks the pain behavior associated with peripheral inflammation by restoring the threshold of nociceptive neurons (Takeda and others 2008).
Some reports have suggested that following peripheral inflammation or nerve injury, activated macrophages take up position around the perikarya of sensory neurons, displacing or replacing SGCs (Dubovy and others 2007; Mori and others 2003). The basis for this interpretation is the increase in perineuronal cells expressing ED-1, an activating macrophage marker (Fig. 5E) in nerve-injured ganglia (Dubovy and others 2007), or Iba1 (ionized calcium binding adapter molecule 1, a specific marker of microglia/macrophages; Mori and others 2003) following herpes simplex virus (HSV) injection. However, neither study included specific immunocytochemical markers to identify SCGs, so it is not clear whether the SGCs had been replaced by macrophages or whether the resident SGCs are themselves capable of up-regulating ED-1 and Iba1. We think the latter possibility is more likely, but in either case the close proximity to neurons of cells that can release a number of inflammatory mediators (Moalem and Tracey 2006) will decrease the nociceptive threshold and cause an increase in spontaneous activity of small fibers in the dorsal root (Xie and others 2006). Increased neuronal activity is probably mediated, in part, by an increase in extracellular glutamate because of a secondary down-regulation of SGC glutamate transporters (Sung and others 2003).
Endothelial Cells
The vascular endothelium also plays a critical role in the immune response impacting glial function and pain through the release of pro-nociceptive mediators and by attracting inflammatory cells (Willis and Davis 2008). Capillaries in sensory ganglia lie in close proximity to SGCs (Fig. 5A–C). Following nerve injury, angiogenic factors such as calcitonin gene-related peptide, nerve growth factor, and vascular endothelial growth factor released by neurons and SGCs (Chao and others 2008; Kutcher and others 2004; Pannese and Procacci 2002) cause an increase in vascularization of the sensory ganglion (Lamotte and Ma 2008). In turn, endothelial cells release molecules, such as cytokines and adhesion molecules, that attract macrophages and other leukocytes to the sensory ganglion (Han and Suk 2005). Many of the factors released by the vascular endothelial cells, and possibly by adjacent fibroblasts, will activate SGCs.
Conclusion
It is well established that SGCs undergo phenotypic changes following nerve injury. It is less clear how the observed phenotypic changes in SGCs might affect neuronal function or sensory perception in normal or injured states. The preponderance of experimental evidence and theoretical considerations, however, support the notion that changes in SGCs, alone or in concert with changes in neuronal function, do affect nociception. Thus the evidence that both PNS and CNS glia play an integral, and possibly primary, role in some types of pain leads us to suggest that the term “gliopathic pain” is appropriate for such conditions. The recognition that we must include both central and peripheral glial cells in constructing our models of sensory mechanisms certainly complicates our task in understanding nociception but also provides more targets for therapeutic interventions aimed at reducing pain.
Footnotes
For reprints and permissions queries, please visit SAGE's Web site at http://www.sagepub.com/journalsPermissions.nav.
Contributor Information
Peter T. Ohara, Department of Anatomy, University of California, San Francisco, California
Jean-Philippe Vit, Department of Anatomy, University of California, San Francisco, California.
Aditi Bhargava, Department of Surgery, University of California, San Francisco, California.
Marcela Romero, UCLA Headache Research and Treatment Program, David Geffen School of Medicine, University of California, Los Angeles, California.
Christopher Sundberg, Department of Neurosurgery & Gene Therapeutics Research Institute, Cedars-Sinai Medical Center, Los Angeles, California.
