

Sulfur is an essential component for the biosynthesis of the sulfur-containing amino acids l-methionine and l-cysteine. The crystal structure of the periplasmic aliphatic sulfonate-binding protein SsuA from E. coli has been solved at 1.75 Å resolution in a substrate-free state. Comparison with the substrate-bound protein shows significant movement of one of the two domains.
Keywords: periplasmic binding proteins, aliphatic sulfonates, substrate-free state, extended C-terminus tail, intermediate open state
Abstract
Sulfur is an essential component for the biosynthesis of the sulfur-containing amino acids l-methionine and l-cysteine. Under sulfur-starvation conditions, bacteria are capable of scavenging sulfur from sulfur-containing compounds and transporting it across membranes. Here, the crystal structure of the periplasmic aliphatic sulfonate-binding protein SsuA from Escherichia coli is reported at 1.75 Å resolution in the substrate-free state. The overall structure of SsuA resembles the structures of other periplasmic binding proteins and contains two globular domains that form a cleft. Comparison with other periplasmic binding proteins revealed that one of the domains has been displaced by a rigid movement of 17°. Interestingly, the tight crystal packing appears to be mediated by a 13-amino-acid tail from the cloning that folds within the cleft of the next monomer.
1. Introduction
Bacteria have developed a series of survival mechanisms during nutrient limitation. They can synthesize alternative compounds, they can utilize high-affinity binding proteins and enzymes for the assimilation of the essential element from other sources or they can express unused genes. Sulfur is essential for bacterial growth and bacteria favour the utilization of cysteine or inorganic sulfur when they are present in the medium. When sulfur sources are at very low levels or are absent, bacteria express genes that are involved in sulfur assimilation. Two systems, tauABCD and ssuABCDE (van der Ploeg et al., 1996 ▶, 1998 ▶, 1999 ▶, 2001 ▶), have been identified as being upregulated during sulfur limitation and are capable of sulfur assimilation from organosulfur compounds such as sulfate esters, sulfamates, sulfonates and alkanesulfonates. The tau gene products can assimilate sulfur from taurine: tauA encodes a periplasmic binding protein (PBP), TauA, that binds taurine (Javaux et al., 2007 ▶) and brings it to TauBC, an ATP-binding cassette-dependent ABC-transporter system, for uptake into the cytoplasm. The taurine is oxidized to sulfite and aminoacetaldehyde by the TauD protein (an α-ketoglutarate-dependent dioxygenase; Eichhorn et al., 1997 ▶; Elkins et al., 2002 ▶; O’Brien et al., 2003 ▶). The ssu system is able to assimilate sulfur from aliphatic sulfonates such as HEPES, MOPS etc. Like tauA, ssuA encodes a periplasmic aliphatic sulfonate-binding protein and ssuBC encodes the ABC transporter responsible for the uptake. The release of the sulfur from the aliphatic sulfonates is catalysed by SsuD, an FMNH2-dependent sulfonate monooxygenase (Eichhorn et al., 1999 ▶, 2002 ▶), and the flavoprotein SsuE (Eichhorn et al., 1999 ▶).
The first step in the assimilation of sulfur from organic compounds is the binding of the compounds by the periplasmic aliphatic sulfonate-binding protein SsuA. Here, we report the crystal structure of Escherichia coli SsuA at 1.75 Å in the substrate-free state and in a comparison with the substrate-bound SsuA structure (PDB code 3e4r; A. Balan, F. T. Araujo, M. Sanches, D. Y. Chirgadze, T. L. Blundell & J. A. R. G. Barbosa, unpublished work) we show a hinge-like movement of the protein cleft for substrate binding/release.
2. Materials and methods
2.1. Cloning and expression
The mature SsuA protein (residues 23–319) from E. coli K12 MG1655 without the signal peptide was cloned into the GFPd vector (Drew et al., 2006 ▶). There is a linker and a TEV recognition site between the ssuA gene and GFP. The restriction sites used were NdeI and EcoRI.
The plasmid containing ssuA was transformed into BL21 (DE3) plysS cells and plated on an LB–agar plate containing kanamycin. A single colony was picked and was used to inoculate 20 ml LB containing 3 µg ml−1 kanamycin. 2 l LB was inoculated with the starter culture and the cells were grown to an OD600 of 0.6 at 310 K before induction with IPTG at 295 K for 24 h. The cells were harvested by centrifugation at 6000g for 10 min. The expression levels of the protein were monitored by measuring the GFP fluorescence (excitation at 488 nm and emission at 512 nm) using a Spectramax plate reader.
