

Abstract
Neutrophils are phagocytes whose principal function is to maintain anti-bacterial immunity. Neutrophils ingest and kill invading bacteria, releasing cytotoxic, chemotactic and inflammatory mediators at sites of infection. This serves to control the immediate host immune response and attract other cells, such as macrophages and dendritic cells, which are important for establishing long-term adaptive immunity. Neutrophils thus contribute to both the initiation and the maintenance of inflammation at sites of infection. Aberrant neutrophil activity is deleterious; suppressed responses can cause extreme susceptibility to infection while overactivation can lead to excessive inflammation and tissue damage. This review will focus on neutrophil regulation by granulocyte colony-stimulating factor (G-CSF), the principal cytokine controlling neutrophil development and function. The review will emphasize the molecular aspects of G-CSF-driven granulopoiesis in steady state (healthy) conditions and during demand-driven or ‘emergency’ conditions elicited by infection or clinical administration of G-CSF. Understanding the molecular control of granulopoiesis will aid in the development of new approaches designed to treat disorders of neutrophil production and function.
Keywords: G-CSF, neutrophils, granulopoiesis, STAT3, infection
General features of cytokines, neutrophils and their regulation by G-CSF
Infections create physiologic stress in host organisms, threatening both the quality and duration of life. Successful resolution of infection requires a functional immune system to recognize and destroy the invading microbe, without eliciting unnecessary damage to the host. Cytokines are proteins found in vertebrates [1, 2] that regulate the development and homeostasis of the immune system. Cytokine production is induced during infection to control the body’s immune response. A great deal of attention has focused upon the characterization of cytokines, from their three-dimensional atomic structures to whole animal responses. In many cases, however, we lack a detailed understanding of the molecular pathways that mediate a cytokine response under physiologic conditions. The elucidation of these pathways is important for developing future therapies to modulate host immunity during infection and to discover new ways to treat immune system disease.
Granulocyte colony-stimulating factor (G-CSF) is the major cytokine regulator of neutrophilic granulocytes [3]. Neutrophils are an essential cell type in the innate immune system necessary for the clearance of bacterial pathogens. Their critical role is demonstrated by the profound immunodeficiency of individuals with neutropenia, or low circulating neutrophil numbers, a condition that can stem from acquired or congenital origin [4–6]. Recombinant G-CSF has become the main therapeutic agent for the treatment of neutropenia, although recently its use has come under scrutiny for potential connections with leukemia [7–11]. This review will present basic features of G-CSF and its signaling mechanisms in steady state and ‘demand-driven’ granulopoiesis, and will conclude with a brief summary of the clinical aspects of G-CSF therapy.
Identification of G-CSF as a neutrophil growth factor
G-CSF was discovered by virtue of its ability to stimulate the growth of neutrophil colonies from human bone marrow progenitor cells ex vivo. Human G-CSF (molecular weight ~30,000) was purified from placental-conditioned medium [12] and murine G-CSF (molecular weight ~25,000) was subsequently isolated from medium conditioned by mouse lung samples following in vivo endotoxin treatment [13]. The use of distinct cell types for G-CSF isolation reveals the diversity of tissues that express this cytokine, which include fibroblasts, endothelial cells, monocytes, macrophages and bone marrow stromal cells (reviewed in [14]). The cDNA for human G-CSF was cloned in 1986 using partial amino acid sequence information and oligonucleotide probes to screen the CHU-2 and 5637 tumor cell line libraries [15, 16]. The murine G-CSF cDNA was cloned by cross-reactivity to the human cDNA [17], confirming a close relationship between human and mouse proteins.
Isolation of the G-CSF cDNA enabled the production of recombinant protein, which greatly facilitated the study of its biologic properties. Purified and recombinant G-CSF stimulate the proliferation of neutrophil precursors and other myeloid progenitor cells in vitro [12, 13, 18–21]. G-CSF, however, selectively supports the terminal differentiation of neutrophils relative to other myeloid lineages. G-CSF has also been shown to promote the differentiation of certain myeloid leukemia cell lines [13], but the profound maturation block of most human leukemias precludes its use as an anti-leukemia therapy. Deletion of the murine G-CSF gene revealed physiologic roles for this cytokine in steady state granulopoiesis and in the production of neutrophils under ‘emergency’ conditions of infection [3, 22, 23]. A comprehensive review of these topics can be found in references [14, 24].
Expression and action of G-CSF in vivo
Healthy individuals express low G-CSF levels in serum (range, <30–163pg/mL) [25–27]. By comparison, infection induces a substantial elevation in serum G-CSF in humans (range, 30–3,199pg/mL) and mice [25–27]. The high levels of G-CSF produced during infection are coupled with an increase in hematopoietic growth factor activity in serum, as judged by colony-stimulating assays and 3H thymidine uptake experiments with bone marrow samples ex vivo [27]. The receptor for G-CSF is expressed in granulocytic precursors and mature neutrophils, with higher receptor levels detected at later stages of maturation [28]. Therefore, the induction of G-CSF serum levels during infection directly stimulates increased proliferation of granulocytic progenitor cells. Committed neutrophil precursors at the myeloblast, promyelocyte and myelocyte stages show elevated rates of proliferation during exposure to G-CSF in vivo [29, 30], indicating that G-CSFR expression in the neutrophil lineage correlates with G-CSF sensitivity. Granulocytic progenitors respond to G-CSF by shortening their passage through the cell cycle, and therefore dividing more frequently [30]. G-CSF is also required for the survival of granulocytic lineage cells [22]. The increased proliferation rate of granulocytic precursors in response to G-CSF results in an elevated proportion of early neutrophil forms (e.g., myeloblast and promyelocyte) in the bone marrow relative to steady state conditions [29, 30]. This ability of the bone marrow to respond to the physiologic demand of infection or pharmacological levels of G-CSF during clinical administration is termed ‘emergency’ or ‘demand-driven’ granulopoiesis. This response is necessary for a sustained output of circulating neutrophils during infection or G-CSF therapy [3, 6, 23, 31].
In addition to its proliferation- and survival-promoting activity, G-CSF treatment induces a rapid (within 4–24 h) and sustained elevation in absolute peripheral neutrophil numbers. G-CSF shortens the transit time of developing granulocytes through the bone marrow compartment and accelerates the release of neutrophils that have undergone recent cell division [18, 29, 30, 32]. The properties of G-CSF were quickly recognized for their clinical value [33–40] (for reviews, see [6, 24, 31]). Recombinant G-CSF was shown to decrease the duration of neutropenia in cancer patients receiving chemotherapy [36, 37] (for reviews, see [6, 31]), which permits more frequent chemotherapeutic treatments to promote cancer regression. G-CSF also served to accelerate the resolution of foot infections in diabetics, thus shortening antibiotic use and hospital stay [41]. Moreover, G-CSF has been found to stimulate an increase in the absolute number of hematopoietic progenitor cells (HPC) in peripheral blood in a process that is known as “stem cell mobilization” [38, 39]. Hence, G-CSF is used to treat myelosuppression and its resulting immunodeficiency, as well as to mobilize HPC to peripheral blood, facilitating their collection for transplantation. In this review, we focus solely on neutrophil regulation by G-CSF; excellent reviews of G-CSF-mediated HPC mobilization and clinical ramifications can be found in references [42–45].
Pathways regulating G-CSF expression
G-CSF production is induced by several inflammatory stimuli that become rapidly elevated during infection such as interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNFα) and lipopolysaccaride (LPS) [24]. Therefore, pathogen-mediated activation of host pattern recognition receptors via LPS, and the host cytokine response to infection, serve to induce circulating levels of G-CSF (Fig. 1). This in turn stimulates neutrophil production in the bone marrow and neutrophil mobilization to the peripheral circulation. The synthesis of G-CSF appears to be controlled via post-transcriptional and transcriptional mechanisms [46, 47]. The G-CSF promoter contains binding sites for NF-κB p65 and NF-IL6/C/EBPβ [48], which are well-characterized transcription factors that participate in many inflammatory and immune ‘stress’ response pathways. These promoter elements mediate inducible expression of G-CSF, as well as its constitutive regulation in carcinoma cells [49–51]. A recent profiling of signaling protein activation and cytokine release in RAW 264.7 cells led to the prediction that the p38 MAP kinase pathway also plays an important role in G-CSF production [52], a hypothesis that requires formal examination via gene knockdown or deletion experiments.
Figure 1.
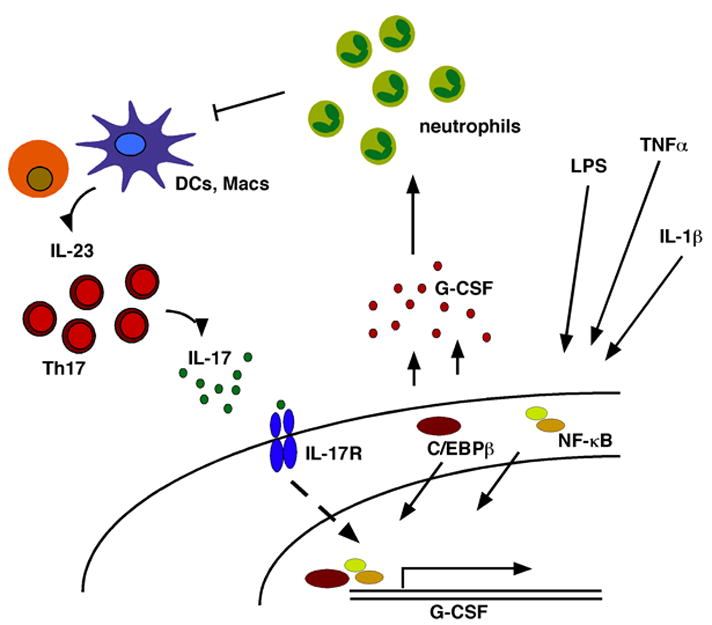
Schematic diagram of pathways regulating G-CSF and neutrophil production. (Right) Inflammatory stimuli in the extracellular microenvironment, such as LPS, TNFα and IL-1β act on target cells (not to scale) to induce G-CSF expression via intracellular signaling molecules such as NF-κB and C/EBPβ. (Left) IL-17 production by Th17 cells activates IL-17R signal transduction, which promotes G-CSF expression. G-CSF can be regulated by transcriptional and post-transcriptional mechanisms. (Center) The increase in G-CSF synthesis stimulates neutrophil production in the bone marrow (anatomical details are not shown). Circulating neutrophils negatively regulate the production of IL-23 and Th17 cells, providing a feedback system to control G-CSF synthesis.
Interleukin-17 (IL-17) has emerged as an important regulator of G-CSF and neutrophil production in vivo. IL-17 is synthesized by a specialized class of activated CD4+ T cells (Th17) that are best known for their contribution to pathologic inflammation [53–56]. Th17 cells reside in the intestinal lamina propria under steady state conditions [57], and can be induced during infection with Candida albicans [58]. Recently a specialized subset of intestinal dendritic cells was found to elicit Th17 differentiation via TGFβ and IL-10 [59], indicating that the gut may be a key source of IL-17 production in basal conditions.
IL-17 signaling through the IL-17 receptor induces expression of G-CSF (Fig. 1), as well as certain CXC-related chemokines [60]. IL-17 stimulates G-CSF- and stem cell factor- (SCF) dependent granulopoiesis and activates neutrophil recruitment during bacterial infection [60–62] (Fig. 1). Thus, in Th17-mediated pathologic inflammation, the neutrophil response elicited by IL-17-dependent regulation of G-CSF may play a role in the initiation and maintenance of inflammation. It is less clear how IL-17 production by resident intestinal Th17 cells affects G-CSF synthesis and neutrophil production under steady state conditions. This is an area that should be explored to determine the contribution of IL-17-elicited neutrophils to mucosal immunity. Regardless, a major function of the IL-17/G-CSF/neutrophil axis is likely to operate during the adaptive phase of an immune response since it is elicited by activated CD4+ Th17 cells. This cytokine cascade may provide a mechanism to stimulate neutrophils during chronic or unresolved infections.
