

A novel family GH16 β-agarase from the marine bacterium Zobellia galactanivorans was expressed, purified and crystallized. Hexagonal crystals belonging to space group P3121 diffracted to 2.2 Å resolution, whereas orthorhombic crystals belonging to space group P212121 diffracted to 1.5–Å resolution.
Keywords: β-agarases, glycoside hydrolases, heterologous expression, Zobellia galactanivorans
Abstract
Marine bacteria secrete specific glycoside hydrolases such as agarases to access polysaccharides from algal cell walls as a carbon and energy source. In an attempt to identify agarases with variable degradation patterns, a novel family GH16 β-agarase from the marine bacterium Zobellia galactanivorans was expressed, purified and crystallized. The purified enzyme crystallized in two distinct forms that were grown by the hanging-drop vapour-diffusion method using polyethylene glycol as a precipitant. Hexagonal crystals belonging to space group P3121 diffracted to 2.2 Å resolution, whereas orthorhombic crystals belonging to space group P212121 diffracted to 1.5 Å resolution.
1. Introduction
Agarose is a polysaccharide that is present as a cell-wall matrix compound in red algae (Craigie & Leigh, 1978 ▶) and can be described chemically as a neutral unbranched polysaccharide composed of repeating neoagarobiose units [(3,6)-anhydro-l-galactose-α(1,3)-d-galactose] joined by β(1,4)-bonds (Rees, 1969 ▶). Industrial processes use alkaline treatment to produce almost pure agarose with high gelling quality and without chemical modifications. In contrast, the galactose subunits of the chemically variable class of natural agarocolloids (Craigie, 1990 ▶), alternatively also named agarans (Knutsen et al., 1994 ▶), are methylated, pyruvated, sulfated or glycosylated to form various derivatives with different gelling properties and solubility characteristics (Morrice et al., 1983 ▶; Rees & Conway, 1962 ▶; Rochas et al., 1986 ▶). These variations occur in algal cell walls as a function of species, geographical origin, stage of the life cycle, physiological state or as a function of age (Lahaye, 2001 ▶). To degrade these chemically different polysaccharide structures to completion, marine bacteria must secrete a set of agar-degrading enzymes, the precise interplay and synergy of which has not yet been studied in detail. A recent genomic and proteomic analysis of Saccharophagus degradans, a rod-shaped aerobic bacterium isolated from the surface of decomposing saltwater cordgrass in marshlands (Ekborg et al., 2005 ▶), has revealed the presence of an agarolytic system with five agarases belonging to three different glycoside hydrolase (GH) families (Ekborg et al., 2006 ▶). Several enzymes belonging to families GH50 and GH86 and one endolytic β-agarase from family GH16 (http://www.cazy.org/Cazy; Henrissat & Davies, 1997 ▶) were identified; however, only unmodified neutral agarose degradation activity was described (Ekborg et al., 2006 ▶). In contrast, Zobellia galactanivorans (formerly Cytophaga drobachiensis) is a marine heterotrophic flavobacterium that is capable of degrading more complex polysaccharides such as agarocolloids (Michel et al., 2006 ▶) using GH16 enzymes only. Indeed, the ongoing genome project of this marine bacterium (G. Michel, personal communication) has revealed a high number of family GH16 enzymes, some of which are clearly related to the previously characterized β-agarases A and B (AgaA and AgaB; Allouch et al., 2003 ▶; Jam et al., 2005 ▶). No family GH50 or GH86 β-agarases have been detected. Nevertheless, the large number of family GH16 agarase-like sequences suggests a finely tuned system for complex agarocolloid degradation. Sequence analysis of one of these GH16 β-agarases (AgaD) revealed that although closely related to AgaA and AgaB (32 and 39% sequence identity, respectively), the sequence of AgaD represents a third family of GH16 β-agarases that contains several large insertions with respect to AgaA and AgaB (Fig. 1 ▶). The sequence variations raised the question of whether AgaD would perform catalysis using a different mode of action (i.e. exolytic or processive) or even display a variation of substrate specificity with respect to the chemical modifications present in the naturally occurring substrate.
Figure 1.

Sequence alignment of three β-agarases (AgaA, AgaB and AgaD) from Z. galactanivorans. The secondary-structure elements of β-agarase A are indicated above the alignment. The alignment was produced using MultAlin (Corpet, 1988 ▶) and the figure was drawn using ESPript (Gouet et al., 2003 ▶). The following sequence identifiers are used: AgaA, β-agarase A from Z. galactanivorans; AgaB, β-agarase B from Z. galactanivorans; AgaD, β-agarase D from Z. galactanivorans.