2.2. Purification
The cells were resuspended in 200 ml PBS buffer containing 0.1 mg ml−1 DNAse (Sigma), 0.1 mg ml−1 Pefablock (Sigma) and 5 mM MgCl2 and passed twice through a cell disruptor (Constant Systems) at 172 MPa. Unbroken cells and cell debris were spun down at 30 000g for 1 h. The clarified supernatant was supplemented with 20 mM imidazole and passed through a His-Trap column (GE Healthcare). The column was washed with ten column volumes of PBS containing 20 mM imidazole and ten column volumes of PBS containing 40 mM imidazole. The SsuA-GFP protein was eluted with PBS containing 500 mM imidazole. TEV protease was added at 1 mg per 10 mg of fusion protein and dialysed overnight at 277 K against 2 l gel-filtration buffer, 20 mM Tris base pH 7.5 and 150 mM NaCl. The cleaved material was passed through a His-Trap column equilibrated with gel-filtration buffer to remove the TEV, GFP and any uncleaved protein. The protein was concentrated and injected onto a Superdex 75 10/300 column (GE Healthcare) equilibrated in gel-filtration buffer. A single peak corresponding to SsuA was observed on the chromatogram. The fraction containing SsuA was analysed on SDS–PAGE and concentrated to 20 mg ml−1 using a 30 kDa concentrator (Millipore). The protein was desalted in crystallization buffer (20 mM Tris base pH 7.5 and 10 mM NaCl).
2.3. Crystallization and data collection
Crystallization conditions were screened using sparse-matrix screens in 96-well plates with protein at a concentration of 12.5 mg ml−1 in crystallization buffer at 277 and 293 K. Drops consisted of 100 nl protein solution and 100 nl precipitant solution and were prepared using a Cartesian robot. Initial crystals were grown overnight and the best-looking crystals were optimized in hanging-drop plates. The best crystals were grown from 22% PEG 3350 and 0.2 M sodium formate at 293 K in 2 d.
Prior to data collection, the crystals were transferred into mother liquor containing 20% glycerol and flash-frozen in liquid nitrogen. A full data set was collected to 1.75 Å resolution on beamline I04 at Diamond Light Source (Table 1 ▶) at 100 K. The crystals belonged to the orthorhombic space group P212121. The data were integrated with XDS (Kabsch, 1988 ▶) and scaled with SCALA (Evans, 1993 ▶).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P212121 |
| Beamline | Diamond I04 |
| Unit-cell parameters (Å, °) | a = 40.9, b = 96.0, c = 142.4, α = β = γ = 90 |
| Resolution (Å) | 71.2–1.75 (1.84–1.75) |
| Rmerge† | 5.7 (41.3) |
| I/σ(I) | 9.7 (1.8) |
| Completeness (%) | 99.1 (99.2) |
| Redundancy | 3.4 (3.4) |
| Refinement | |
| Resolution (Å) | 79.62–1.75 |
| No. of reflections | 54060 |
| Rwork/Rfree‡ (%) | 18.5/22.5 |
| Average B factor (Å2) | 13.74 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (°) | 1.619 |
R
merge = 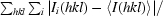
 , where Ii(hkl) is the intensity of an individual reflection and 〈I(hkl)〉 is the average intensity.
, where Ii(hkl) is the intensity of an individual reflection and 〈I(hkl)〉 is the average intensity.
R
work and R
free = 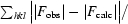
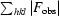 . R
free was calculated using 5% of the data
. R
free was calculated using 5% of the data
2.4. Structure determination and model building
The SsuA structure was solved by molecular replacement using BALBES (Long et al., 2008 ▶). The search model that produced a solution was domain I of SsuA from Xanthomonas axonopodis pv. citri (PDB entry 3e4r; A. Balan, F. T. Araujo, M. Sanches, D. Y. Chirgadze, T. L. Blundell & J. A. R. G. Barbosa, unpublished work). Two copies were located and were used as a fixed-input model in MOLREP (Vagin & Teplyakov, 1997 ▶) to locate the second domain (residues 110–205 from 3e4r). After rigid-body refinement in REFMAC5 (Murshudov et al., 1997 ▶), R work and R free were 48.7% and 47.9%, respectively. Restrained refinement lowered R work to 41.1% and R free to 45.6%. The phases were input into ARP/wARP (Langer et al., 2008 ▶) and an almost complete model was produced (sequence coverage of >98%). Manual electron-density inspection and building in Coot (Emsley & Cowtan, 2004 ▶) and addition of water molecules with ARP/wARP (Langer et al., 2008 ▶) resulted in a complete model with a final R work of 18.5% and R free of 22.5% (Table 1 ▶). Figures were prepared using PyMOL (DeLano, 2002 ▶). Coordinates and structure factors have been deposited in the RCSB Protein Data Bank with PDB code 2x26.