Andrew C. Charles, UCLA Headache Research and Treatment Program, David Geffen School of Medicine, University of California, Los Angeles, California
Luc Jasmin, Department of Anatomy, University of California, San Francisco, California, Los Angeles Neurosurgical Institute, Beverly Hills, California.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–52. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir R, Devor M. Electrical excitability of the soma of sensory neurons is required for spike invasion of the soma, but not for through-conduction. Biophys J. 2003a;84:2181–91. doi: 10.1016/S0006-3495(03)75024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir R, Devor M. Extra spike formation in sensory neurons and the disruption of afferent spike patterning. Biophys J. 2003b;84:2700–8. doi: 10.1016/S0006-3495(03)75075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98:641–53. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- Bhatheja K, Field J. Schwann cells: origins and role in axonal maintenance and regeneration. Int J Biochem Cell Biol. 2006;38:1995–9. doi: 10.1016/j.biocel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87:659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- Campana WM. Schwann cells: activated peripheral glia and their role in neuropathic pain. Brain Behav Immun. 2007;21:522–7. doi: 10.1016/j.bbi.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Zhang YQ. Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev. 2008;32:972–83. doi: 10.1016/j.neubiorev.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Hargett GL. Colocalization of metabotropic glutamate receptors in rat dorsal root ganglion cells. J Comp Neurol. 2007;501:780–9. doi: 10.1002/cne.21285. [DOI] [PubMed] [Google Scholar]
- Chao T, Pham K, Steward O, Gupta R. Chronic nerve compression injury induces a phenotypic switch of neurons within the dorsal root ganglia. J Comp Neurol. 2008;506:180–93. doi: 10.1002/cne.21537. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li GW, Wang C, Gu Y, Huang LY. Mechanisms underlying enhanced P2X receptor-mediated responses in the neuropathic pain state. Pain. 2005;119:38–48. doi: 10.1016/j.pain.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang X, Wang C, Li G, Gu Y, Huang LY. Activation of P2X7 receptors in glial satellite cells reduces pain through downregulation of P2X3 receptors in nociceptive neurons. Proc Natl Acad Sci U S A. 2008;105:16773–8. doi: 10.1073/pnas.0801793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkas PS, Huang TY, Pannicke T, Tal M, Reichenbach A, Hanani M. The effects of axotomy on neurons and satellite glial cells in mouse trigeminal ganglion. Pain. 2004;110:290–8. doi: 10.1016/j.pain.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Cook SP, Vulchanova L, Hargreaves KM, Elde R, McCleskey EW. Distinct ATP receptors on pain-sensing and stretch-sensing neurons. Nature. 1997;387:505–8. doi: 10.1038/387505a0. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–8. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M. Chronic pain in the aged: possible relation between neurogenesis, involution and pathophysiology in adult sensory ganglia. J Basic Clin Physiol Pharmacol. 1991;2:1–15. doi: 10.1515/jbcpp.1991.2.1-2.1. [DOI] [PubMed] [Google Scholar]
- Dijkstra CD, Damoiseaux JG. Macrophage heterogeneity established by immunocytochemistry. Prog Histochem Cytochem. 1993;27:1–65. doi: 10.1016/s0079-6336(11)80067-7. [DOI] [PubMed] [Google Scholar]
- Dina OA, Hucho T, Yeh J, Malik-Hall M, Reichling DB, Levine JD. Primary afferent second messenger cascades interact with specific integrin subunits in producing inflammatory hyperalgesia. Pain. 2005;115:191–203. doi: 10.1016/j.pain.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–65. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doya H, Ohtori S, Takahashi K, Aoki Y, Ino H, Takahashi Y, et al. Extracellular signal-regulated kinase mitogen-activated protein kinase activation in the dorsal root ganglion (DRG) and spinal cord after DRG injury in rats. Spine. 2005;30:2252–6. doi: 10.1097/01.brs.0000182091.53834.08. [DOI] [PubMed] [Google Scholar]