Neutrophil turnover has been shown recently to play an important role in the homeostatic regulation of IL-17 and its control of granulopoiesis [56]. When neutrophil migration into tissues is blocked by adhesion molecule deficiency, macrophages and dendritic cells secrete excessive levels of IL-23, a key cytokine that drives Th17 development and IL-17 production [53–55], leading to increased G-CSF-dependent granulopoiesis (Fig. 1). Neutrophil phagocytosis by macrophages and dendritic cells suppresses their production of IL-23, thus decreasing IL-17 synthesis and G-CSF-dependent granulopoiesis [56] (Fig. 1). Therefore, IL-17 regulates granulopoiesis by its control of G-CSF expression; circulating neutrophils can act in a negative feedback loop to block excessive production of Th17 cells and IL-17. These studies demonstrate that G-CSF synthesis and neutrophil production are under exquisite control. Moreover, G-CSF-dependent neutrophil production is linked to responses in both the innate and adaptive immune systems (Fig. 1). The molecular details of the pathways leading to G-CSF production remain poorly understood, and particularly how these pathways operate in vivo in steady state and infection settings. For example, the signaling mechanisms that are used by IL-17, and its function in natural infection settings, are unresolved. Similarly, we have little knowledge of the mechanisms controlling the transcriptional activation of the G-CSF promoter or G-CSF mRNA turnover, despite the importance of this cytokine in immunity.
Characterization of the G-CSF receptor (G-CSFR) and its signal transduction mechanisms
The G-CSFR (molecular weight ~100,000–130,000) shares structural similarity with Type I cytokine receptors, containing a conserved cytokine receptor homologous (CRH) domain, an Ig-like domain and three fibronectin type III-like domains in the extracellular region; a transmembrane region; and an intracellular region lacking intrinsic catalytic activity [63–66]. Atomic resolution of the complex between human G-CSF and the ligand-binding domain of the murine or human G-CSFRs revealed a 2:2 ligand:receptor stoichiometry. The human G-CSFR forms a “cross-over” complex in association with G-CSF, which is tethered at the membrane distal Ig-like domains [67]. Ligand binding is mediated by contacts with residues in the Ig domain of one receptor monomer and the membrane-proximal CRH region from the second receptor monomer [67]. Although the G-CSFR operates as a homo-oligomer, the 3-dimensional structure of the human G-CSF/G-CSFR complex shows similarity with the IL-6/IL-6Rα/gp130 complex rather than other homodimeric type I cytokine receptor structures such as the erythropoietin (Epo)/Epo receptor or growth hormone receptor complexes [67–69]. Moreover, the structural configuration for the human G-CSF/human G-CSFR complex differs from the human G-CSF/murine G-CSFR complex [67, 70], although both are capable of eliciting biologic responses. Thus, the high degree of primary structure homology between human and murine ligands renders distinct structures of an activated receptor complex, indicating flexibility in the mechanisms to initiate the G-CSFR signal transduction cascade. This is useful knowledge for the prospective design and characterization of small molecule G-CSF mimetics.
G-CSFR expression has been detected in both hematopoietic and non-hematopoietic cell types including hematopoietic stem and progenitor cells, the placenta, neurons within the central nervous system, adult neural stem cells, endothelial cells, and cardiomyocytes [63, 71–74]. In the hematopoietic system, the G-CSFR shows predominant expression within the myeloid lineage [63–65]. The G-CSFR is detected on granulocytic lineage cells, as well as a subset of monocytes and blast cells, but is not found on erythrocytes, lymphocytes or eosinophils [28]. Activation of G-CSFR signal transduction proceeds by a mechanism that is shared with other cytokine receptors, in which associated Jak protein tyrosine kinases undergo trans-phosphorylation and full kinase activation upon cytokine binding. Jak1 and Jak2 are activated by the G-CSFR [75, 76]. While individual deletion of the Jak1 or Jak2 genes does not affect the level of fetal liver granulocytes or granulocytic progenitors (e.g., CFU-G), respectively, [77, 78], Jak1, but not Jak2, appears to play a non-redundant role in the G-CSFR signaling pathway [79]. Early structure/function studies of the G-CSFR showed that the cytoplasmic domain contained regions with distinct signaling functions. The membrane-proximal region, which is now known to couple to the Jak kinases, is essential for G-CSF-mediated proliferation. The membrane-distal region of the G-CSFR regulates inducible gene expression, neutrophil marker protein induction and granulocytic differentiation [80–82]. This region of the G-CSFR contains 4 tyrosine residues (in humans, Y704, Y729, Y744, Y764; in mice, Y703, Y728, Y743, Y763; see Fig. 2), which undergo G-CSF-dependent phosphorylation mediated by the receptor-associated Jak kinases. The G-CSFR phosphotyrosines serve to recruit intracellular signaling molecules to the ligand-activated receptor complex via SH2 domain/phosphotyrosine interactions [83–90] (for review, see [91]) (Fig. 2).
Figure 2.
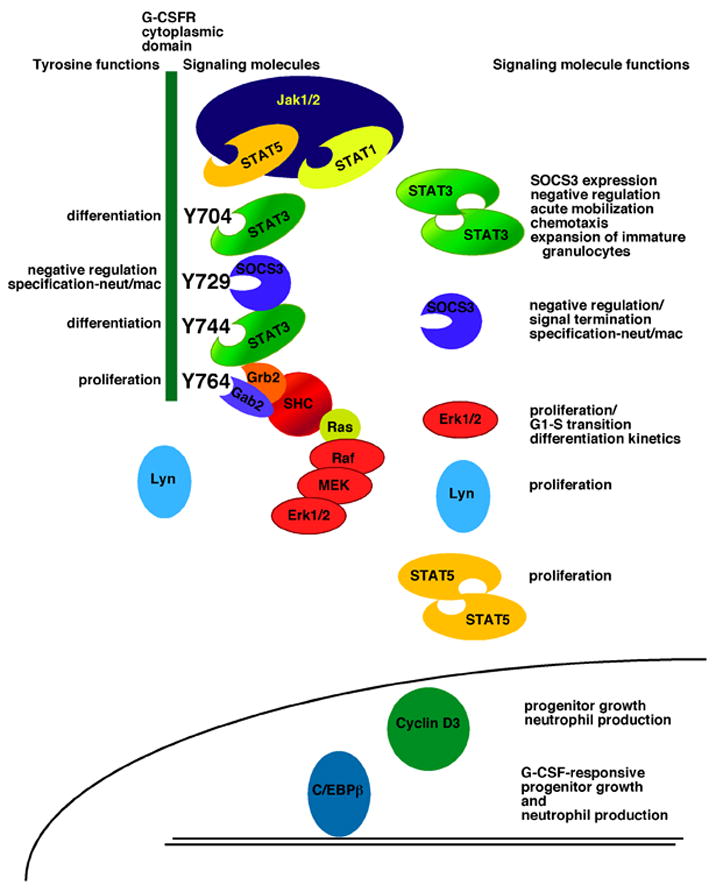
Functions of G-CSFR tyrosine residues and receptor-activated signaling pathways. The intracellular region of the G-CSFR is illustrated (narrow green box, left; not drawn to scale), with the four tyrosine residues highlighted in black. On the left of each tyrosine residue are functions that have been assigned by in vitro and in vivo studies, including proliferation, differentiation and macrophage/granulocyte lineage-specification. On the right of each tyrosine, representations of signaling proteins that couple to specific residues are shown. The function of these molecules is listed on the right side of the figure. At the lower portion of the figure, a schematic diagram of the nucleus of a granulocytic progenitor is shown, along with additional signaling molecules that are required for ‘emergency’ granulopoiesis. References for tyrosine and signal protein function can be found in the text.
The precise role of G-CSFR tyrosine residues in mediating proliferation and differentiation signals from the receptor has been examined by enforced expression of engineered G-CSFRs in myeloid tissue culture cells and primary bone marrow progenitors [84–86, 92], as well as experiments with genetically recombined G-CSFR loci expressing mutated receptors [93–96]. Figure 2 summarizes the results of studies that have examined the role of G-CSFR-coupled signaling cascades, the majority of which become activated through their association with receptor phosphotyrosines. The G-CSFR cytoplasmic tyrosines are particularly crucial in delivering the G-CSF signal at sub-saturating ligand concentrations [88]. In saturating amounts of G-CSF, the receptor cytoplasmic tyrosine residues appear to be dispensable ex vivo [88], although it is not clear if this is the case in vivo since knock-in studies with a mutated G-CSFR lacking all 4 cytoplasmic tyrosines in the context of the full-length receptor have not been reported. In the background of a truncated G-CSFR, which lacks sequences encoding the 3 C-terminal tyrosine residues, mutation of the remaining tyrosine residue at position 704 (G-CSFR d715F) caused defects in neutrophil production and mobilization [94]. Minimally, it is possible that the G-CSFR tyrosine residues enable an amplification or enhancement of signaling responses in the context of the full-length receptor; this may be particularly crucial in delivering the G-CSF signal under conditions of demand or cellular stress. By contrast, it would not be surprising if the receptor tyrosines are absolutely necessary for G-CSF-responsive neutrophil regulation in vivo. Future studies to determine the function of G-CSFR tyrosines in the context of the full-length receptor are important for elucidating the structural requirements of a functional G-CSFR.
The G-CSFR stimulates numerous signal transduction proteins, including the STAT 1, 3 and 5 transcription factors, the Ras/Mek/Erk1/2 pathway, the Src-related kinases Lyn and Hck, the serine/threonine kinase Akt, and the Syk tyrosine kinase [76, 87, 90, 97–102] reviewed in [91] (see Fig. 2). These pathways are generally considered to transmit signals that induce and/or enhance granulocytic proliferation and differentiation [83, 85, 87, 102–106]. In addition, the G-CSFR physically associates with integrin α9β1, which augments receptor signaling and granulopoiesis in vivo [107]. The G-CSFR also couples to pathways that negatively regulate its signaling output and granulopoiesis-inducing function, including those mediated by the SHP-1 tyrosine phosphatase (e.g., hematopoietic cell phosphatase, HCP) and the suppressor of cytokine signaling 3 (SOCS3) protein [108, 109] (Fig. 2). Negative regulation of G-CSFR signaling is also accomplished by ligand-mediated internalization and degradation of the receptor [110].
Aside from immediate signaling cascades elicited by G-CSF, investigators have elucidated G-CSF-responsive genes. Several of these have established roles in cellular proliferation and/or myeloid biology. G-CSF-responsive genes include Mac1 (CD18/CD11b), FcγII/III, and neutrophil secondary granule proteins, which are important for neutrophil function [92, 104, 111]; immediate early genes including c-fos, Oncostatin M (OSM), IRF-1, egr-1, fibrinogen and SOCS3 [99]; and the myeloid transcription factors PU.1, C/EBPα, C/EBPβ and Gfi-1 [104, 112–114]. Surprisingly, however, in most cases we have a poor understanding of the mechanisms that connect the immediate G-CSFR signaling responses to the induction of G-CSF-responsive gene expression, and therefore to the precise cellular functions that are driven by specific G-CSFR signaling cascades. Certain genes have been identified as direct STAT-responsive genes, including SOCS3 and OSM [99, 115, 116]. A putative STAT binding site has been reported in the human Gfi-1 promoter but its function is unknown [117]. In other cases, such as PU.1 and C/EBPα, STATs are known to play a role but their exact mechanism is less clear [104, 114]. Beyond the STATs, the function of Ras/Mek/Erk1/2, Lyn, Hck, Akt, and Syk in G-CSF-dependent gene expression is even less well understood. In addition, many studies have used tissue culture systems to study candidate gene expression, and therefore we lack a comprehensive view of G-CSF-dependent transcriptional responses in primary granulocytes. Furthermore, our knowledge of the specific role of individual signaling pathways in steady state and ‘emergency’ conditions of granulopoiesis remains quite limited due to the lack of appropriate model systems to explore in vivo functions. The following section will discuss the available information.