2.5. Structure analysis
Initial superpositions were carried out using the program SUPERPOSE (Krissinel & Henrick, 2004 ▶) in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). Improvement of the superpositions was carried out using the program O (Jones et al., 1991 ▶) with an algorithm that searches for structure fragments that can be aligned within less than 3.8 Å of all matching Cα pairs.
3. Results
3.1. Overall structure
The structure of SsuA from E. coli was determined at 1.75 Å using molecular replacement. The asymmetric unit contains two SsuA protomers that can be superimposed with a root-mean-square deviation (r.m.s.d.) of 0.34 Å over 285 Cα atoms. The protein that was used for crystallization (residues 23–319) lacked the signal peptide (residues 1–22) and contained an additional 13 amino acids at the C-terminus after the mature protein owing to the linker and the TEV recognition protease site. Chain A consists of residues 24–332, whereas the C-terminal extension in chain B is disordered (only the last three residues could be located in the electron-density maps). Further discussion will be based on the structure of SsuA from chain A unless stated otherwise.
The overall structure of SsuA resembles the structures of other PBPs. SsuA is an α/β protein that belongs to the class II PBPs (Dwyer & Hellinga, 2004 ▶) and contains two globular domains that form a cleft; domain I consists of residues 25–106 and 206–315 and domain II consists of residues 107–207, with domain I being larger in size than domain II (Fig. 1 ▶). The two domains exhibit similar folds consisting of five mixed β-sheets flanked by α-helices (Fig. 1 ▶). The two domains are connected by two short peptides (hinge region) composed of residues 105–108 and 204–209.
Figure 1.

Ribbon representation of the SsuA monomer from E. coli. The view is from the front of the cleft. The structure has been coloured and labelled according to the secondary structure (α-helices in red, β-sheets in yellow and loops in green). The C-terminal extended tail is coloured black. The glycerol molecule is shown as pink spheres.
A glycerol molecule is bound in domain I just below the cleft. It is only stabilized by interactions from domain I: a hydrogen bond from Arg269, van der Waals interactions with Gly36 and Glu104 and water-mediated hydrogen bonds.
3.2. Linker region and crystal packing
Additionally, there is a long tail of 13 amino acids at the C-terminus after the mature protein that consists of the linker (EcoRI, KpnI and BamHI residues from the vector) and the TEV recognition-site residues (Glu327-Asn328-Leu329-Tyr330-Phe331-Gln332). Interestingly, application of the symmetry-related monomers revealed that the TEV recognition site folds and is stabilized within the cleft of the symmetry-related monomer (Figs. 2 ▶ a and 2 ▶ b). Gln332 forms a salt bridge to the side chain and main chain of Ser139′, Gln34′ and Thr83′ from the symmetry-related monomer (residues from the symmetry-related monomer are indicated by a prime). The rest of the residues form hydrogen bonds either to the main chain or side chains: Tyr330 forms a hydrogen bond to Gly36′, Leu329 forms hydrogen bonds to Trp183′ and Pro64′, Asn328 forms a hydrogen bond to Tyr186′ and Glu327 forms hydrogen bonds to Ala166′ and Thr164′. The rest of the linker is stabilized by interactions with β-sheet β2 of domain I: Asn326 interacts with Gln68′, Val323 forms a hydrogen bond to Glu62′, Phe321 forms hydrogen bonds to Glu62′ and Trp60′ and the side chain of Glu320 is stabilized by a hydrogen bond to the side chain of Lys44′. The linker in the second monomer is disordered (only three residues could be modelled) and adopts two conformations: one within the cleft and one close to domain I of the first monomer. This interaction appears to be a consequence of the crystallization environment as the protein migrates as a monomer in size-exclusion chromatography (data not shown).
Figure 2.

(a) The tail is stabilized against the symmetry-related monomer (ribbon representation in grey) and folds within the cleft. The side chains of the extended tail of chain A are shown as yellow sticks (O atoms in red and N atoms in blue). The electron-density map around the tail (2F o − F c contoured at 1.3σ) is shown as a blue mesh. (b) Stereo representation of the residues from the symmetry-related monomer that are involved in stabilization of the extended tail (shown as yellow sticks); the symmetry-related molecule is shown in grey. The electron-density map around the tail (2F o − F c contoured at 1.3σ) is shown as a blue mesh.