Agar is not only of great ecological importance: owing to its gelling properties it is also widely used in the food, cosmetics, pharmaceutical and biotechnological industries. Access to multiple active agarose enzymes with varying substrate specificities would therefore be biotechnologically advantageous. In contrast to the impact of its substrate, structural details describing substrate recognition and specificity have to date only been determined for the two true endo-β-agarases AgaA and AgaB (Allouch et al., 2003 ▶, 2004 ▶; Jam et al., 2005 ▶).
In order to analyse and understand the variability of substrate specificity in family GH16 β-agarase, we have produced and crystallized this third β-agarase AgaD from Z. galactanivorans. Moreover, structural and biochemical comparison with the existing β-agarase structures will lead to an improved understanding of the agarolytic system of the heterotrophic marine bacterium Z. galactanivorans, which is a model organism for transformation of marine organic matter.
2. Material and methods
2.1. Expression and purification
Bioinformatic analysis (Fig. 1 ▶) of the open reading frame coding for β-agarase D revealed that similar to agarase A and in contrast to agarase B it is a bimodular protein that is composed of a C-terminal domain of unknown function and an N-terminal catalytic domain which belongs to the GH16 family of glycoside hydrolases. In the past, bimodular glycoside hydrolases have been found to be recalcitrant to crystallization; we therefore cloned a construct containing the catalytic domain only (residues 21–377 of the full-length sequence, hereafter referred to as AgaD_cat).
The nucleotide sequence corresponding to the catalytic domain was amplified by PCR from Z. galactanivorans genomic DNA using a set of primers (forward, 5′-GGGGGGAGATCTCAATACGATTGGGACAACGTGCC-3′; reverse, 5′-CCCCCCCAATTGTTAGTTCACAGGTTTGTAAACCCGGAT-3′). The PCR product obtained was purified using a Qiagen QIAquick purification kit and digested with BglII/EcoRI (5′/3′ ends) in NEB2 buffer (BioLabs) at 310 K for 3 h. Digested and purified PCR product was ligated with a similarly digested and purified pFO4 vector (a derivative of pET15 that adds an N-terminal tag consisting of six histidine residues) at 277 K using T4 DNA Ligase (overnight). The ligation mixture was transformed into Escherichia coli DH5α strain by chemical transformation. The colonies obtained were screened by colony PCR and a positive colony with the correct fragment length was used to prepare the plasmid for expression studies. The plasmid was extracted with a Wizard Plus SV Minipreps kit (Promega) and used to transform chemically competent E. coli BL21 (DE3) cells. One fresh colony was picked to inoculate a preculture consisting of 5 ml LB medium containing 100 µg ml−1 ampicillin, which was then incubated overnight at 310 K. 1 ml of the preculture was used to inoculate 1 l ZYP-5052 expression culture (Studier, 2005 ▶; 100 µg ml−1 ampicillin), which was grown at 293 K until saturation of the culture (final OD600nm of ∼16) was achieved. The cells were harvested by centrifugation (4000g, 277 K, 20 min) and the cell pellet was resuspended in buffer A (20 mM Tris, 200 mM NaCl, 20 mM imidazole pH 7.5). The cells were treated with 500 µl lysis buffer containing 1 mg ml−1 lysozyme and 1 mg ml−1 DNAse for 30 min on ice followed by sonication; the lysate was then cleared by centrifugation (50 000g, 277 K, 30 min) and subsequent filtration using a 0.2 µm filter (Millipore). The filtrated solution was loaded onto a 10 ml IMAC HyperCell resin column (Pall Corporation) charged with NiSO4. The column was equilibrated with buffer A without lysozyme and DNAse. After washing with buffer A (ten column volumes), the protein was eluted with a 60 ml linear gradient from buffer A to 60% buffer B (20 mM Tris, 200 mM NaCl and 500 mM imidazole) at a flow rate of 1 ml min−1. The fractions were analysed by SDS–PAGE and the fractions which contained AgaD_cat were pooled. The volume was reduced to 4 ml by ultrafiltration on an Amicon membrane (polyethersulfone, 30 kDa cutoff). Preliminary trials to concentrate the enzyme to beyond a nominal concentration of 2 mg ml−1 failed because the protein precipitated from the solution. Since efficient crystal growth depends on initial protein concentration and solubility, we used a solubility screen (Collins et al., 2004 ▶) to find an optimal buffer combination for crystallization and protein storage. A buffer composed of 100 mM sodium citrate containing 1 mM CaCl2 and 1 mM MgCl2 pH 8 (buffer C) was used in the final gel-filtration polishing step as well as a protein-storage buffer. The protein solutions were systematically checked for monodispersity using a Malvern Zetasizer instrument to perform dynamic light-scattering measurements.