G-CSF-responsive signaling pathways that control steady state and G-CSF-driven ‘emergency’ granulopoiesis
G-CSF and its receptor are essential for basal and stress-induced granulopoiesis [3, 23, 118, 119]. Mice deficient in G-CSF or G-CSFR have severe neutropenia and reduced levels (~50%) of late-stage neutrophil precursors in bone marrow under resting conditions [3, 118]. G-CSF−/− mice are unable to respond to infection with Listeria monocytogenes by stimulating granulopoiesis and induction of circulating neutrophils, thereby resulting in a markedly reduced ability to control the infection [3]. G-CSFR−/− mice fail to mount an increase in bone marrow or circulating neutrophils in response to G-CSF administration [93, 120]. These data highlight the necessity of the G-CSF/G-CSFR pathway in the anti-bacterial immune response, and furthermore demonstrate that the G-CSFR is solely responsible for transmitting G-CSF signals in vivo. A residual level of neutrophil production does continue in G-CSF−/− or G-CSFR−/− mice [3, 118], indicating separate mechanisms to induce neutrophil production. Interleukin-6 was identified as an independent regulator of granulopoiesis, as loss of IL-6 function worsened the neutropenia observed in G-CSFR−/− animals [121]. Conversely, G-CSF was found to provide a compensatory signal for the myeloid progenitor deficiency in mice lacking thrombopoietin [122]. It is presently unclear whether and how additional cytokine-dependent or –independent pathways contribute to the residual granulopoiesis found in G-CSF−/− or G-CSFR−/− mice. Regardless, the G-CSFR signaling pathway is absolutely crucial for regulating terminal neutrophil differentiation since the residual neutrophils found in G-CSFR−/− animals exhibit defective functional responses including impaired adhesion and chemoattractant-induced migration [123]. G-CSFR function is required throughout development of the neutrophil lineage, beginning with myeloid-committed progenitors in vivo [124]. The G-CSFR elicits specific signals to regulate granulopoiesis; for example, these can not be replaced by signaling through the cytoplasmic region of the related cytokine receptor, EpoR [93]. The identity of G-CSFR-mediated signaling cascades important for granulopoiesis have been relatively elusive, although a first glimpse came through experiments with G-CSFR d715F mice. These experiments revealed a role for the G-CSF-responsive transcription factor STAT3 in neutrophil production and function [94].
Initial studies investigating STAT3 function in vivo demonstrated an essential role in embryonic development and viability [125], therefore it was necessary to generate conditional knockout mice to examine STAT3 function in granulopoiesis. Several groups accomplished this using tissue-specific or inducible Cre recombinase mouse strains and mice carrying floxed STAT3 alleles. Surprisingly deletion of STAT3 in the hematopoietic compartment led to neutrophilia [103, 126–128] rather than neutrophil deficiency. Hematopoietic-specific STAT3 deletion results in a specific increase in late stage neutrophils (e.g., band/segmented) in bone marrow and elevated uptake of 3H-thymidine in G-CSF-stimulated granulocytes ex vivo [126, 127]. By contrast, normal levels of G-CSF-responsive colony-forming progenitor cells and immature granulocytic precursors, including promyelocytes, myelocytes and metamyelocytes, were found in STAT3-deficient bone marrow [126, 127]. These results point to a specific role for STAT3 in negatively regulating the terminal stages of bone marrow granulopoiesis and circulating neutrophil levels.
This negative regulatory role for STAT3 was surprising in light of studies that had pointed to its important function during neutrophil development [94, 104, 105], yet it meshed well with the finding that STAT3 controlled the expression of SOCS3 [115, 126], an inducible negative regulator of cytokine signaling responses [129, 130]. STAT3 deletion abrogates the ability of G-CSF to activate SOCS3 and causes enhanced G-CSFR signaling via STAT1 and Erk1/2 [126, 127]. Conditional deletion of SOCS3 in bone marrow progenitors leads to a phenotype in older mice (aged >17 weeks) that resembles aspects of the phenotype of hematopoietic conditional STAT3 knockout mice, albeit one that is observed much earlier in development (ages 3–8 weeks) [109, 128]. Aged bone marrow SOCS3-deficient mice contain elevated levels of late stage bone marrow granulocytes (e.g., metamyelocytes and neutrophils) and exhibit peripheral neutrophilia under steady state conditions. This is similar to the increase in late stage bone marrow granulocytes and circulating neutrophils found in bone marrow STAT3-deficient mice [103, 126–128]. Furthermore, SOCS3-deficient bone marrow cells show prolonged G-CSF-dependent STAT3 activation and enhanced responses to G-CSF ex vivo (e.g., proliferation, cloning frequency) [109], resembling STAT3-deficient granulocytes [126]. Following G-CSF injection, mice carrying hematopoietic SOCS3 deletion have elevated neutrophil production and mobilization in vivo, and they develop an inflammatory neutrophil infiltration into tissues, indicative of enhanced neutrophil activity [109]. Bone marrow STAT3-deficient mice also have increased levels of mature neutrophils in the peripheral blood after prolonged exposure (5 d) to G-CSF in vivo [103, 127]. These data collectively indicate that SOCS3 is a critical G-CSF/STAT3 target gene that controls the magnitude and duration of signaling from the G-CSFR, thereby regulating the production of neutrophils from the bone marrow. The conserved SOCS box domain of SOCS3, which mediates binding to the elongin B/C ubiquitin ligase complex, is responsible for controlling STAT3 activation via the G-CSFR and for maintaining regulated output of neutrophils during exposure to pharmacologic levels of G-CSF in vivo [131]. The SOCS box is dispensable for regulating steady state circulating neutrophil levels, although increased levels of G-CSF-responsive progenitors were found in bone marrow samples of mice expressing the SOCS box-deficient SOCS3 allele (SOCS3ΔSB) suggesting that the SOCS box does control homeostatic progenitor growth/survival mechanisms [131]. SOCS3 attenuates G-CSFR-mediated signals by interacting with a membrane-proximal lysine residue in the receptor cytoplasmic region, to regulate receptor ubiquitination and routing to lysosomes [132].
Elucidation of the G-CSFR/STAT3/SOCS3 axis did not explain the previously identified roles for STAT3 in granulocytic differentiation, myeloid transcription factor expression, and neutrophil function [94, 104, 112, 114, 133]. Moreover, the role of STAT3 in G-CSF-driven ‘emergency’ granulopoiesis and neutrophil function had not been examined thoroughly. To address these questions, we utilized mice with bone marrow conditional STAT3 deletion (Tie2cre/STAT3Δ/flox mice, referred to herein as STAT3-deficient) [103]. We found that STAT3 was critical for neutrophil chemotaxis toward ligands for the CXCR2 receptor (e.g., MIP-2 and KC) and for regulating chemoattractant-stimulated actin polymerization, demonstrating essential roles for STAT3 in cellular migration pathways that are vital to the mature neutrophil. Furthermore, STAT3-deficient mice fail to induce acute neutrophil mobilization from the bone marrow in response to G-CSF or MIP-2 (reference [103] and ADP and SSW, unpublished results). Following prolonged exposure to G-CSF (e.g., 5 d), STAT3-deficient mice are unable to upregulate the frequency or absolute numbers of immature neutrophils in the bone marrow or peripheral blood, unlike wild type animals. Rather, STAT3-deficient mice demonstrate a skewed long-term mobilization response which is characterized by excessive mature neutrophil numbers and reduced immature neutrophils in the peripheral circulation [103]. We found that these functions of STAT3 were independent of SOCS3, as mice with myeloid-specific deletion of SOCS3 exhibit a normal frequency of immature neutrophils in response to G-CSF, normal G-CSF-driven neutrophil mobilization and intact neutrophil chemotactic function [103]. In addition, estimation of total neutrophil numbers in hematopoietic tissues indicated that STAT3 regulates the quality of the neutrophil response (e.g., excessive mature neutrophils and impaired immature neutrophils relative to wild type), but does not control the total neutrophil output following exposure to G-CSF [103]. These data indicate that G-CSF and STAT3 control additional genes beyond SOCS3 that regulate crucial steps in G-CSF-driven granulopoiesis and neutrophil function. The identity of these genes is important for understanding the molecular mechanisms of G-CSF action, which may lead to novel approaches for diseases associated with neutrophil deregulation.
Additional transcription factors and cell cycle regulators controlling G-CSF-driven granulopoiesis
Steady state granulopoiesis is dependent upon an array of transcription factors that function at distinct developmental stages (e.g., RUNX1, SCL, PU.1, IRF8, LEF-1, Gfi-1, C/EBPα and C/EBPε) [134–137]. The developmental expression of these transcription factors [138], and their role in basal myelopoiesis has been discussed elsewhere [134–136] and therefore will not be reviewed at length herein.
In comparison, the pathway(s) that control G-CSF-driven ‘emergency’ granulopoiesis at the transcriptional level have been relatively understudied. This relates, in part, to the early developmental defects in the hematopoietic system or the neutrophil lineage in the absence of key myeloid transcriptional regulators such as PU.1 or C/EBPα [135]. Analysis of the role of these factors in ‘emergency’ granulopoiesis requires developmentally staged gene deletion/knockdown to bypass the early developmental block and enable the maturation of G-CSF-responsive precursor cells [139]. Certain key myeloid transcriptional regulators such as Gfi-1 and C/EBPε have late-stage developmental defects, which allows investigation into their role in G-CSF-driven ‘emergency’ granulopoiesis. For example, under steady state conditions, Gfi-1−/− mice lack morphologically mature neutrophils and instead show accumulation of ‘atypical’ Gr-1+/CD11b+ myeloid cells. These atypical myeloid cells lack granules and neutrophil granule protein transcripts, yet possess limited neutrophil functions such as oxidative burst and phagocytosis [140]. Gfi-1−/− mice respond to G-CSF treatment by inducing the level of ‘atypical’ myeloid cells, yet they fail to upregulate circulating mature neutrophils. Thus, Gfi-1 appears dispensable for a G-CSF-dependent expansion/proliferative phase in myelopoiesis. By contrast, Gfi-1 is critical for regulating the terminal stages of neutrophil maturation during basal and G-CSF-driven granulopoiesis.
The transcription factor C/EBPα is essential for the transition between the common myeloid progenitor (CMP) stage and the granulocyte-macrophage progenitor stage, yet appears dispensable for later developmental stages of granulopoiesis under steady state conditions [139, 141]. In addition, neutrophil development could be driven in C/EBPα−/− hematopoietic progenitors or C/EBPα−/− myeloid cell lines in response to other myeloid cytokines such as interleukin-3 and/or granulocyte-macrophage colony-stimulating factor [142–144], indicating the presence of alternative pathways to induce granulopoiesis. To examine the underlying cause, expression of other C/EBP family members was examined in primary myeloid progenitors during cytokine exposure ex vivo [113]. These studies revealed that C/EBPβ mRNA levels were selectively upregulated in granulocytic-macrophage progenitors (GMPs) in response to G-CSF. By contrast, C/EBPα was not induced by G-CSF in hematopoietic stem cells (HSCs), CMPs or GMPs [113]. G-CSF-dependent induction of C/EBPβ suggested it may control important aspects of emergency ‘granulopoiesis’, a prediction that was confirmed using mice deficient in C/EBPβ [113]. C/EBPβ is required for G-CSF-driven expansion of the granulocytic compartment, G-CSF-dependent induction of circulating neutrophil levels and efficient bacterial clearance in vivo [113], yet is strikingly dispensable for granulopoiesis under basal conditions [145–147]. These results highlight the importance of G-CSF-dependent transcriptional response pathways under conditions of ‘demand-driven’ granulopoiesis, and furthermore indicate that new studies should not remain limited to previously identified myeloid transcription factors. Moreover, it is well established that the expression level of lineage-specification transcription factors, and the ratio between key regulators within an individual lineage, is essential for directing correct hematopoietic lineage maturation and suppressing malignant transformation [134, 135]. While G-CSF has been shown to influence the expression of several important myeloid transcription factors [104, 112–114], it is not known if this contributes to neutrophil maturation in basal or ‘demand-driven’ conditions. This question can be addressed by deletion of the G-CSF-responsive signaling pathways that are necessary for transcription factor regulation, and/or by the genetic manipulation of transcription factor levels in developing granulocytes.
An important consequence of the G-CSF-dependent transcriptional response is how it impinges upon the cell cycle machinery to control the proliferation of myeloid progenitor cells and granulocytic precursors. Cyclin D3−/− mice have a profound defect in neutrophil production, exhibiting neutropenia in steady state conditions and an inability to mount an increase in circulating neutrophil levels in response to G-CSF administration, rendering extreme susceptibility to infection with Listeria monocytogenes [148]. Cyclin D3 expression is induced in bone marrow cells upon G-CSF administration in vivo [148], suggesting it is transcriptionally regulated by a G-CSF-dependent signaling cascade. Elucidation of the key molecular elements of G-CSF-mediated cyclin D3 regulation will provide an important step toward understanding the mechanisms that regulate G-CSF-driven granulopoiesis. Unraveling these is crucial for determining the host pathways that mount a functional neutrophil response during infection.
