

Abstract
c-Myc is a transcription factor that is implicated in many cellular processes including proliferation, apoptosis and cancers. Recently, c-Myc was shown to be involved in regulation of glutamate cysteine ligase through E-box sequences. This investigation examined whether c-Myc also regulates phase II genes through interaction with the electrophile response element (EpRE). Experiments were conducted in human bronchial epithelial cells using si-RNA to knock down c-Myc. RT-PCR and reporter assays were used to measure transcription and promoter activity. c-Myc down-regulated transcription and promoter activity of phase II genes. Chromatin immunoprecipitation verified binding of c-Myc to EpRE while co-immunoprecipitation demonstrated interaction of c-Myc with Nrf2. c-Myc also forms a ternary complex with Nrf2 and p-c-Jun. Finally, c-Myc decreased Nrf2 stability. Thus, our results suggest regulation of the EpRE/Nrf2 signaling pathway by c-Myc through both interaction with the EpRE binding complex and increased degradation of Nrf2.
Keywords: c-Myc, Nrf2, EpRE, Phase II genes
Introduction
c-Myc is a transcription factor implicated in many cellular processes such as proliferation, and apoptosis (1) and is over expressed in most cancers (2). Phase II genes play a major role in chemoresistance by detoxifying anticancer drugs. Understanding the role of c-Myc in regulation of phase II genes was the goal of this work. In particular, we tried to shed light on the functional consequence of silencing of c-Myc expression on phase II gene expression.
The c-Myc protein belongs to the basic helix-loop-helix leucine-zipper protein family (b/HLH/Z), and forms a dimer with the ubiquitously expressed protein Max. The dimer binds to the specific CAC(G/A)TG motif designated as E- boxes and thereby regulates transcription of target genes both positively and negatively (3,4). Complex regulatory networks control c-Myc activity resulting in up-regulation of its expression in response to stimuli (1).
In addition to the well-established involvement of c-Myc in cellular processes, c-Myc is involved in synthesis of amino acids and nucleotides, and regulation of lipid metabolism, glycolysis, and mitochondrial homeostasis (5). Whether c-Myc regulates cellular homeostasis in response to oxidative stress is also under investigation. Glutathione (GSH), the most abundant non protein thiol in cells, is involved in cellular detoxification, redox balance, stress responses and cell cycle regulation (6,7). Glutamate cysteine ligase (GCL) catalyzes the first rate-limiting step in GSH biosynthesis. GCL is a heterodimer composed of catalytic (GCLC) and regulatory (GCLM) subunit, which are encoded by two separate genes (8). The GCL enzyme plays a key role in the maintenance of intracellular redox balance and in determining cellular response to several different stimuli, including oxidative stress, xenobiotics, drugs and growth factors (9-11).
It is known that c-Myc modulates the expression of GCL genes and thereby affects the intracellular GSH content (12). Functional c-Myc binding consensus sites were identified on both GCL subunits promoters (13). Exposure to H2O2 enhances c-Myc recruitment to GCL regulatory regions through an ERK-dependent pathway (13,14).
The electrophile responsive element (EpRE), containing the TGANNNNGC sequence is a cis acting regulatory element that plays an important role in the gene expression of phase II detoxification enzymes and antioxidant proteins including GCL (15-17), NAD(P)H:quinone oxidoreductase-1 (NQO1) (18), and NAD(P)H:quinone oxidoreductase 2 (NQO2) (19). Regardless, not all EpRE-driven gene expression responds equally to stimulation and not all putative EpRE sequences are functional. For instance there are several putative EpRE sequences in the GCLC promoter but only EpRE4 is involved in response to stimulation (15). Furthermore, there may be differences in responses of the same EpRE sequence in different cells within the same species (20). Part of these differences may be in the proteins that form the binding complexes to the EpRE sequences and in the signaling for their activation.
In the present work, our results emphasize a central role of c-Myc in the regulation of phase II genes through the EpRE/Nrf2- dependent pathway. We have shown that c-Myc down-regulated Nrf2-dependent cytoprotective genes under both basal and induced conditions. Silencing c-Myc expression increased both endogenous mRNA expression as well as promoter activity of phase II genes. Moreover, c-Myc directly bound to EpRE sequences of gclc, gclm and nqo1. We further demonstrated that c-Myc interacted with Nrf2 and p-c-Jun to form ternary complex in vivo. Finally, we have shown that c-Myc decreased the stability of Nrf2.
Material and Methods
Materials
All chemicals and reagents were obtained form Sigma Chemical (St. Louis, MO) unless stated otherwise. HNE was purchased from Cayman Chemical (Ann Arbor, MI). TaqMan reverse transcription reagent and SYBR Green Master Mix were from Applied Biosystems (Foster City, CA). FuGENE 6 transfection reagent was from Roche (Indianapolis, IN). Anti-actin antibody was from BD transduction laboratories (San Jose, CA). Anti-c-Myc, anti-p-c-Myc, anti-p-c-Jun, anti-c-Jun, anti- Nrf2 and anti-lamin antibodies were from Santa Cruz Biotechnology, as well as c-Myc- and Nrf2- siRNA (Santa Cruz, CA). Chromatin immunoprecipitation (ChIP) assay kit was from Upstate (Chicago, IL). M-PER mammalian protein extraction reagent and NE-PER nuclear extraction reagent were from Pierce (Rockford, IL).