For gel filtration, a Sephacryl S-200 column (GE Healthcare) pre-equilibated with buffer C at a flow rate of 1 ml min−1 (Fig. 2 ▶) was used. The purified enzyme was concentrated to ∼8 mg ml−1 by ultrafiltration on an Amicon membrane (10 kDa cutoff). All chromatography was carried out on an ÄKTA Explorer chromatography system (GE Healthcare). The final concentration of the protein solution was estimated by measuring the absorbance at 280 nm using a theoretical extinction coefficient of 95 465 M −1 cm−1.
Figure 2.
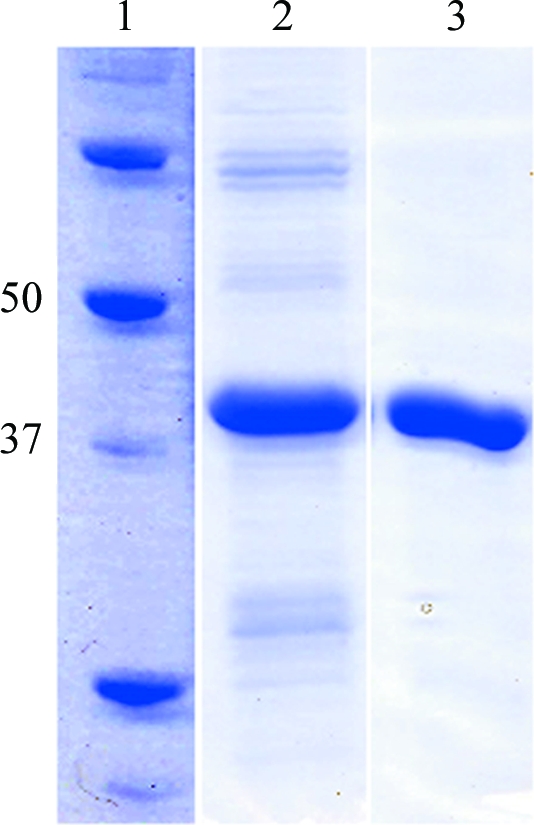
SDS–PAGE analysis of purified recombinant β-agarase D (AgaD_cat). Lane 1, molecular-weight markers (kDa). Lane 2, AgaD_cat after nickel-affinity chromatography (IMAC-cellulose). Lane 3, AgaD_cat after gel-filtration chromatography on Sephacryl (S-200) resin.
2.2. Crystallization
Initial crystallization screening experiments were carried out at 292 K using the sitting-drop vapour-diffusion method in 96-well Corning plates. Crystallization screening was performed using the JCSG+, PACT and PEG I screens from Qiagen, corresponding to a total of 288 conditions in three 96-well plates (Corning No. 3551, one-well sitting drop). A crystallization robot (Proteomics Solutions Honeybee961) was used for pipetting and the drops contained 300 nl protein solution (∼8 mg ml−1 in buffer C) mixed with 150 nl reservoir solution. After visual identification of initial crystallization conditions, these conditions were further optimized in 24-well Linbro plates using the hanging-drop vapour-diffusion method. The drops were prepared on siliconized cover slips by mixing 2 µl protein solution with 1 µl well solution and equilibration was performed against 500 µl reservoir solution at room temperature.
2.3. Data collection and X-ray diffraction analysis
X-ray diffraction data were collected from a hexagonal crystal of recombinant AgaD_cat at 100 K and a wavelength of 0.933 Å on beamline ID14-EH2 at the ESRF (Grenoble, France) using an ADSC Quantum 4 detector and a crystal-to-detector distance of 222.9 mm and were collected from the orthorhombic crystal form at 100 K and a wavelength of 0.934 Å on the ESRF beamline ID14-EH1 using an ADSC Quantum Q210 detector and a crystal-to-detector distance of 142.88 mm. For cryoprotection, crystals were soaked for 1 min in crystallization solution complemented with 6% glycerol before being flash-cooled in the cryostream. All raw data were processed using the program MOSFLM (Leslie, 1992 ▶). The data were then merged and scaled using the program SCALA (Collaborative Computational Project, Number 4, 1994 ▶). Detailed data statistics for both crystal forms are reported in Table 1 ▶.