G-CSF function in infection
Perhaps not surprisingly, G-CSF has distinct functions depending on the type of infection and its route of delivery. Considering the vast array of microbes that elicit disease, and the number of host-derived molecules that are involved in the immune response, it is reasonable to expect that individual cytokines will demonstrate selective and unique functions. Studies with G-CSF have utilized well-characterized models that mimic natural and harmful infections. G-CSF has a critical role in the ‘emergency’ response to infection with Listeria monocytogenes, a bacterial pathogen that propagates within macrophages. In an intravenous infection model using G-CSF-deficient mice, G-CSF was required for the induction of circulating neutrophil and monocyte levels elicited by L. monocytogenes. G-CSF also plays a critical role in suppressing the bacterial load in spleen and liver [3]. Following intraperitoneal infection, G-CSF was required for maintaining total bone marrow cell numbers, a response that is likely to be related to its ability to preserve bone marrow colony-forming (e.g., progenitor) cells in steady state and during L. monocytogenes exposure [23]. In addition, G-CSF is necessary for macrophage phagocytosis function and the induction of absolute peritoneal macrophage numbers elicited by L. monocytogenes [23, 149]. Certain aspects of the response to L. monocytogenes are G-CSF-independent, for example neutrophil recruitment into the infected peritoneum is unaffected by G-CSF deficiency [23]. Thus, with L. monocytogenes, G-CSF plays a major role in controlling hematopoietic progenitor and macrophage responses, as well as circulating neutrophil levels, which are collectively required for bacterial clearance.
Studies with G-CSFR-deficient mice have also emphasized the importance of the G-CSF/receptor axis in infection. Gregory et al. [119] recently investigated the role of G-CSFR in response to infection with Pseudomonas aeruginosa, a Gram-negative bacteria that poses a major risk to hospital and cystic fibrosis patients. Using an intratracheal delivery model to mimic the pulmonary infection in cystic fibrosis patients, G-CSFR was found to be a necessary component of the host response to P. aeruginosa. G-CSFR is required for the systemic induction of circulating neutrophil levels, bacterial clearance from the lung, and the prevention of extensive pulmonary damage during infection. Furthermore, neutrophil survival in the infected lung tissue is impaired in the absence of G-CSFR, suggesting that G-CSF signals are important for controlling neutrophil survival at the site of infection. The neutrophil response to P. aeruginosa is thus dependent upon G-CSFR signal transduction, and this response is necessary for the resolution of infection.
G-CSF has more modest function during infection with Candida albicans, a fungal pathogen that frequently causes severe infection in immunocompromised individuals. Following intravenous C. albicans infection, circulating neutrophil levels were induced in both wild type and G-CSF-deficient animals and the latter showed elevated levels of myeloid progenitors in bone marrow [150]. As suggested by the authors of this report [150], these data strongly imply the presence of additional growth factors or alternative mechanisms that generate neutrophil responses in certain stress situations. This idea is underscored by the fact that G-CSF-deficiency does not cause an absolute blockade in steady state neutrophil production [3]. The identity of G-CSF-independent neutrophil regulatory pathways are unknown, with the exception of IL-6 [121], and are a subject of much interest as they could provide novel approaches for the treatment of neutropenia. In summary, therefore, the function of G-CSF is dependent upon the type and route of infection. Moreover, it is important to consider that any functions revealed are those that have been assayed directly; G-CSF is likely to have broad and varied activity for a number of bacterial pathogens, which future research should reveal.
Clinical use of G-CSF and future avenues for research and development
G-CSF is used to treat neutropenia resulting from immunosuppressive chemotherapy or severe congenital disorders [6, 36, 37, 40, 151–154], which cause extreme sensitivity to life-threatening bacterial infection. G-CSF reduces the degree and duration of neutropenia in individuals who receive chemotherapy, and protects patients with congenital neutropenia from acute infection and early lethality [6, 36, 37, 40, 151–154] (Fig. 3). Concerns have been raised, however, about a potential association between the clinical use of G-CSF and development of acute myeloid leukemia (AML) [7–11]. This was first questioned in individuals with severe congenital neutropenia (SCN) (Fig. 3), as a subset have acquired a mutant G-CSFR allele that encodes a truncated protein lacking sequences necessary for negative regulation and induction of differentiation signaling; the presence of the mutant G-CSFR correlated with the development of AML in these patients [110, 155–162]. The mutant G-CSFR was originally suggested as a potential cause of the neutrophil maturation arrest at the promyelocyte/myelocyte stage of development and the profound absolute neutropenia observed in SCN patients, as well as a potential contributing factor to leukemic transformation [155, 157, 163, 164] (Fig. 3). It is now recognized that distinct molecular mechanisms underlie the neutrophil maturation arrest in SCN, including mutations in the genes for neutrophil elastase and Hax-1, as well as reduced expression of the transcription factor LEF-1 [4, 137, 164–169]. Thus, the acquired G-CSFR mutation may provide a growth advantage to myeloid progenitor cells that already have a maturation defect, increasing the risk of additional genetic mutations and leukemic transformation [170]. A single nucleotide polymorphism in the G-CSFR gene that encodes Lys785, in place of Glu785, is associated with high-risk myelodysplastic syndrome [171], underscoring the idea that perturbations in G-CSFR signaling function contribute to hematological disease. Importantly, in SCN patients, G-CSF therapy significantly reduces the risk of mortality from sepsis, from 6–7% per year in the pre-G-CSF era to 0.9% per year with G-CSF treatment [9]. SCN patients do have an increased risk of developing MDS/AML over time on G-CSF therapy, with 2.9% per year incidence rate after 6 years of treatment compared with 8.0% per year after 12 years [9]. The risk of sepsis mortality remains constant over time [9]. Moreover, a subset of SCN patients that demonstrated less responsiveness to G-CSF, and have thus been treated with higher levels of the cytokine, have an elevated risk of MDS/AML as well as mortality from sepsis [9]. SCN is a pre-leukemic condition; the extension of lifespan with G-CSF therapy may therefore contribute to the increased risk of MDS/AML transformation. Moreover, inherent differences may exist between SCN patients that show robust or poor responses to G-CSF. More severe forms of the disease (i.e., reduced ability to overcome the neutrophil maturation defect upon G-CSF therapy) may have even greater predisposition to leukemic transformation. G-CSF remains the therapy of choice for SCN since it dramatically prevents the likelihood of mortality from sepsis. Stem cell transplantation may provide an alternative treatment for individuals that show poor responsiveness to G-CSF, as these patients are at the highest risk of developing MDS/AML [9].
Figure 3.

A timeline of major events in the development and use of G-CSF in the clinic. Left to right, the timeline highlights principal findings that are relevant to the therapeutic use of G-CSF including the initial description of congenital neutropenia [163] and leukemia in SCN [184], purification and cloning of human G-CSF [12, 15, 16], first clinical use of G-CSF for neutropenia resulting from chemotherapy or congenital origin [36, 37, 152–154], cloning of G-CSFR and identification of receptor functional domains [63, 64, 80–82], initial reports of G-CSFR mutations in SCN [155–157], functional studies of G-CSFR mutants found in SCN [95, 96, 110, 161], and associations between the therapeutic use of G-CSF and the development of MDS/AML [9, 170, 185]. Due to space limitations, all significant references could not be included. (Timeline: Purple, 1956-1959; Pink, 1960-1969; Yellow, 1970-1979; Green, 1980-1989; Blue, 1990-1999; Red, 2000-present)
Summary and future prospectives
While this review focused solely upon neutrophil regulation by G-CSF, it is important to highlight recent discoveries that show G-CSF operates in other immune cell types and non-hematopoietic tissues. These include a role for G-CSF in mediating T cell tolerance by influencing the development and/or function of antigen-presenting cells, an activity that could prove useful in multiple clinical applications such as stem cell transplantation to enhance graft-versus-leukemia and concomitantly suppress graft-versus-host responses [45, 172–176]. G-CSF has also been shown to improve cardiomyocyte survival and cardiac repair following myocardial infarction in animal models, via a process that appears to require the transcriptional activity of STAT3 [74]. Neuronal survival and recovery from cortical ischemia is similarly protected by G-CSF, which is capable of crossing the blood-brain barrier and of stimulating neuronal progenitor responses in vivo [73, 177]. These findings have suggested a potential for broadening the clinical uses of G-CSF; careful in vivo experimentation with animal models and appropriately designed preclinical/clinical studies are required to thoroughly evaluate these possibilities [177–181]. Lastly, within the neutrophil lineage, G-CSF can exacerbate both acute and chronic arthritis, raising the possibility that G-CSF antagonists may be useful for the clinical treatment of joint inflammation [182, 183].
The advent of precise genetic tools to investigate G-CSF signaling molecule function and new methods to visualize immune cells in vivo, such as multi-photon microscopy, should yield a thorough understanding of the molecular mechanisms utilized by G-CSF during an active immune response. In the near future, it will be important to study the function of G-CSF in the host response to pathogens that are prevalent in both healthy and immunocompromised populations, and not to limit investigation to a small number of pathogens that are readily handled in the laboratory. Of course, this will require collaborative arrangements between immunologists, signal transduction experts and microbiologists; however, the efforts should yield information that will facilitate the development of new agents to treat neutropenic individuals and those who are refractory to G-CSF, and may provide novel approaches to common and deadly pathogens. Moreover, a thorough understanding of G-CSF function is necessary to reduce any potential risks that arise from its long-term clinical use and to develop methods to regulate neutrophil-mediated inflammatory disease.