- Dublin P, Hanani M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav Immun. 2007;21:592–8. doi: 10.1016/j.bbi.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Dubovy P, Jancalek R, Klusakova I, Svizenska I, Pejchalova K. Intra- and extraneuronal changes of immunofluorescence staining for TNF-alpha and TNFR1 in the dorsal root ganglia of rat peripheral neuropathic pain models. Cell Mol Neurobiol. 2006;26:1205–17. doi: 10.1007/s10571-006-9006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubovy P, Tuckova L, Jancalek R, Svizenska I, Klusakova I. Increased invasion of ED-1 positive macrophages in both ipsi- and contralateral dorsal root ganglia following unilateral nerve injuries. Neurosci Lett. 2007;427:88–93. doi: 10.1016/j.neulet.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Elson K, Simmons A, Speck P. Satellite cell proliferation in murine sensory ganglia in response to scarification of the skin. Glia. 2004;45:105–9. doi: 10.1002/glia.10294. [DOI] [PubMed] [Google Scholar]
- Elson K, Speck P, Simmons A. Herpes simplex virus infection of murine sensory ganglia induces proliferation of neuronal satellite cells. J Gen Virol. 2003;84:1079–84. doi: 10.1099/vir.0.19035-0. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Fernandez-Fernandez JM, Marsh SJ, Simon J, Barnard EA, Brown DA. Activation and inhibition of neuronal G protein-gated inwardly rectifying K(+) channels by P2Y nucleotide receptors. Mol Pharmacol. 2004;66:468–77. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- Griffin JW, Thompson WJ. Biology and pathology of nonmyelinating Schwann cells. Glia. 2008;56:1518–31. doi: 10.1002/glia.20778. [DOI] [PubMed] [Google Scholar]
- Hagedorn L, Paratore C, Brugnoli G, Baert JL, Mercader N, Suter U, et al. The Ets domain transcription factor Erm distinguishes rat satellite glia from Schwann cells and is regulated in satellite cells by neuregulin signaling. Dev Biol. 2000;219:44–58. doi: 10.1006/dbio.1999.9595. [DOI] [PubMed] [Google Scholar]
- Hamilton N, Vayro S, Kirchhoff F, Verkhratsky A, Robbins J, Gorecki DC, et al. Mechanisms of ATP- and glutamate-mediated calcium signaling in white matter astrocytes. Glia. 2008;56:734–49. doi: 10.1002/glia.20649. [DOI] [PubMed] [Google Scholar]
- Han HS, Suk K. The function and integrity of the neurovascular unit rests upon the integration of the vascular and inflammatory cell systems. Curr Neurovasc Res. 2005;2:409–23. doi: 10.2174/156720205774962647. [DOI] [PubMed] [Google Scholar]
- Hanani M, Huang TY, Cherkas PS, Ledda M, Pannese E. Glial cell plasticity in sensory ganglia induced by nerve damage. Neuroscience. 2002;114:279–83. doi: 10.1016/s0306-4522(02)00279-8. [DOI] [PubMed] [Google Scholar]
- Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience. 2002;112:23–38. doi: 10.1016/s0306-4522(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Huang TY, Cherkas PS, Rosenthal DW, Hanani M. Dye coupling among satellite glial cells in mammalian dorsal root ganglia. Brain Res. 2005;1036:42–9. doi: 10.1016/j.brainres.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Inoue K. The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol Ther. 2006;109:210–26. doi: 10.1016/j.pharmthera.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Jacobs JM, Macfarlane RM, Cavanagh JB. Vascular leakage in the dorsal root ganglia of the rat, studied with horseradish peroxidase. J Neurol Sci. 1976;29:95–107. doi: 10.1016/0022-510x(76)90083-6. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Janni G, Moallem TM, Lappi DA, Ohara PT. Schwann cells are removed from the spinal cord after effecting recovery from paraplegia. J Neurosci. 2000;20:9215–23. doi: 10.1523/JNEUROSCI.20-24-09215.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Lee KM, Park ES, Jeon YH, Cho HJ. Monocyte chemoattractant protein-1 immunoreactivity in sensory ganglia and hindpaw after adjuvant injection. Neuroreport. 2008;19:183–6. doi: 10.1097/WNR.0b013e3282f3c781. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–82. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Pavlov I, Voikar V, Lauri SE, Hienola A, Riekki R, et al. Syndecan-3-deficient mice exhibit enhanced LTP and impaired hippocampus-dependent memory. Mol Cell Neurosci. 2002;21:158–72. doi: 10.1006/mcne.2002.1167. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Fukuoka T, Yamanaka H, Dai Y, Obata K, Tokunaga A, et al. Differential expression patterns of mRNAs for P2X receptor subunits in neurochemically characterized dorsal root ganglion neurons in the rat. J Comp Neurol. 2005;481:377–90. doi: 10.1002/cne.20393. [DOI] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, et al. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–81. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- Kutcher ME, Klagsbrun M, Mamluk R. VEGF is required for the maintenance of dorsal root ganglia blood vessels but not neurons during development. FASEB J. 2004;18:1952–4. doi: 10.1096/fj.04-2320fje. [DOI] [PubMed] [Google Scholar]