Table 1. Data-collection statistics for the two crystal forms of AgaD_cat.
| Trigonal | Orthorhombic | |
|---|---|---|
| Beamline | ID14-EH2 | ID14-EH1 |
| Wavelength (Å) | 0.933 | 0.934 |
| Space group | P3121 | P212121 |
| Unit-cell parameters (Å) | a = 98.49, b = 98.49, c = 181.21 | a = 53.25, b = 77.27, c = 83.7 |
| Resolution range (Å) | 62.14–2.20 (2.32–2.20) | 43.85–1.38 (1.46–1.38) |
| No. of observations | 302205 (30196) | 385296 (18314) |
| No of. unique reflections | 49464 (6817) | 55977 (5908) |
| Redundancy | 6.1 (4.4) | 6.9 (3.1) |
| Completeness (%) | 96.3 (91.8) | 99.9 (67.3†) |
| Mean I/σ(I) | 15.4 (3.5) | 21.3 (3.1) |
| Rmerge (%) | 10.5 (27.1) | 6.1 (35.7) |
The highest resolution data extended into the corners of the detector, leading to lower completeness. The data were more than 99% complete to 1.50 Å resolution.
3. Results and discussion
After initial purification and concentration, a tendency to low solubility (<2 mg ml−1) associated with high aggregation was observed for AgaD_cat. Therefore, we performed a solubility screen following the strategy of Collins et al. (2004 ▶) and a buffer combination that strongly improved the solubility was identified. This allowed us to obtain a monodisperse solution of AgaD_cat at a concentration of 8 mg ml−1 (Fig. 2 ▶). The recombinant protein was tested for agarolytic activity by depositing one drop of protein solution at 8 mg ml−1 on an agarose gel, where it clearly dug a hole into the gel. Subsequently, three crystallization screens were used for screening crystallization conditions (PEG I, PACT and JCSG+). Hexagonal and orthorhombic single crystals grew in a total of 18 different conditions (Supplementary Table 1 ▶ 1), generally within one or two weeks at 292 K. Subsequently, the conditions which produced the largest single crystals (JCSG+ condition No. 60) were scaled up in 24-well Linbro plates using the hanging-drop vapour-diffusion method. Hexagonal crystals suitable for diffraction studies could be grown after further optimization trials using a crystallization solution consisting of 28–34%(w/v) PEG 8000 and 0.1 M imidazole pH 8 at 292 K.
The crystals used for diffraction studies grew within one week, were of hexagonal shape with dimensions of about 0.3 × 0.3 × 0.05 mm and diffracted to 2.2 Å resolution, while the orthogonal crystals had dimensions of about 0.6 × 0.1 × 0.08 mm. A complete data set was collected from these crystals and the data statistics are reported in Table 1 ▶. We succeeded in obtaining phases by molecular replacement using β-agarase B from Z. galactanivorans (PDB code 1o4z; Allouch et al., 2003 ▶) as a search model with two molecules per asymmetrical unit. However, the cumulative intensity distribution given by SCALA (Collaborative Computational Project, Number 4, 1994 ▶) indicated twinning and the R free factor did not decrease even after several rounds of refinement. To avoid the complex refinement of twinned data and having identified numerous crystallization conditions, we first attempted to obtain a variant crystal form of AgaD_cat. Volatile additives such as dioxane are known to reduce twinning (Bergfors, 2003 ▶). Therefore, we used 4% dioxane or 2-propanol together with the initial optimized crystallization condition (28–34% PEG 8000 and 0.1 M imidazole pH 8). Both additives changed the crystal morphology from hexagonal plates to needle-shaped or rod-shaped crystals (Fig. 3 ▶). Several crystals of the new crystal form were tested and one crystal (grown using 2-propanol as an additive) diffracted to 1.5 Å resolution. The data set was treated as described above and all crystallographic data are listed in Table 1 ▶.
Figure 3.

Crystals of AgaD_cat. (a) Crystals of AgaD_cat displaying a trigonal crystal form with space group P3121. The average dimensions of the crystals are 0.3 × 0.3 × 0.05 mm. (b) Crystals of AgaD_cat obtained by the addition of dioxane to the crystallization condition, leading to orthorhombic crystals belonging to space group P212121. The average dimensions of the crystals are 0.6 × 0.1 × 0.08 mm.
The addition of dioxane or 2-propanol changed the space group from hexagonal P3121 to orthorhombic P212121, with unit-cell parameters a = 53.25, b = 77.27, c = 83.7 Å. Moreover, the data quality strongly improved. Assuming a molecular weight of 40.24 kDa, calculation of the Matthews coefficient resulted in a V M value of 2.14 Å3 Da−1 and a solvent content of 42.2% assuming the presence of one molecule in the asymmetric unit. Molecular replacement was carried out once more against this orthorhombic crystal form using the program AMoRe (Navaza, 2001 ▶) with the β-agarase B protein structure from Z. galactanivorans (PDB code 1o4z) as the search model, leading to one contrasted solution with a correlation factor of 25.4 and an R factor of 50.9%.