3.3. Structure comparison
In order to identify other close homologues to SsuA, the structure was submitted to the DALI server (Holm & Sander, 1995 ▶) and the closest homologues found were SsuA from X. axonopodis pv. citri (PDB code 3e4r; Z score = 38.6%; A. Balan, F. T. Araujo, M. Sanches, D. Y. Chirgadze, T. L. Blundell & J. A. R. G. Barbosa, unpublished work), the bicarbonate transporter CmpA from Synechocystis PCC 6803 (PDB code 2i48; Z score = 23.8%; Koropatkin et al., 2007 ▶), the nitrate transporter NrtA from Synechocystis PCC 6803 (PDB code 2g29; Z score = 23.3%; Koropatkin et al., 2006 ▶) and the desulfurization enzyme DszB from Rhodococcus sp. strain IGTS8 (PDB code 2de4; Z score = 21.2%; Lee et al., 2006 ▶). It also shows similarity to other PBPs (Z > 13%), but with very low identity (∼10–14%). The closest structural homologue of SsuA is SsuA from X. axonopodis pv. citri with a HEPES molecule bound in the cleft. The structures share 58% identity, with most of the residues in the α-helices and β-sheets being conserved.
3.4. Domain movement
The two SsuA structures can be aligned with an r.m.s.d. deviation of 6.3 Å over 280 Cα atoms, suggesting conformational changes. The superposition of the two domains was improved using the program O (see §2.4) and resulted in an r.m.s.d. of only 1.7 Å over 250 Cα atoms (Fig. 3 ▶ a). Alignment of the two structures shows a hinge movement of domain II relative to domain I (Fig. 3 ▶ a), corresponding to a rigid-body rotation of 17° (Fig. 3 ▶ b). In the HEPES-bound structure the whole of domain II moves towards domain I for substrate binding (Fig. 3 ▶ a). Structure alignment of domain II only shows an r.m.s.d. of 0.96 Å (Fig. 3 ▶ c). A very similar hinge movement of domain II relative to domain I has been observed in other PBPs, with the most significant movement being found for the lysine/arginine/ornithine PBP (PDB code 1lst) from Salmonella typhimurium (Oh et al., 1993 ▶). The rigid-body movement between the apo and substrate-bound protein is a rotation of 52°. Unlike other apo-state periplasmic binding proteins, SsuA is in a substrate-free state since the cleft is occupied by the extended C-terminus.
Figure 3.

(a) Ribbon representation of the superimposed SsuAs from E. coli (blue) and X. axonopodis pv. citri (magenta). The HEPES molecule is shown as black sticks. The loops from domain I have been truncated for visualization. (b) Close-up view of the superimposed domains II. The rotation axis is shown as a solid black line. (c) Superimposition of the two domains from (b) along the rotation axis. They superimpose with an r.m.s.d. of 0.96 Å.
The HEPES molecule in SsuA from X. axonopodis pv. citri is bound by residues from both domains: residues from helix α6 of domain II and from helix α2 and β3 of domain I. All of the interactions are between the sulfate group of HEPES and the conserved Ser141, Gln36 and Gly68 residues. These residues are also involved in the binding of the TEV recognition residues within the cleft. In E. coli SsuA helix α4 has been displaced backwards by 5.3 Å relative to domain I. The other large displacement is of helix α5 (domain II), which does not make any interactions with the HEPES molecule but narrows the entrance to the cleft. In the substrate-free form it has been displaced by 5.6 Å. These kind of displacements have also been reported for apo and substrate-bound lysine/arginine/ornithine PBP from S. typhimurium (Oh et al., 1993 ▶). There is also a subtle movement of helices α1, α2 and α10 of domain I of the E. coli SsuA protein. The displacement of domain II is stabilized by the long tail of the symmetry-related monomer. Even though it is in a substrate-free state, both domains provide strong interactions for stabilization of the tail within the cleft. This could potentially be an intermediate state between the apo and substrate-bound forms of the protein. Such an intermediate state has been reported for the choline-binding protein ChoX from Sinorhizobium meliloti (Oswald et al., 2009 ▶). The ChoX structure is in the semi-closed form and shows movement of domain I rather than domain II compared with ligand-bound (closed-state) and ligand-free (open-state) ChoX (Oswald et al., 2008 ▶).