Acknowledgments
We thank Drs. Seth Corey, Huiyuan Zhang and Chen Dong for their critical review of this manuscript. We thank members of the Watowich lab for stimulating discussions that contributed to ideas presented in this review. ADP acknowledges support from an NIH predoctoral training grant in Cancer Immunology (T32-CA-09598-16) and an American Legion Auxiliary Award during her graduate studies at UT MD Anderson Cancer Center and the UT Graduate School of Biomedical Sciences. Related research in SSW’s lab has been supported by grants from the NIH (CA77447), AHA Texas Affiliate (0255899Y, 0455143Y), Gillson Longenbaugh Foundation, the MDACC Institutional Grants Program and a Preclinical Research Agreement with Amgen, Inc. Due to space limitations, the authors would like to apologize for the fact that they have not been able to include all relevant references.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huising MO, Kruiswijk CP, Flik G. Phylogeny and evolution of class-I helical cytokines. J Endocrinol. 2006;189:1–25. doi: 10.1677/joe.1.06591. [DOI] [PubMed] [Google Scholar]
- 2.Lutfalla G, Roest Crollius H, Stange-Thomann N, Jaillon O, Mogensen K, Monneron D. Comparative genomic analysis reveals independent expansion of a lineage-specific gene family in vertebrates: the class II cytokine receptors and their ligands in mammals and fish. BMC Genomics. 2003;4:29. doi: 10.1186/1471-2164-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–46. [PubMed] [Google Scholar]
- 4.Zeidler C, Schwinzer B, Welte K. Congenital neutropenias. Rev Clin Exp Hematol. 2003;7:72–83. [PubMed] [Google Scholar]
- 5.Lakshman R, Finn A. Neutrophil disorders and their management. J Clin Pathol. 2001;54:7–19. doi: 10.1136/jcp.54.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welte K, Gabrilove J, Bronchud MH, Platzer E, Morstyn G. Filgrastim (r-metHuG-CSF): the first 10 years. Blood. 1996;88:1907–29. [PubMed] [Google Scholar]
- 7.Smith RE, Bryant J, DeCillis A, Anderson S. Acute myeloid leukemia and myelodysplastic syndrome after doxorubicin-cyclophosphamide adjuvant therapy for operable breast cancer: the National Surgical Adjuvant Breast and Bowel Project Experience. J Clin Oncol. 2003;21:1195–204. doi: 10.1200/JCO.2003.03.114. [DOI] [PubMed] [Google Scholar]
- 8.Socie G, Mary JY, Schrezenmeier H, Marsh J, Bacigalupo A, Locasciulli A, Fuehrer M, Bekassy A, Tichelli A, Passweg J. Granulocyte-stimulating factor and severe aplastic anemia: a survey by the European Group for Blood and Marrow Transplantation (EBMT) Blood. 2007;109:2794–6. doi: 10.1182/blood-2006-07-034272. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Fier C, Freedman M, Kannourakis G, Kinsey S, Schwinzer B, Zeidler C, Welte K, Dale DC. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107:4628–35. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hershman D, Neugut AI, Jacobson JS, Wang J, Tsai WY, McBride R, Bennett CL, Grann VR. Acute myeloid leukemia or myelodysplastic syndrome following use of granulocyte colony-stimulating factors during breast cancer adjuvant chemotherapy. J Natl Cancer Inst. 2007;99:196–205. doi: 10.1093/jnci/djk028. [DOI] [PubMed] [Google Scholar]
- 11.Freedman MH, Bonilla MA, Fier C, Bolyard AA, Scarlata D, Boxer LA, Brown S, Cham B, Kannourakis G, Kinsey SE, Mori PG, Cottle T, Welte K, Dale DC. Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood. 2000;96:429–36. [PubMed] [Google Scholar]
- 12.Nicola NA, Metcalf D, Johnson GR, Burgess AW. Separation of functionally distinct human granulocyte-macrophage colony-stimulating factors. Blood. 1979;54:614–27. [PubMed] [Google Scholar]
- 13.Nicola NA, Metcalf D, Matsumoto M, Johnson GR. Purification of a factor inducing differentiation in murine myelomonocytic leukemia cells. Identification as granulocyte colony-stimulating factor. J Biol Chem. 1983;258:9017–23. [PubMed] [Google Scholar]
- 14.Roberts AW, Nicola NA. Granulocyte colony-stimulating factor. In: Garland JM, Quesenberry PJ, Hilton DJ, editors. Colony-Stimulating Factors, Molecular and Cellular Biology. 2. New York: Marcel Dekker, Inc; 1997. pp. 203–225. [Google Scholar]
- 15.Nagata S, Tsuchiya M, Asano S, Kaziro Y, Yamazaki T, Yamamoto O, Hirata Y, Kubota N, Oheda M, Nomura H, et al. Molecular cloning and expression of cDNA for human granulocyte colony-stimulating factor. Nature. 1986;319:415–8. doi: 10.1038/319415a0. [DOI] [PubMed] [Google Scholar]
- 16.Souza LM, Boone TC, Gabrilove J, Lai PH, Zsebo KM, Murdock DC, Chazin VR, Bruszewski J, Lu H, Chen KK, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science. 1986;232:61–5. doi: 10.1126/science.2420009. [DOI] [PubMed] [Google Scholar]
- 17.Tsuchiya M, Asano S, Kaziro Y, Nagata S. Isolation and characterization of the cDNA for murine granulocyte colony-stimulating factor. Proc Natl Acad Sci U S A. 1986;83:7633–7. doi: 10.1073/pnas.83.20.7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zsebo KM, Cohen AM, Murdock DC, Boone TC, Inoue H, Chazin VR, Hines D, Souza LM. Recombinant human granulocyte colony stimulating factor: molecular and biological characterization. Immunobiology. 1986;172:175–84. doi: 10.1016/S0171-2985(86)80097-3. [DOI] [PubMed] [Google Scholar]
- 19.Strife A, Lambek C, Wisniewski D, Gulati S, Gasson JC, Golde DW, Welte K, Gabrilove JL, Clarkson B. Activities of four purified growth factors on highly enriched human hematopoietic progenitor cells. Blood. 1987;69:1508–23. [PubMed] [Google Scholar]
- 20.Metcalf D, Nicola NA. Proliferative effects of purified granulocyte colony-stimulating factor (G-CSF) on normal mouse hemopoietic cells. J Cell Physiol. 1983;116:198–206. doi: 10.1002/jcp.1041160211. [DOI] [PubMed] [Google Scholar]
- 21.Platzer E, Welte K, Gabrilove JL, Lu L, Harris P, Mertelsmann R, Moore MA. Biological activities of a human pluripotent hemopoietic colony stimulating factor on normal and leukemic cells. J Exp Med. 1985;162:1788–801. doi: 10.1084/jem.162.6.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood. 2002;100:854–861. doi: 10.1182/blood.v100.3.854. [DOI] [PubMed] [Google Scholar]
- 23.Zhan Y, Lieschke GJ, Grail D, Dunn AR, Cheers C. Essential roles for granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF in the sustained hematopoietic response of Listeria monocytogenes-infected mice. Blood. 1998;91:863–9. [PubMed] [Google Scholar]
- 24.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78:2791–808. [PubMed] [Google Scholar]
- 25.Watari K, Asano S, Shirafuji N, Kodo H, Ozawa K, Takaku F, Kamachi S. Serum granulocyte colony-stimulating factor levels in healthy volunteers and patients with various disorders as estimated by enzyme immunoassay. Blood. 1989;73:117–22. [PubMed] [Google Scholar]
- 26.Kawakami M, Tsutsumi H, Kumakawa T, Abe H, Hirai M, Kurosawa S, Mori M, Fukushima M. Levels of serum granulocyte colony-stimulating factor in patients with infections. Blood. 1990;76:1962–4. [PubMed] [Google Scholar]
- 27.Cheers C, Haigh AM, Kelso A, Metcalf D, Stanley ER, Young AM. Production of colony-stimulating factors (CSFs) during infection: separate determinations of macrophage-, granulocyte-, granulocyte-macrophage-, and multi-CSFs. Infect Immun. 1988;56:247–51. doi: 10.1128/iai.56.1.247-251.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicola NA, Metcalf D. Binding of 125I-labeled granulocyte colony-stimulating factor to normal murine hemopoietic cells. J Cell Physiol. 1985;124:313–21. doi: 10.1002/jcp.1041240222. [DOI] [PubMed] [Google Scholar]
- 29.Lord BI, Bronchud MH, Owens S, Chang J, Howell A, Souza L, Dexter TM. The kinetics of human granulopoiesis following treatment with granulocyte colony-stimulating factor in vivo. Proc Natl Acad Sci U S A. 1989;86:9499–503. doi: 10.1073/pnas.86.23.9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lord BI, Molineux G, Pojda Z, Souza LM, Mermod JJ, Dexter TM. Myeloid cell kinetics in mice treated with recombinant interleukin-3, granulocyte colony-stimulating factor (CSF), or granulocyte-macrophage CSF in vivo. Blood. 1991;77:2154–9. [PubMed] [Google Scholar]
- 31.Sung L, Dror Y. Clinical applications of granulocyte-colony stimulating factor. Front Biosci. 2007;12:1988–2002. doi: 10.2741/2204. [DOI] [PubMed] [Google Scholar]
- 32.Welte K, Bonilla MA, Gillio AP, Boone TC, Potter GK, Gabrilove JL, Moore MA, O’Reilly RJ, Souza LM. Recombinant human granulocyte colony-stimulating factor. Effects on hematopoiesis in normal and cyclophosphamide-treated primates. J Exp Med. 1987;165:941–8. doi: 10.1084/jem.165.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bensinger W, Singer J, Appelbaum F, Lilleby K, Longin K, Rowley S, Clarke E, Clift R, Hansen J, Shields T, et al. Autologous transplantation with peripheral blood mononuclear cells collected after administration of recombinant granulocyte stimulating factor. Blood. 1993;81:3158–63. [PubMed] [Google Scholar]
- 34.Weaver CH, Buckner CD, Longin K, Appelbaum FR, Rowley S, Lilleby K, Miser J, Storb R, Hansen JA, Bensinger W. Syngeneic transplantation with peripheral blood mononuclear cells collected after the administration of recombinant human granulocyte colony-stimulating factor. Blood. 1993;82:1981–4. [PubMed] [Google Scholar]
- 35.Baumann I, Testa NG, Lange C, de Wynter E, Luft T, Dexter TM, van Hoef ME, Howell A. Haemopoietic cells mobilised into the circulation by lenograstim as alternative to bone marrow for allogeneic transplants. Lancet. 1993;341:369. doi: 10.1016/0140-6736(93)90166-e. [DOI] [PubMed] [Google Scholar]
- 36.Bronchud MH, Scarffe JH, Thatcher N, Crowther D, Souza LM, Alton NK, Testa NG, Dexter TM. Phase I/II study of recombinant human granulocyte colony-stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. Br J Cancer. 1987;56:809–13. doi: 10.1038/bjc.1987.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morstyn G, Campbell L, Souza LM, Alton NK, Keech J, Green M, Sheridan W, Metcalf D, Fox R. Effect of granulocyte colony stimulating factor on neutropenia induced by cytotoxic chemotherapy. Lancet. 1988;1:667–72. doi: 10.1016/s0140-6736(88)91475-4. [DOI] [PubMed] [Google Scholar]
- 38.Duhrsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood. 1988;72:2074–81. [PubMed] [Google Scholar]
- 39.Molineux G, Pojda Z, Hampson IN, Lord BI, Dexter TM. Transplantation potential of peripheral blood stem cells induced by granulocyte colony-stimulating factor. Blood. 1990;76:2153–8. [PubMed] [Google Scholar]
- 40.Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, Wang W, Jakubowski A, Winton E, Lalezari P, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81:2496–502. [PMC free article] [PubMed] [Google Scholar]
- 41.Gough A, Clapperton M, Rolando N, Foster AV, Philpott-Howard J, Edmonds ME. Randomised placebo-controlled trial of granulocyte-colony stimulating factor in diabetic foot infection. Lancet. 1997;350:855–9. doi: 10.1016/S0140-6736(97)04495-4. [DOI] [PubMed] [Google Scholar]
- 42.Nervi B, Link DC, DiPersio JF. Cytokines and hematopoietic stem cell mobilization. J Cell Biochem. 2006;99:690–705. doi: 10.1002/jcb.21043. [DOI] [PubMed] [Google Scholar]
- 43.Winkler IG, Levesque JP. Mechanisms of hematopoietic stem cell mobilization: when innate immunity assails the cells that make blood and bone. Exp Hematol. 2006;34:996–1009. doi: 10.1016/j.exphem.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 44.Cashen AF, Link D, Devine S, DiPersio J. Cytokines and stem cell mobilization for autologous and allogeneic transplantation. Curr Hematol Rep. 2004;3:406–12. [PubMed] [Google Scholar]
- 45.Morris ES, MacDonald KP, Hill GR. Stem cell mobilization with G-CSF analogs: a rational approach to separate GVHD and GVL? Blood. 2006;107:3430–5. doi: 10.1182/blood-2005-10-4299. [DOI] [PubMed] [Google Scholar]