The β-agarase cloned, expressed, purified and described here contains three sequence insertions which do not occur within the previously described β-agarases A and B from the same organism. The enzyme showed significant activity towards agarose (data not shown) and the sequence divergence may be explained by a different mode of action or a different specificity for natural agarocolloids. The systemic characterization of agarase-like enzymes from Z. galactanivorans will extend our knowledge of the degradation of marine organic matter by glycoside hydrolases.
Supplementary Material
Supplementary material file. DOI: 10.1107/S174430911000429X/hc5097sup1.pdf
Acknowledgments
JHH was supported by an FP-6 Marie Curie Fellowship (MEST-CT-2005-020737). This work was also supported by the ‘Region Bretagne’ through the Marine3D program. We are indebted to the European Synchrotron Radiation Facility (ESRF, Grenoble, France) for access to beam time through a beam-allocation group and we thank all the beamline scientists and staff for help on the beamlines.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: HC5097).
References
- Allouch, J., Helbert, W., Henrissat, B. & Czjzek, M. (2004). Structure, 12, 623–632. [DOI] [PubMed]
- Allouch, J., Jam, M., Helbert, W., Barbeyron, T., Kloareg, B., Henrissat, B. & Czjzek, M. (2003). J. Biol. Chem.278, 47171–47180. [DOI] [PubMed]
- Bergfors, T. (2003). J. Struct. Biol.142, 66–76. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Collins, B. K., Tomanicek, S. J., Lyamicheva, N., Kaiser, M. W. & Mueser, T. C. (2004). Acta Cryst. D60, 1674–1678. [DOI] [PubMed]
- Corpet, F. (1988). Nucleic Acids Res.16, 10881–10890. [DOI] [PMC free article] [PubMed]
- Craigie, J. S. (1990). Biology of the Red Algae, edited by K. Cole & R. Sheath, pp. 221–257. Cambridge University Press.
- Craigie, J. S. & Leigh, C. (1978). Handbook of Phycological Methods, edited by J. A. Hellebust & J. S. Craigie, pp. 109–133. Cambridge University Press.
- Ekborg, N. A., Gonzalez, J. M., Howard, M. B., Taylor, L. E., Hutcheson, S. W. & Weiner, R. M. (2005). Int. J. Syst. Evol. Microbiol.55, 1545–1549. [DOI] [PubMed]
- Ekborg, N. A., Taylor, L. E., Longmire, A. G., Henrissat, B., Weiner, R. M. & Hutcheson, S. W. (2006). Appl. Environ. Microbiol.72, 3396–3405. [DOI] [PMC free article] [PubMed]
- Gouet, P., Robert, X. & Courcelle, E. (2003). Nucleic Acids Res.31, 3320–3323. [DOI] [PMC free article] [PubMed]
- Henrissat, B. & Davies, G. (1997). Curr. Opin. Struct. Biol.7, 637–644. [DOI] [PubMed]
- Jam, M., Flament, D., Allouch, J., Potin, P., Thion, L., Kloareg, B., Czjzek, M., Helbert, W., Michel, G. & Barbeyron, T. (2005). Biochem. J.385, 703–713. [DOI] [PMC free article] [PubMed]
- Knutsen, S., Myslabodski, D., Larsen, B. & Usov, A. (1994). Botanica Marina, 37, 163–169.
- Lahaye, M. (2001). J. Appl. Phycol.13, 173–184.
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Michel, G., Nyval-Collen, P., Barbeyron, T., Czjzek, M. & Helbert, W. (2006). Appl. Microbiol. Biotechnol.71, 23–33. [DOI] [PubMed]
- Morrice, L. M., McLean, M. W., Long, W. F. & Williamson, F. B. (1983). Eur. J. Biochem.133, 673–684. [DOI] [PubMed]
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed]
- Rees, D. A. (1969). Adv. Carbohydr. Chem. Biochem.24, 267–332. [DOI] [PubMed]
- Rees, D. A. & Conway, E. (1962). Biochem. J.84, 411–416. [DOI] [PMC free article] [PubMed]
- Rochas, C., Lahaye, M. & Yaphe, W. (1986). Botanica Marina, 29, 335–340.
- Studier, F. W. (2005). Protein Expr. Purif.41, 207–234. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material file. DOI: 10.1107/S174430911000429X/hc5097sup1.pdf
Supplementary material file. DOI: 10.1107/S174430911000429X/hc5097sup1.pdf