- Kuwabara S. Guillain-Barre syndrome: epidemiology, pathophysiology and management. Drugs. 2004;64:597–610. doi: 10.2165/00003495-200464060-00003. [DOI] [PubMed] [Google Scholar]
- Lamotte RH, Ma C. Hyperexcitable neurons and altered non-neuronal cells in the compressed spinal ganglion. Sheng Li Xue Bao. 2008;60:597–602. [PMC free article] [PubMed] [Google Scholar]
- Lan L, Yuan H, Duan L, Cao R, Gao B, Shen J, et al. Blocking the glial function suppresses subcutaneous formalin-induced nociceptive behavior in the rat. Neurosci Res. 2007;57:112–9. doi: 10.1016/j.neures.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Papastefanaki F, Thomaidou D, Matsas R. Schwann cell transplantation for CNS repair. Curr Med Chem. 2008;15:151–60. doi: 10.2174/092986708783330593. [DOI] [PubMed] [Google Scholar]
- Le Douarin N, Dulac C, Dupin E, Cameron-Curry P. Glial cell lineages in the neural crest. Glia. 1991;4:175–84. doi: 10.1002/glia.440040209. [DOI] [PubMed] [Google Scholar]
- Liu T, van Rooijen N, Tracey DJ. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain. 2000;86:25–32. doi: 10.1016/s0304-3959(99)00306-1. [DOI] [PubMed] [Google Scholar]
- Lo JC, Huang WC, Chou YC, Tseng CH, Lee WL, Sun SH. Activation of P2X(7) receptors decreases glutamate uptake and glutamine synthetase activity in RBA-2 astrocytes via distinct mechanisms. J Neurochem. 2008;105:151–64. doi: 10.1111/j.1471-4159.2007.05119.x. [DOI] [PubMed] [Google Scholar]
- Lu X, Richardson PM. Inflammation near the nerve cell body enhances axonal regeneration. J Neurosci. 1991;11:972–8. doi: 10.1523/JNEUROSCI.11-04-00972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Richardson PM. Responses of macrophages in rat dorsal root ganglia following peripheral nerve injury. J Neurocytol. 1993;22:334–41. doi: 10.1007/BF01195557. [DOI] [PubMed] [Google Scholar]
- Martini R, Fischer S, Lopez-Vales R, David S. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia. 2008;56:1566–77. doi: 10.1002/glia.20766. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006;51:240–64. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Mori I, Goshima F, Koshizuka T, Imai Y, Kohsaka S, Koide N, et al. Iba1-expressing microglia respond to herpes simplex virus infection in the mouse trigeminal ganglion. Brain Res Mol Brain Res. 2003;120:52–6. doi: 10.1016/j.molbrainres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Morin N, Owolabi SA, Harty MW, Papa EF, Tracy TF, Jr, Shaw SK, et al. Neutrophils invade lumbar dorsal root ganglia after chronic constriction injury of the sciatic nerve. J Neuroimmunol. 2007;184:164–71. doi: 10.1016/j.jneuroim.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Murphy P, Topilko P, Schneider-Maunoury S, Seitanidou T, Baron-Van Evercooren A, Charnay P. The regulation of Krox-20 expression reveals important steps in the control of peripheral glial cell development. Development. 1996;122:2847–57. doi: 10.1242/dev.122.9.2847. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Ochalski PA, Li J, Hertzberg EL. Evidence for the co-localization of another connexin with connexin-43 at astrocytic gap junctions in rat brain. Neuroscience. 1997;78:533–48. doi: 10.1016/s0306-4522(96)00584-2. [DOI] [PubMed] [Google Scholar]
- Nave KA, Sereda MW, Ehrenreich H. Mechanisms of disease: inherited demyelinating neuropathies—from basic to clinical research. Nat Clin Pract Neurol. 2007;3:453–64. doi: 10.1038/ncpneuro0583. [DOI] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–67. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Ochalski PA, Frankenstein UN, Hertzberg EL, Nagy JI. Connexin-43 in rat spinal cord: localization in astrocytes and identification of heterotypic astro-oligodendrocytic gap junctions. Neuroscience. 1997;76:931–45. doi: 10.1016/s0306-4522(96)00394-6. [DOI] [PubMed] [Google Scholar]
- Ohara PT, Vit JP, Bhargava A, Jasmin L. Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. J Neurophysiol. 2008;100:3064–73. doi: 10.1152/jn.90722.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem. 2008;107:589–601. doi: 10.1111/j.1471-4159.2008.05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi WW, Srinivasan J. Leprosy and the peripheral nervous system: basic and clinical aspects. Muscle Nerve. 2004;30:393–409. doi: 10.1002/mus.20113. [DOI] [PubMed] [Google Scholar]