4. Conclusion
Structural analysis of SsuA from E. coli revealed that the protein adopts a PBP-type fold. This is the first structural report of a PBP that is involved in the assimilation pathway of sulfur from aliphatic sulfonates. The substrate-free form of the protein shows significant domain movement relative to the substrate-bound form of SsuA from X. axonopodis pv. citri. The aliphatic sulfonate compounds are relatively large in size and the large domain movements are essential for binding or release of the substrate.
Supplementary Material
PDB reference: SsuA, 2x26
Acknowledgments
We would like to thank Dr Alexander Cameron for useful discussions and Tian Geng for technical assistance. KB is a RCUK Fellow. SI is funded by the Wellcome Trust. Data were collected on beamline I04 at Diamond Light Source (UK). We would like to thank the beamline scientists and Diamond Light Source for allocating beamtime. We would also like to acknowledge the Membrane Protein Laboratory.
References
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Viewer. http://www.pymol.org.
- Drew, D., Lerch, M., Kunji, E., Slotboom, D. J. & de Gier, J. W. (2006). Nature Methods, 3, 303–313. [DOI] [PubMed]
- Dwyer, M. A. & Hellinga, H. W. (2004). Curr. Opin. Struct. Biol.14, 495–504. [DOI] [PubMed]
- Eichhorn, E., Davey, C. A., Sargent, D. F., Leisinger, T. & Richmond, T. J. (2002). J. Mol. Biol.324, 457–468. [DOI] [PubMed]
- Eichhorn, E., van der Ploeg, J. R., Kertesz, M. A. & Leisinger, T. (1997). J. Biol. Chem.272, 23031–23036. [DOI] [PubMed]
- Eichhorn, E., van der Ploeg, J. R. & Leisinger, T. (1999). J. Biol. Chem.274, 26639–26646. [DOI] [PubMed]
- Elkins, J. M., Ryle, M. J., Clifton, I. J., Dunning Hotopp, J. C., Lloyd, J. S., Burzlaff, N. I., Baldwin, J. E., Hausinger, R. P. & Roach, P. L. (2002). Biochemistry, 41, 5185–5192. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. R. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122. Warrington: Daresbury Laboratory.
- Holm, L. & Sander, C. (1995). Trends Biochem. Sci.20, 478–480. [DOI] [PubMed]
- Javaux, C., Joris, B. & De Witte, P. (2007). Protein J.26, 231–238. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kabsch, W. (1988). J. Appl. Cryst.21, 916–924.
- Koropatkin, N. M., Koppenaal, D. W., Pakrasi, H. B. & Smith, T. J. (2007). J. Biol. Chem.282, 2606–2614. [DOI] [PubMed]
- Koropatkin, N. M., Pakrasi, H. B. & Smith, T. J. (2006). Proc. Natl Acad. Sci. USA, 103, 9820–9825. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc.3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Lee, W. C., Ohshiro, T., Matsubara, T., Izumi, Y. & Tanokura, M. (2006). J. Biol. Chem.281, 32534–32539. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- O’Brien, J. R., Schuller, D. J., Yang, V. S., Dillard, B. D. & Lanzilotta, W. N. (2003). Biochemistry, 42, 5547–5554. [DOI] [PubMed]
- Oh, B.-H., Pandit, J., Kang, C.-H., Nikaido, K., Gokcen, S., Ames, G. F.-L. & Kim, S.-H. (1993). J. Biol. Chem.268, 11348–11355. [PubMed]
- Oswald, C., Smits, S. H., Höing, M., Bremer, E. & Schmitt, L. (2009). Biol. Chem.390, 1163–1170. [DOI] [PubMed]
- Oswald, C., Smits, S. H., Höing, M., Sohn-Bösser, L., Dupont, L., Le Rudulier, D., Schmitt, L. & Bremer, E. (2008). J. Biol. Chem.283, 32848–32859. [DOI] [PubMed]
- Ploeg, J. R. van der, Cummings, N. J., Leisinger, T. & Connerton, I. F. (1998). Microbiology, 144, 2555–2561. [DOI] [PubMed]
- Ploeg, J. R. van der, Eichhorn, E. & Leisinger, T. (2001). Arch. Microbiol.176, 1–8. [DOI] [PubMed]
- Ploeg, J. R. van der, Iwanicka-Nowicka, R., Bykowski, T., Hryniewicz, M. M. & Leisinger, T. (1999). J. Biol. Chem.274, 29358–29365. [DOI] [PubMed]
- Ploeg, J. R. van der, Weiss, M. A., Saller, E., Nashimoto, H., Saito, N., Kertesz, M. A. & Leisinger, T. (1996). J. Bacteriol.178, 5438–5446. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: SsuA, 2x26
PDB reference: SsuA, 2x26