- 46.Falkenburg JH, Harrington MA, de Paus RA, Walsh WK, Daub R, Landegent JE, Broxmeyer HE. Differential transcriptional and posttranscriptional regulation of gene expression of the colony-stimulating factors by interleukin-1 and fetal bovine serum in murine fibroblasts. Blood. 1991;78:658–65. [PubMed] [Google Scholar]
- 47.Ernst TJ, Ritchie AR, Demetri GD, Griffin JD. Regulation of granulocyte- and monocyte-colony stimulating factor mRNA levels in human blood monocytes is mediated primarily at a post-transcriptional level. J Biol Chem. 1989;264:5700–3. [PubMed] [Google Scholar]
- 48.Dunn SM, Coles LS, Lang RK, Gerondakis S, Vadas MA, Shannon MF. Requirement for nuclear factor (NF)-kappa B p65 and NF-interleukin-6 binding elements in the tumor necrosis factor response region of the granulocyte colony-stimulating factor promoter. Blood. 1994;83:2469–79. [PubMed] [Google Scholar]
- 49.Nishizawa M, Tsuchiya M, Watanabe-Fukunaga R, Nagata S. Multiple elements in the promoter of granulocyte colony-stimulating factor gene regulate its constitutive expression in human carcinoma cells. J Biol Chem. 1990;265:5897–902. [PubMed] [Google Scholar]
- 50.Nishizawa M, Nagata S. Regulatory elements responsible for inducible expression of the granulocyte colony-stimulating factor gene in macrophages. Mol Cell Biol. 1990;10:2002–11. doi: 10.1128/mcb.10.5.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shannon MF, Coles LS, Fielke RK, Goodall GJ, Lagnado CA, Vadas MA. Three essential promoter elements mediate tumour necrosis factor and interleukin-1 activation of the granulocyte-colony stimulating factor gene. Growth Factors. 1992;7:181–93. doi: 10.3109/08977199209046923. [DOI] [PubMed] [Google Scholar]
- 52.Pradervand S, Maurya MR, Subramaniam S. Identification of signaling components required for the prediction of cytokine release in RAW 264.7 macrophages. Genome Biol. 2006;7:R11. doi: 10.1186/gb-2006-7-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 56.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–94. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 57.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 58.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–8. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 59.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–94. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 60.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–27. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwarzenberger P, La Russa V, Miller A, Ye P, Huang W, Zieske A, Nelson S, Bagby GJ, Stoltz D, Mynatt RL, Spriggs M, Kolls JK. IL-17 stimulates granulopoiesis in mice: use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J Immunol. 1998;161:6383–9. [PubMed] [Google Scholar]
- 62.Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z, Bagby G, Nelson S, Kolls JK. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. 2000;164:4783–9. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 63.Larsen A, Davis T, Curtis BM, Gimpel S, Sims JE, Cosman D, Park L, Sorensen E, March CJ, Smith CA. Expression cloning of a human granulocyte colony-stimulating factor receptor: a structural mosaic of hematopoietin receptor, immunoglobulin, and fibronectin domains. J Exp Med. 1990;172:1559–70. doi: 10.1084/jem.172.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukunaga R, Seto Y, Mizushima S, Nagata S. Three different mRNAs encoding human granulocyte colony-stimulating factor receptor. Proc Natl Acad Sci U S A. 1990;87:8702–6. doi: 10.1073/pnas.87.22.8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fukunaga R, Ishizaka-Ikeda E, Seto Y, Nagata S. Expression cloning of a receptor for murine granulocyte colony-stimulating factor. Cell. 1990;61:341–50. doi: 10.1016/0092-8674(90)90814-u. [DOI] [PubMed] [Google Scholar]
- 66.Bazan JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci U S A. 1990;87:6934–8. doi: 10.1073/pnas.87.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tamada T, Honjo E, Maeda Y, Okamoto T, Ishibashi M, Tokunaga M, Kuroki R. Homodimeric cross-over structure of the human granulocyte colony-stimulating factor (GCSF) receptor signaling complex. Proc Natl Acad Sci U S A. 2006;103:3135–40. doi: 10.1073/pnas.0511264103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Vos AM, Ultsch M, Kossiakoff AA. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science. 1992;255:306–12. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 69.Syed RS, Reid SW, Li C, Cheetham JC, Aoki KH, Liu B, Zhan H, Osslund TD, Chirino AJ, Zhang J, Finer-Moore J, Elliott S, Sitney K, Katz BA, Matthews DJ, Wendoloski JJ, Egrie J, Stroud RM. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature. 1998;395:511–6. doi: 10.1038/26773. [DOI] [PubMed] [Google Scholar]
- 70.Aritomi M, Kunishima N, Okamoto T, Kuroki R, Ota Y, Morikawa K. Atomic structure of the GCSF-receptor complex showing a new cytokine-receptor recognition scheme. Nature. 1999;401:713–7. doi: 10.1038/44394. [DOI] [PubMed] [Google Scholar]
- 71.Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci U S A. 2002;99:11872–7. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bussolino F, Wang JM, Defilippi P, Turrini F, Sanavio F, Edgell CJ, Aglietta M, Arese P, Mantovani A. Granulocyte- and granulocyte-macrophage-colony stimulating factors induce human endothelial cells to migrate and proliferate. Nature. 1989;337:471–3. doi: 10.1038/337471a0. [DOI] [PubMed] [Google Scholar]
- 73.Schneider A, Kruger C, Steigleder T, Weber D, Pitzer C, Laage R, Aronowski J, Maurer MH, Gassler N, Mier W, Hasselblatt M, Kollmar R, Schwab S, Sommer C, Bach A, Kuhn HG, Schabitz WR. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest. 2005;115:2083–98. doi: 10.1172/JCI23559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, Ohtsuka M, Matsuura K, Sano M, Nishi J, Iwanaga K, Akazawa H, Kunieda T, Zhu W, Hasegawa H, Kunisada K, Nagai T, Nakaya H, Yamauchi-Takihara K, Komuro I. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med. 2005;11:305–11. doi: 10.1038/nm1199. [DOI] [PubMed] [Google Scholar]
- 75.Nicholson SE, Oates AC, Harpur AG, Ziemiecki A, Wilks AF, Layton JE. Tyrosine kinase JAK1 is associated with the granulocyte-colony-stimulating factor receptor and both become tyrosine-phosphorylated after receptor activation. Proc Natl Acad Sci U S A. 1994;91:2985–8. doi: 10.1073/pnas.91.8.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian SS, Lamb P, Seidel HM, Stein RB, Rosen J. Rapid activation of the STAT3 transcription factor by granulocyte colony-stimulating factor. Blood. 1994;84:1760–4. [PubMed] [Google Scholar]
- 77.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–95. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 78.Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, Johnson EM, Jr, Schreiber RD. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93:373–83. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 79.Shimoda K, Feng J, Murakami H, Nagata S, Watling D, Rogers NC, Stark GR, Kerr IM, Ihle JN. Jak1 plays an essential role for receptor phosphorylation and Stat activation in response to granulocyte colony-stimulating factor. Blood. 1997;90:597–604. [PubMed] [Google Scholar]
- 80.Fukunaga R, Ishizaka-Ikeda E, Nagata S. Growth and differentiation signals mediated by different regions in the cytoplasmic domain of granulocyte colony-stimulating factor receptor. Cell. 1993;74:1079–87. doi: 10.1016/0092-8674(93)90729-a. [DOI] [PubMed] [Google Scholar]
- 81.Ziegler SF, Bird TA, Morella KK, Mosley B, Gearing DP, Baumann H. Distinct regions of the human granulocyte-colony-stimulating factor receptor cytoplasmic domain are required for proliferation and gene induction. Mol Cell Biol. 1993;13:2384–90. doi: 10.1128/mcb.13.4.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dong F, van Buitenen C, Pouwels K, Hoefsloot LH, Lowenberg B, Touw IP. Distinct cytoplasmic regions of the human granulocyte colony-stimulating factor receptor involved in induction of proliferation and maturation. Mol Cell Biol. 1993;13:7774–81. doi: 10.1128/mcb.13.12.7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nicholson SE, Starr R, Novak U, Hilton DJ, Layton JE. Tyrosine residues in the granulocyte colony-stimulating factor (G-CSF) receptor mediate G-CSF-induced differentiation of murine myeloid leukemic (M1) cells. J Biol Chem. 1996;271:26947–53. doi: 10.1074/jbc.271.43.26947. [DOI] [PubMed] [Google Scholar]
- 84.de Koning JP, Schelen AM, Dong F, van Buitenen C, Burgering BM, Bos JL, Lowenberg B, Touw IP. Specific involvement of tyrosine 764 of human granulocyte colony-stimulating factor receptor in signal transduction mediated by p145/Shc/GRB2 or p90/GRB2 complexes. Blood. 1996;87:132–40. [PubMed] [Google Scholar]
- 85.Akbarzadeh S, Ward AC, McPhee DO, Alexander WS, Lieschke GJ, Layton JE. Tyrosine residues of the granulocyte colony-stimulating factor receptor transmit proliferation and differentiation signals in murine bone marrow cells. Blood. 2002;99:879–87. doi: 10.1182/blood.v99.3.879. [DOI] [PubMed] [Google Scholar]
- 86.Hermans MH, van de Geijn GJ, Antonissen C, Gits J, van Leeuwen D, Ward AC, Touw IP. Signaling mechanisms coupled to tyrosines in the granulocyte colony-stimulating factor receptor orchestrate G-CSF-induced expansion of myeloid progenitor cells. Blood. 2003;101:2584–90. doi: 10.1182/blood-2002-07-2062. [DOI] [PubMed] [Google Scholar]
- 87.de Koning JP, Soede-Bobok AA, Schelen AM, Smith L, van Leeuwen D, Santini V, Burgering BM, Bos JL, Lowenberg B, Touw IP. Proliferation signaling and activation of Shc, p21Ras, and Myc via tyrosine 764 of human granulocyte colony-stimulating factor receptor. Blood. 1998;91:1924–33. [PubMed] [Google Scholar]
- 88.Ward AC, Hermans MH, Smith L, van Aesch YM, Schelen AM, Antonissen C, Touw IP. Tyrosine-dependent and -independent mechanisms of STAT3 activation by the human granulocyte colony-stimulating factor (G-CSF) receptor are differentially utilized depending on G-CSF concentration. Blood. 1999;93:113–24. [PubMed] [Google Scholar]
- 89.Dong F, Liu X, de Koning JP, Touw IP, Hennighausen L, Larner A, Grimley PM. Stimulation of Stat5 by granulocyte colony-stimulating factor (G-CSF) is modulated by two distinct cytoplasmic regions of the G-CSF receptor. J Immunol. 1998;161:6503–9. [PubMed] [Google Scholar]
- 90.Zhu QS, Robinson LJ, Roginskaya V, Corey SJ. G-CSF-induced tyrosine phosphorylation of Gab2 is Lyn kinase dependent and associated with enhanced Akt and differentiative, not proliferative, responses. Blood. 2004;103:3305–12. doi: 10.1182/blood-2003-06-1861. [DOI] [PubMed] [Google Scholar]
- 91.Avalos BR. Molecular analysis of the granulocyte colony-stimulating factor receptor. Blood. 1996;88:761–77. [PubMed] [Google Scholar]
- 92.Koay DC, Sartorelli AC. Functional differentiation signals mediated by distinct regions of the cytoplasmic domain of the granulocyte colony-stimulating factor receptor. Blood. 1999;93:3774–84. [PubMed] [Google Scholar]
- 93.Semerad CL, Poursine-Laurent J, Liu F, Link DC. A role for G-CSF receptor signaling in the regulation of hematopoietic cell function but not lineage commitment or differentiation. Immunity. 1999;11:153–161. doi: 10.1016/s1074-7613(00)80090-4. [DOI] [PubMed] [Google Scholar]
- 94.McLemore ML, Grewal S, Liu F, Archambault A, Poursine-Laurent J, Haug J, Link DC. Stat-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity. 2001;14:193–204. doi: 10.1016/s1074-7613(01)00101-7. [DOI] [PubMed] [Google Scholar]