- Pannese E. The satellite cells of the sensory ganglia. Adv Anat Embryol Cell Biol. 1981;65:1–111. doi: 10.1007/978-3-642-67750-2. [DOI] [PubMed] [Google Scholar]
- Pannese E, Bianchi R, Calligaris B, Ventura R, Weibel ER. Quantitative relationships between nerve and satellite cells in spinal ganglia. An electron microscopical study. I. Mammals. Brain Res. 1972;46:215–34. doi: 10.1016/0006-8993(72)90017-0. [DOI] [PubMed] [Google Scholar]
- Pannese E, Procacci P. Ultrastructural localization of NGF receptors in satellite cells of the rat spinal ganglia. J Neurocytol. 2002;31:755–63. doi: 10.1023/a:1025708132119. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Astrocytic neurotransmitter receptors in situ and in vivo. Prog Neurobiol. 1997;51:439–55. doi: 10.1016/s0301-0082(96)00068-8. [DOI] [PubMed] [Google Scholar]
- Previtali SC, Malaguti MC, Riva N, Scarlato M, Dacci P, Dina G, et al. The extracellular matrix affects axonal regeneration in peripheral neuropathies. Neurology. 2008;71:322–31. doi: 10.1212/01.wnl.0000319736.43628.04. [DOI] [PubMed] [Google Scholar]
- Procacci P, Magnaghi V, Pannese E. Perineuronal satellite cells in mouse spinal ganglia express the gap junction protein connexin43 throughout life with decline in old age. Brain Res Bull. 2008;75:562–9. doi: 10.1016/j.brainresbull.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Qin M, Wang JJ, Cao R, Zhang H, Duan L, Gao B, et al. The lumbar spinal cord glial cells actively modulate subcutaneous formalin induced hyperalgesia in the rat. Neurosci Res. 2006;55:442–50. doi: 10.1016/j.neures.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–30. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Romao LF, Sousa Vde O, Neto VM, Gomes FC. Glutamate activates GFAP gene promoter from cultured astrocytes through TGF-beta1 pathways. J Neurochem. 2008;106:746–56. doi: 10.1111/j.1471-4159.2008.05428.x. [DOI] [PubMed] [Google Scholar]
- Schreiber RC, Vaccariello SA, Boeshore K, Shadiack AM, Zigmond RE. A comparison of the changes in the non-neuronal cell populations of the superior cervical ganglia following decentralization and axotomy. J Neurobiol. 2002;53:68–79. doi: 10.1002/neu.10093. [DOI] [PubMed] [Google Scholar]
- Siddall PJ, Loeser JD. Pain following spinal cord injury. Spinal Cord. 2001;39:63–73. doi: 10.1038/sj.sc.3101116. [DOI] [PubMed] [Google Scholar]
- Simons M, Trotter J. Wrapping it up: the cell biology of myelination. Curr Opin Neurobiol. 2007;17:533–40. doi: 10.1016/j.conb.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Smith SB, Marker CL, Perry C, Liao G, Sotocinal SG, Austin JS, et al. Quantitative trait locus and computational mapping identifies Kcnj9 (GIRK3) as a candidate gene affecting analgesia from multiple drug classes. Pharmacogenet Genomics. 2008;18:231–41. doi: 10.1097/FPC.0b013e3282f55ab2. [DOI] [PubMed] [Google Scholar]
- Souslova V, Cesare P, Ding Y, Akopian AN, Stanfa L, Suzuki R, et al. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–7. doi: 10.1038/35039526. [DOI] [PubMed] [Google Scholar]
- Spataro LE, Sloane EM, Milligan ED, Wieseler-Frank J, Schoeniger D, Jekich BM, et al. Spinal gap junctions: potential involvement in pain facilitation. J Pain. 2004;5:392–405. doi: 10.1016/j.jpain.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Vizi ES, Wirkner K, Illes P. P2X7 receptors in the nervous system. Prog Neurobiol. 2006;78:327–46. doi: 10.1016/j.pneurobio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Stangel M, Hartung HP. Remyelinating strategies for the treatment of multiple sclerosis. Prog Neurobiol. 2002;68:361–76. doi: 10.1016/s0301-0082(02)00105-3. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Brosnan CF, Scemes E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci. 2006;26:1378–85. doi: 10.1523/JNEUROSCI.3902-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SM, Lee A, Bjorkman ST, Miller SM, Sullivan RK, Poronnik P, et al. Cytoskeletal anchoring of GLAST determines susceptibility to brain damage: an identified role for GFAP. J Biol Chem. 2007;282:29414–23. doi: 10.1074/jbc.M704152200. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer SM, Schubert P, DeLeo JA. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001;297:1210–7. [PubMed] [Google Scholar]
- Takahashi N, Kikuchi S, Shubayev VI, Campana WM, Myers RR. TNF-alpha and phosphorylation of ERK in DRG and spinal cord: insights into mechanisms of sciatica. Spine. 2006;31:523–9. doi: 10.1097/01.brs.0000201305.01522.17. [DOI] [PubMed] [Google Scholar]
- Takeda M, Takahashi M, Matsumoto S. Contribution of activated interleukin receptors in trigeminal ganglion neurons to hyperalgesia via satellite glial interleukin-1-beta paracrine mechanism. Brain Behav Immun. 2008;22:1016–23. doi: 10.1016/j.bbi.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Kadoi J, Nasu M, Takahashi M, Kitagawa J, et al. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain. 2007;129:155–66. doi: 10.1016/j.pain.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Nasu M, Ikeda M, Kadoi J, Matsumoto S. Activation of NK1 receptor of trigeminal root ganglion via substance P paracrine mechanism contributes to the mechanical allodynia in the temporomandibular joint inflammation in rats. Pain. 2005;116:375–85. doi: 10.1016/j.pain.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–9. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Vit JP, Jasmin L, Bhargava A, Ohara PT. Satellite glial cells in the trigeminal ganglion as a determinant of orofacial neuropathic pain. Neuron Glia Biol. 2006a;2:247–257. doi: 10.1017/s1740925x07000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vit JP, Ohara PT, Bhargava A, Kelley K, Jasmin L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in painlike behavior in the absence of nerve injury. J Neurosci. 2008;28:4161–71. doi: 10.1523/JNEUROSCI.5053-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vit JP, Ohara PT, Tien DA, Fike JR, Eikmeier L, Beitz A, et al. The analgesic effect of low dose focal irradiation in a mouse model of bone cancer is associated with spinal changes in neuro-mediators of nociception. Pain. 2006b;120:188–201. doi: 10.1016/j.pain.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–35. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- Weick M, Cherkas PS, Hartig W, Pannicke T, Uckermann O, Bringmann A, et al. P2 receptors in satellite glial cells in trigeminal ganglia of mice. Neuroscience. 2003;120:969–77. doi: 10.1016/s0306-4522(03)00388-9. [DOI] [PubMed] [Google Scholar]
- Weng HR, Aravindan N, Cata JP, Chen JH, Shaw AD, Dougherty PM. Spinal glial glutamate transporters downregulate in rats with taxol-induced hyperalgesia. Neurosci Lett. 2005;386:18–22. doi: 10.1016/j.neulet.2005.05.049. [DOI] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005;102:14092–7. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis CL, Davis TP. Chronic inflammatory pain and the neurovascular unit: a central role for glia in maintaining BBB integrity? Curr Pharm Des. 2008;14:1625–43. doi: 10.2174/138161208784705414. [DOI] [PubMed] [Google Scholar]
- Woodhoo A, Sahni V, Gilson J, Setzu A, Franklin RJ, Blakemore WF, et al. Schwann cell precursors: a favourable cell for myelin repair in the Central Nervous System. Brain. 2007;130:2175–85. doi: 10.1093/brain/awm125. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Bo X, Burnstock G. Localization of ATP-gated P2X receptor immunoreactivity in rat sensory and sympathetic ganglia. Neurosci Lett. 1998;256:105–8. doi: 10.1016/s0304-3940(98)00774-5. [DOI] [PubMed] [Google Scholar]
- Xie WR, Deng H, Li H, Bowen TL, Strong JA, Zhang JM. Robust increase of cutaneous sensitivity, cytokine production and sympathetic sprouting in rats with localized inflammatory irritation of the spinal ganglia. Neuroscience. 2006;142:809–22. doi: 10.1016/j.neuroscience.2006.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Huang LY. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J Neurosci. 2002;22:93–102. doi: 10.1523/JNEUROSCI.22-01-00093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Ochalski A, Hertzberg EL, Nagy JI. On the organization of astrocytic gap junctions in rat brain as suggested by LM and EM immunohisto chemistry of connexin43 expression. J Comp Neurol. 1990;302:853–83. doi: 10.1002/cne.903020414. [DOI] [PubMed] [Google Scholar]
- Zarei MM, Toro B, McCleskey EW. Purinergic synapses formed between rat sensory neurons in primary culture. Neuroscience. 2004;126:195–201. doi: 10.1016/j.neuroscience.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Jones G, Tsutsui S, Opii W, Liu S, Silva C, et al. Lentivirus infection causes neuroinflammation and neuronal injury in dorsal root ganglia: pathogenic effects of STAT-1 and inducible nitric oxide synthase. J Immunol. 2005;175:1118–26. doi: 10.4049/jimmunol.175.2.1118. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]