- 95.McLemore ML, Poursine-Laurent J, Link DC. Increased granulocyte colony-stimulating factor responsiveness but normal resting granulopoiesis in mice carrying a targeted granulocyte colony-stimulating factor receptor mutation derived from a patient with severe congenital neutropenia. J Clin Invest. 1998;102:483–492. doi: 10.1172/JCI3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hermans MH, Antonissen C, Ward AC, Mayen AE, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999;189:683–92. doi: 10.1084/jem.189.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Corey SJ, Burkhardt AL, Bolen JB, Geahlen RL, Tkatch LS, Tweardy DJ. Granulocyte colony-stimulating factor receptor signaling involves the formation of a three-component complex with Lyn and Syk protein-tyrosine kinases. Proc Natl Acad Sci U S A. 1994;91:4683–7. doi: 10.1073/pnas.91.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nicholson SE, Novak U, Ziegler SF, Layton JE. Distinct regions of the granulocyte colony-stimulating factor receptor are required for tyrosine phosphorylation of the signaling molecules JAK2, Stat3, and p42, p44MAPK. Blood. 1995;86:3698–704. [PubMed] [Google Scholar]
- 99.Tian SS, Tapley P, Sincich C, Stein RB, Rosen J, Lamb P. Multiple signaling pathways induced by granulocyte colony-stimulating factor involving activation of JAKs, STAT5, and/or STAT3 are required for regulation of three distinct classes of immediate early genes. Blood. 1996;88:4435–44. [PubMed] [Google Scholar]
- 100.Ward AC, Monkhouse JL, Csar XF, Touw IP, Bello PA. The Src-like tyrosine kinase Hck is activated by granulocyte colony-stimulating factor (G-CSF) and docks to the activated G-CSF receptor. Biochem Biophys Res Commun. 1998;251:117–23. doi: 10.1006/bbrc.1998.9441. [DOI] [PubMed] [Google Scholar]
- 101.Dong F, Larner AC. Activation of Akt kinase by granulocyte colony-stimulating factor (G-CSF): evidence for the role of a tyrosine kinase activity distinct from the Janus kinases. Blood. 2000;95:1656–62. [PubMed] [Google Scholar]
- 102.Corey SJ, Dombrosky-Ferlan PM, Zuo S, Krohn E, Donnenberg AD, Zorich P, Romero G, Takata M, Kurosaki T. Requirement of Src kinase Lyn for induction of DNA synthesis by granulocyte colony-stimulating factor. J Biol Chem. 1998;273:3230–5. doi: 10.1074/jbc.273.6.3230. [DOI] [PubMed] [Google Scholar]
- 103.Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–90. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Panopoulos AP, Bartos D, Zhang L, Watowich SS. Control of myeloid-specific integrin αMβ2 (CD11b/CD18) expression by cytokines is regulated by Stat3-dependent activation of PU.1. J Biol Chem. 2002;277:19001–19007. doi: 10.1074/jbc.M112271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shimozaki K, Nakajima K, Hirano T, Nagata S. Involvement of STAT3 in the granulocyte colony-stimulating factor-induced differentiation of myeloid cells. J Biol Chem. 1997;272:25184–9. doi: 10.1074/jbc.272.40.25184. [DOI] [PubMed] [Google Scholar]
- 106.Grishin A, Sinha S, Roginskaya V, Boyer MJ, Gomez-Cambronero J, Zuo S, Kurosaki T, Romero G, Corey SJ. Involvement of Shc and Cbl-PI 3-kinase in Lyn-dependent proliferative signaling pathways for G-CSF. Oncogene. 2000;19:97–105. doi: 10.1038/sj.onc.1203254. [DOI] [PubMed] [Google Scholar]
- 107.Chen C, Huang X, Atakilit A, Zhu QS, Corey SJ, Sheppard D. The Integrin alpha9beta1 contributes to granulopoiesis by enhancing granulocyte colony-stimulating factor receptor signaling. Immunity. 2006;25:895–906. doi: 10.1016/j.immuni.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 108.Tapley P, Shevde NK, Schweitzer PA, Gallina M, Christianson SW, Lin IL, Stein RB, Shultz LD, Rosen J, Lamb P. Increased G-CSF responsiveness of bone marrow cells from hematopoietic cell phosphatase deficient viable motheaten mice. Exp Hematol. 1997;25:122–31. [PubMed] [Google Scholar]
- 109.Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, Cluse LA, Sutherland KD, Hartley L, Williams E, Zhang J-G, Hilton DJ, Nicola NA, Alexander WS, Roberts AW. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity. 2004;20:153–165. doi: 10.1016/s1074-7613(04)00022-6. [DOI] [PubMed] [Google Scholar]
- 110.Hunter MG, Avalos BR. Deletion of a critical internalization domain in the G-CSFR in acute myelogenous leukemia preceded by severe congenital neutropenia. Blood. 1999;93:440–6. [PubMed] [Google Scholar]
- 111.Wang L, Arcasoy MO, Watowich SS, Forget BG. Cytokine signals through STAT3 promote expression of granulocyte secondary granule proteins in 32D cells. Exp Hematol. 2005;33:308–17. doi: 10.1016/j.exphem.2004.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.De La Luz Sierra M, Gasperini P, McCormick PJ, Zhu J, Tosato G. Transcription factor Gfi-1 induced by G-CSF is a negative regulator of CXCR4 in myeloid cells. Blood. 2007;110:2276–85. doi: 10.1182/blood-2007-03-081448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, Tenen DG. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 114.Numata A, Shimoda K, Kamezaki K, Haro T, Kakumitsu H, Shide K, Kato K, Miyamoto T, Yamashita Y, Oshima Y, Nakajima H, Iwama AKA, Takase K, Gondo H, Mano H, Harada M. Signal transducers and activators of transcription 3 augments the transcriptional activity of CCAAT/enhancer-binding protein alpha in granulocyte colony-stimulating factor signaling pathway. J Biol Chem. 2005;280:12621–12629. doi: 10.1074/jbc.M408442200. [DOI] [PubMed] [Google Scholar]
- 115.Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci USA. 1999;96:6964–6969. doi: 10.1073/pnas.96.12.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yoshimura A, Ichihara M, Kinjyo I, Moriyama M, Copeland NG, Gilbert DJ, Jenkins NA, Hara T, Miyajima A. Mouse oncostatin M: an immediate early gene induced by multiple cytokines through the JAK-STAT5 pathway. Embo J. 1996;15:1055–63. [PMC free article] [PubMed] [Google Scholar]
- 117.Liu S, Cowell JK. Cloning and characterization of the TATA-less promoter from the human GFI1 proto-oncogene. Ann Hum Genet. 2000;64:83–6. doi: 10.1017/S0003480000007971. [DOI] [PubMed] [Google Scholar]
- 118.Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity. 1996;5:491–501. doi: 10.1016/s1074-7613(00)80504-x. [DOI] [PubMed] [Google Scholar]
- 119.Gregory AD, Hogue LA, Ferkol TW, Link DC. Regulation of systemic and local neutrophil responses by G-CSF during pulmonary Pseudomonas aeruginosa infection. Blood. 2007;109:3235–43. doi: 10.1182/blood-2005-01-015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17:413–423. doi: 10.1016/s1074-7613(02)00424-7. [DOI] [PubMed] [Google Scholar]
- 121.Liu F, Poursine-Laurent J, Wu HY, Link DC. Interleukin-6 and the granulocyte colony-stimulating factor receptor are major independent regulators of granulopoiesis in vivo but are not required for lineage commitment or terminal differentiation. Blood. 1997;90:2583–90. [PubMed] [Google Scholar]
- 122.Kaushansky K, Fox N, Lin NL, Liles WC. Lineage-specific growth factors can compensate for stem and progenitor cell deficiencies at the postprogenitor cell level: an analysis of doubly TPO- and G-CSF receptor-deficient mice. Blood. 2002;99:3573–8. doi: 10.1182/blood.v99.10.3573. [DOI] [PubMed] [Google Scholar]
- 123.Betsuyaku T, Liu F, Senior RM, Haug JS, Brown EJ, Jones SL, Matsushima K, Link DC. A functional granulocyte colony-stimulating factor receptor is required for normal chemoattractant-induced neutrophil activation. J Clin Invest. 1999;103:825–32. doi: 10.1172/JCI5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Richards MK, Liu F, Iwasaki H, Akashi K, Link DC. Pivotal role of granulocyte colony-stimulating factor in the development of progenitors in the common myeloid pathway. Blood. 2003;102:3562–8. doi: 10.1182/blood-2003-02-0593. [DOI] [PubMed] [Google Scholar]
- 125.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–4. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee C-k, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, DePinho RA, Levy DE. Stat3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17:63–72. doi: 10.1016/s1074-7613(02)00336-9. [DOI] [PubMed] [Google Scholar]
- 127.Kamezaki K, Shimoda K, Numata A, Haro T, Kakumitsu H, Yoshie M, Yamamoto M, Takeda K, Matsuda T, Akira S, Ogawa K, Harada M. Roles of Stat3 and ERK in G-CSF signaling. Stem Cells. 2005;23:252–63. doi: 10.1634/stemcells.2004-0173a. [DOI] [PubMed] [Google Scholar]
- 128.Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, Kano A, Iwamoto Y, Li E, Craft JE, Bothwell AL, Fikrig E, Koni PA, Flavell RA, Fu XY. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kile BT, Alexander WS. The suppressors of cytokine signalling (SOCS) Cell Mol Life Sci. 2001;58:1627–35. doi: 10.1007/PL00000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hilton DJ, Krebs DL. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19:378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 131.Boyle K, Egan P, Rakar S, Willson TA, Wicks IP, Metcalf D, Hilton DJ, Nicola NA, Alexander WS, Roberts AW, Robb L. The SOCS box of suppressor of cytokine signaling-3 contributes to the control of G-CSF responsiveness in vivo. Blood. 2007;110:1466–74. doi: 10.1182/blood-2007-03-079178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Irandoust MI, Aarts LH, Roovers O, Gits J, Erkeland SJ, Touw IP. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. Embo J. 2007;26:1782–93. doi: 10.1038/sj.emboj.7601640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Onai N, Obata-Onai A, Tussiwand R, Lanzavecchia A, Manz MG. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J Exp Med. 2006;203:227–38. doi: 10.1084/jem.20051645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tenen DG, Hromas R, Licht JD, Zhang DE. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489–519. [PubMed] [Google Scholar]
- 135.Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol. 2007;7:105–17. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- 136.Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet. 2000;1:57–64. doi: 10.1038/35049577. [DOI] [PubMed] [Google Scholar]
- 137.Skokowa J, Cario G, Uenalan M, Schambach A, Germeshausen M, Battmer K, Zeidler C, Lehmann U, Eder M, Baum C, Grosschedl R, Stanulla M, Scherr M, Welte K. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat Med. 2006;12:1191–7. doi: 10.1038/nm1474. [DOI] [PubMed] [Google Scholar]
- 138.Bjerregaard MD, Jurlander J, Klausen P, Borregaard N, Cowland JB. The in vivo profile of transcription factors during neutrophil differentiation in human bone marrow. Blood. 2003;101:4322–32. doi: 10.1182/blood-2002-03-0835. [DOI] [PubMed] [Google Scholar]
- 139.Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, Shigematsu H, Levantini E, Huettner CS, Lekstrom-Himes JA, Akashi K, Tenen DG. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21:853–63. doi: 10.1016/j.immuni.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 140.Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, Orkin SH. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. 2003;18:109–20. doi: 10.1016/s1074-7613(02)00501-0. [DOI] [PubMed] [Google Scholar]
- 141.Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;21:569–574. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Collins SJ, Ulmer J, Purton LE, Darlington G. Multipotent hematopoietic cell lines derived from C/EBPalpha(−/−) knockout mice display granulocyte macrophage-colony-stimulating factor, granulocyte- colony-stimulating factor, and retinoic acid-induced granulocytic differentiation. Blood. 2001;98:2382–8. doi: 10.1182/blood.v98.8.2382. [DOI] [PubMed] [Google Scholar]
- 143.Zhang P, Nelson E, Radomska HS, Iwasaki-Arai J, Akashi K, Friedman AD, Tenen DG. Induction of granulocytic differentiation by 2 pathways. Blood. 2002;99:4406–12. doi: 10.1182/blood.v99.12.4406. [DOI] [PubMed] [Google Scholar]
- 144.Zhang P, Iwama A, Datta MW, Darlington GJ, Link DC, Tenen DG. Upregulation of interleukin 6 and granulocyte colony-stimulating factor receptors by transcription factor CCAAT enhancer binding protein alpha (C/EBP alpha) is critical for granulopoiesis. J Exp Med. 1998;188:1173–84. doi: 10.1084/jem.188.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sterneck E, Tessarollo L, Johnson PF. An essential role for C/EBPbeta in female reproduction. Genes Dev. 1997;11:2153–62. doi: 10.1101/gad.11.17.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Screpanti I, Romani L, Musiani P, Modesti A, Fattori E, Lazzaro D, Sellitto C, Scarpa S, Bellavia D, Lattanzio G, et al. Lymphoproliferative disorder and imbalanced T-helper response in C/EBP beta-deficient mice. Embo J. 1995;14:1932–41. doi: 10.1002/j.1460-2075.1995.tb07185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tanaka T, Akira S, Yoshida K, Umemoto M, Yoneda Y, Shirafuji N, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell. 1995;80:353–61. doi: 10.1016/0092-8674(95)90418-2. [DOI] [PubMed] [Google Scholar]
- 148.Sicinska E, Lee YM, Gits J, Shigematsu H, Yu Q, Rebel VI, Geng Y, Marshall CJ, Akashi K, Dorfman DM, Touw IP, Sicinski P. Essential role for cyclin D3 in granulocyte colony-stimulating factor-driven expansion of neutrophil granulocytes. Mol Cell Biol. 2006;26:8052–60. doi: 10.1128/MCB.00800-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhan Y, Basu S, Lieschke GJ, Grail D, Dunn AR, Cheers C. Functional deficiencies of peritoneal cells from gene-targeted mice lacking G-CSF or GM-CSF. J Leukoc Biol. 1999;65:256–64. doi: 10.1002/jlb.65.2.256. [DOI] [PubMed] [Google Scholar]
- 150.Basu S, Hodgson G, Zhang HH, Katz M, Quilici C, Dunn AR. Emergency” granulopoiesis in G-CSF-deficient mice in response to Candida albicans infection. Blood. 2000;95:3725–33. [PubMed] [Google Scholar]
- 151.Welte K, Zeidler C, Reiter A, Muller W, Odenwald E, Souza L, Riehm H. Differential effects of granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor in children with severe congenital neutropenia. Blood. 1990;75:1056–63. [PubMed] [Google Scholar]
- 152.Bonilla MA, Gillio AP, Ruggeiro M, Kernan NA, Brochstein JA, Abboud M, Fumagalli L, Vincent M, Gabrilove JL, Welte K, et al. Effects of recombinant human granulocyte colony-stimulating factor on neutropenia in patients with congenital agranulocytosis. N Engl J Med. 1989;320:1574–80. doi: 10.1056/NEJM198906153202402. [DOI] [PubMed] [Google Scholar]
- 153.Hammond WPt, Price TH, Souza LM, Dale DC. Treatment of cyclic neutropenia with granulocyte colony-stimulating factor. N Engl J Med. 1989;320:1306–11. doi: 10.1056/NEJM198905183202003. [DOI] [PubMed] [Google Scholar]
- 154.Jakubowski AA, Souza L, Kelly F, Fain K, Budman D, Clarkson B, Bonilla MA, Moore MA, Gabrilove J. Effects of human granulocyte colony-stimulating factor in a patient with idiopathic neutropenia. N Engl J Med. 1989;320:38–42. doi: 10.1056/NEJM198901053200107. [DOI] [PubMed] [Google Scholar]
- 155.Dong F, Brynes RK, Tidow N, Welte K, Lowenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333:487–93. doi: 10.1056/NEJM199508243330804. [DOI] [PubMed] [Google Scholar]
- 156.Dong F, Dale DC, Bonilla MA, Freedman M, Fasth A, Neijens HJ, Palmblad J, Briars GL, Carlsson G, Veerman AJ, Welte K, Lowenberg B, Touw IP. Mutations in the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Leukemia. 1997;11:120–5. doi: 10.1038/sj.leu.2400537. [DOI] [PubMed] [Google Scholar]
- 157.Dong F, Hoefsloot LH, Schelen AM, Broeders CA, Meijer Y, Veerman AJ, Touw IP, Lowenberg B. Identification of a nonsense mutation in the granulocyte-colony-stimulating factor receptor in severe congenital neutropenia. Proc Natl Acad Sci U S A. 1994;91:4480–4. doi: 10.1073/pnas.91.10.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dong F, van Paassen M, van Buitenen C, Hoefsloot LH, Lowenberg B, Touw IP. A point mutation in the granulocyte colony-stimulating factor receptor (G-CSF-R) gene in a case of acute myeloid leukemia results in the overexpression of a novel G-CSF-R isoform. Blood. 1995;85:902–11. [PubMed] [Google Scholar]
- 159.van de Geijn GJ, Gits J, Aarts LH, Heijmans-Antonissen C, Touw IP. G-CSF receptor truncations found in SCN/AML relieve SOCS3-controlled inhibition of STAT5 but leave suppression of STAT3 intact. Blood. 2004;104:667–74. doi: 10.1182/blood-2003-08-2913. [DOI] [PubMed] [Google Scholar]
- 160.Hunter MG, Jacob A, O’Donnell LC, Agler A, Druhan LJ, Coggeshall KM, Avalos BR. Loss of SHIP and CIS recruitment to the granulocyte colony-stimulating factor receptor contribute to hyperproliferative responses in severe congenital neutropenia/acute myelogenous leukemia. J Immunol. 2004;173:5036–45. doi: 10.4049/jimmunol.173.8.5036. [DOI] [PubMed] [Google Scholar]
- 161.Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective internalization and sustained activation of truncated granulocyte colony-stimulating factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood. 1999;93:447–58. [PubMed] [Google Scholar]
- 162.Tidow N, Pilz C, Teichmann B, Muller-Brechlin A, Germeshausen M, Kasper B, Rauprich P, Sykora KW, Welte K. Clinical relevance of point mutations in the cytoplasmic domain of the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Blood. 1997;89:2369–75. [PubMed] [Google Scholar]
- 163.Kostmann R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr Suppl. 1956;45:1–78. [PubMed] [Google Scholar]
- 164.Skokowa J, Germeshausen M, Zeidler C, Welte K. Severe congenital neutropenia: inheritance and pathophysiology. Curr Opin Hematol. 2007;14:22–8. doi: 10.1097/00062752-200701000-00006. [DOI] [PubMed] [Google Scholar]
- 165.Bernard T, Gale RE, Evans JP, Linch DC. Mutations of the granulocyte-colony stimulating factor receptor in patients with severe congenital neutropenia are not required for transformation to acute myeloid leukaemia and may be a bystander phenomenon. Br J Haematol. 1998;101:141–9. doi: 10.1046/j.1365-2141.1998.00652.x. [DOI] [PubMed] [Google Scholar]
- 166.Horwitz MS, Duan Z, Korkmaz B, Lee HH, Mealiffe ME, Salipante SJ. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109:1817–24. doi: 10.1182/blood-2006-08-019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zeidler C, Welte K. Kostmann syndrome and severe congenital neutropenia. Semin Hematol. 2002;39:82–8. doi: 10.1053/shem.2002.31913. [DOI] [PubMed] [Google Scholar]
- 168.Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N, Bohn G, Melin M, Carlsson G, Fadeel B, Dahl N, Palmblad J, Henter JI, Zeidler C, Grimbacher B, Welte K. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nat Genet. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- 169.Rezaei N, Moin M, Pourpak Z, Ramyar A, Izadyar M, Chavoshzadeh Z, Sherkat R, Aghamohammadi A, Yeganeh M, Mahmoudi M, Mahjoub F, Germeshausen M, Grudzien M, Horwitz MS, Klein C, Farhoudi A. The clinical, immunohematological, and molecular study of Iranian patients with severe congenital neutropenia. J Clin Immunol. 2007;27:525–33. doi: 10.1007/s10875-007-9106-y. [DOI] [PubMed] [Google Scholar]
- 170.Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood. 2007;109:93–9. doi: 10.1182/blood-2006-02-004275. [DOI] [PubMed] [Google Scholar]
- 171.Wolfler A, Erkeland SJ, Bodner C, Valkhof M, Renner W, Leitner C, Olipitz W, Pfeilstocker M, Tinchon C, Emberger W, Linkesch W, Touw IP, Sill H. A functional single-nucleotide polymorphism of the G-CSF receptor gene predisposes individuals to high-risk myelodysplastic syndrome. Blood. 2005;105:3731–6. doi: 10.1182/blood-2004-06-2094. [DOI] [PubMed] [Google Scholar]
- 172.Rutella S, Zavala F, Danese S, Kared H, Leone G. Granulocyte colony-stimulating factor: a novel mediator of T cell tolerance. J Immunol. 2005;175:7085–91. doi: 10.4049/jimmunol.175.11.7085. [DOI] [PubMed] [Google Scholar]
- 173.Sloand EM, Kim S, Maciejewski JP, Van Rhee F, Chaudhuri A, Barrett J, Young NS. Pharmacologic doses of granulocyte colony-stimulating factor affect cytokine production by lymphocytes in vitro and in vivo. Blood. 2000;95:2269–74. [PubMed] [Google Scholar]
- 174.Morris ES, MacDonald KP, Rowe V, Banovic T, Kuns RD, Don AL, Bofinger HM, Burman AC, Olver SD, Kienzle N, Porcelli SA, Pellicci DG, Godfrey DI, Smyth MJ, Hill GR. NKT cell-dependent leukemia eradication following stem cell mobilization with potent G-CSF analogs. J Clin Invest. 2005;115:3093–103. doi: 10.1172/JCI25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.MacDonald KP, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JL, Thomas R, Hill GR. Cytokine expanded myeloid precursors function as regulatory antigen-presenting cells and promote tolerance through IL-10-producing regulatory T cells. J Immunol. 2005;174:1841–50. doi: 10.4049/jimmunol.174.4.1841. [DOI] [PubMed] [Google Scholar]
- 176.Morris ES, MacDonald KP, Rowe V, Johnson DH, Banovic T, Clouston AD, Hill GR. Donor treatment with pegylated G-CSF augments the generation of IL-10-producing regulatory T cells and promotes transplantation tolerance. Blood. 2004;103:3573–81. doi: 10.1182/blood-2003-08-2864. [DOI] [PubMed] [Google Scholar]
- 177.Schneider A, Kuhn HG, Schabitz WR. A role for G-CSF (granulocyte-colony stimulating factor) in the central nervous system. Cell Cycle. 2005;4:1753–7. doi: 10.4161/cc.4.12.2213. [DOI] [PubMed] [Google Scholar]
- 178.Hill GR, Morris ES, Fuery M, Hutchins C, Butler J, Grigg A, Roberts A, Bradstock K, Szer J, Kennedy G, Morton J, Durrant S. Allogeneic stem cell transplantation with peripheral blood stem cells mobilized by pegylated G-CSF. Biol Blood Marrow Transplant. 2006;12:603–7. doi: 10.1016/j.bbmt.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 179.Ripa RS, Jorgensen E, Wang Y, Thune JJ, Nilsson JC, Sondergaard L, Johnsen HE, Kober L, Grande P, Kastrup J. Stem cell mobilization induced by subcutaneous granulocyte-colony stimulating factor to improve cardiac regeneration after acute ST-elevation myocardial infarction: result of the double-blind, randomized, placebo-controlled stem cells in myocardial infarction (STEMMI) trial. Circulation. 2006;113:1983–92. doi: 10.1161/CIRCULATIONAHA.105.610469. [DOI] [PubMed] [Google Scholar]
- 180.Takano H, Ohtsuka M, Akazawa H, Toko H, Harada M, Hasegawa H, Nagai T, Komuro I. Pleiotropic effects of cytokines on acute myocardial infarction: G-CSF as a novel therapy for acute myocardial infarction. Curr Pharm Des. 2003;9:1121–7. doi: 10.2174/1381612033455008. [DOI] [PubMed] [Google Scholar]
- 181.Ellis SG, Penn MS, Bolwell B, Garcia M, Chacko M, Wang T, Brezina KJ, McConnell G, Topol EJ. Granulocyte colony stimulating factor in patients with large acute myocardial infarction: results of a pilot dose-escalation randomized trial. Am Heart J. 2006;152:1051e9–14. doi: 10.1016/j.ahj.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 182.Lawlor KE, Campbell IK, Metcalf D, O’Donnell K, van Nieuwenhuijze A, Roberts AW, Wicks IP. Critical role for granulocyte colony-stimulating factor in inflammatory arthritis. Proc Natl Acad Sci U S A. 2004;101:11398–403. doi: 10.1073/pnas.0404328101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Eyles JL, Roberts AW, Metcalf D, Wicks IP. Granulocyte colony-stimulating factor and neutrophils--forgotten mediators of inflammatory disease. Nat Clin Pract Rheumatol. 2006;2:500–10. doi: 10.1038/ncprheum0291. [DOI] [PubMed] [Google Scholar]
- 184.Gilman PA, Jackson DP, Guild HG. Congenital agranulocytosis: prolonged survival and terminal acute leukemia. Blood. 1970;36:576–85. [PubMed] [Google Scholar]
- 185.Link DC, Kunter G, Kasai Y, Zhao Y, Miner T, McLellan MD, Ries RE, Kapur D, Nagarajan R, Dale DC, Bolyard AA, Boxer LA, Welte K, Zeidler C, Donadieu J, Bellanne-Chantelot C, Vardiman JW, Caligiuri MA, Bloomfield CD, DiPersio JF, Tomasson MH, Graubert TA, Westervelt P, Watson M, Shannon W, Baty J, Mardis ER, Wilson RK, Ley TJ. Distinct patterns of mutations occurring in de novo AML versus AML arising in the setting of severe congenital neutropenia. Blood. 2007;110:1648–55. doi: 10.1182/blood-2007-03-081216. [DOI] [PMC free article] [PubMed] [Google Scholar]