Cell Culture
The human bronchial epithelial cell line was a gift from Dr James Yankaskas of the University of North Carolina. The cells were maintained at 37°C in a humidified 5% CO2 incubator and grown in serum-free F12 Ham media supplemented with 6 hormones (5μg/ml Human insulin, 1× 10-6 M Hydrocortisone, 3.7 μg/ml endothelial cell growth supplement, 25 ng/ml epidermal growth factor, 3×10-8 M tri-iodothyronine, 5 μg/ml transferrin) 100 units/ml penicillin, 100 μg/ml streptomycin and 40 μg/ml gentamicin on collagen-coated dishes. HNE was dissolved in ethanol, and the final concentration of ethanol in the medium was 0.05%. HBE1 cells were treated at about 85% confluence with 10μM of HNE as indicated in the results session.
Western blot Analysis
Proteins were resolved on a 4–20% Tris-glycine acrylamide gel (Invitrogen, Carlsbad, CA) under denaturing conditions before being transferred electrophoretically onto a polyvinylidene difluoride (PVDF) membrane (Immobilon P; Millipore, Bedford, MA). Membranes were blocked with 5% nonfat dry milk (NFDM) at room temperature for 1 h and then incubated overnight at 4°C with primary antibody diluted in 5% NFDM in Tris buffer saline (TBS) as indicated (1:200 anti-c-Myc; 1:500 anti-p-c-Jun; 1:2500 anti lamin; 1:5000 anti-actin; 1:500 anti Nrf2; 1:1000 anti c-Jun). After being washed with TBS containing 0.05% Tween 20, the membrane was incubated with goat anti-mouse IgG or goat anti-rabbit conjugated to horseradish peroxidase (1:3000) at room temperature for 2 h. The blots were developed by the enhanced chemiluminescence technique (ECL Plus, Amersham, Arlington Heights, IL) according to the manufacturer's instructions. The bands of interest were imaged with and quantified by photon counting using the charged-coupled device camera of a Kodak Image Station 2000R (Kodak, Rochester, NY, USA) and Kodak 1D 3.6 Image Analysis Software. Photon counting was used for graphing and statistical analysis.
Immunoprecipitation Assay
Nuclear extracts contains 300-400 μg of proteins were precleared with protein A-Sepharose beads for 1 h and incubated with 5 μg of antibody to either c-Myc or Nrf2 or p-c-Jun at 4°C overnight. Immunoprecipitated complexes were washed 3 times with lysis buffer and then boiled in SDS sample buffer for 10 minutes. The immunoprecipitation products were run on acrylamide gel and electrophoretically transferred to PVDF as described above.
ChIP Assay
ChIP assays were performed by following a protocol provided with a kit from Upstate. Briefly, cells were incubated with formaldehyde by directly adding it into the medium (1% final concentration) at room temperature (21) for 10 min. The cell pellet was then lysed on ice for 10 min and sonicated under conditions that cause DNA to be broken into 200- to 800-bp fragments. Sonicated cell lysate was pre-cleared with 60 μl of salmon sperm DNA/agarose, and the supernatant was used for immunoprecipitation with antibodies to specific transcription factors overnight at 4°C. The protein/DNA complex was eluted from agarose in elution buffer and the DNA/protein complex was reversed by adding 5 M NaCl and incubating the mixture at 65°C for overnight. The DNA was extracted with phenol: chloroform: isoamyl alcohol (25:24:1). Primers used for PCR in the ChIP assay were: h-NQO1 (181 bp) Sense [-584∼-565] 5′-CCAGGGAAGTGTGTTGTATG-3′; Antisense [-626∼-644] 5′-GAGCAGAAAAAGAGCCGAT-3′; GCLM (137 bp) Sense [-408∼-391] 5′-AAGGGCCAGTCACTTCGG-3′; Antisense [-272∼-293] 5′-AGCTGTTTCCTG GAAGACAATG-3′; GCLC (EpRE4) (127 bp) Sense [-3259∼-3240] 5′-ATCGACTGCGGCAATCCTAG-3′ Antisense [-3151∼-3133] 5′-CGTGACTCAGCGCT TTGTG-3′.
Immunoprecipitation analysis was conducted using control rabbit IgG, anti-Nrf2 (Santa Cruz; H-300, sc-13032, however (c-20) was examined as well), anti-c-Myc (Santa Cruz; 9E10) and anti- RNA polymerase II (Santa Cruz; sc-899) antibodies.
2% of the chromatin DNA was also subjected to PCR analysis and indicated as input. PCR products were quantified using iQ SYBR Green Supermix (Bio-Rad) with the Chromo 4™ Real-Time PCR Detection System (Bio-Rad). Specific enrichment of DNA by anti-Nrf2 and anti-c-Myc antibodies was normalized by the PCR value of certain antibody to the PCR input value.
Transfection of small interfering RNA
The small interfering RNAs (siRNA) directed against human c-Myc (c-Myc-siRNA), human Nrf2 (Nrf2-siRNA) or non-targeting negative control siRNA (NC-siRNA) were purchased from Santa Cruz. Transfection of siRNA was performed using FuGENE 6 transfection reagent following the procedure provided with the transfection reagent. HBE1 cells at about 60-70% of confluence were transfected with 50 nM c-Myc-siRNA or 50nM Nrf2-siRNA. For mRNA expression and ChIP assay, twenty four hours after the transfection in 60 mm or 10 cm dishes, media was replaced and cells were treated with 10 μM HNE or vehicle for different time of exposure as indicated on results section. For western blot analyzes cells were transfected for 48 h and were collected. The whole cell proteins or cytosol and nuclear fraction were extracted using M-PER mammalian protein extraction reagent or NE-PER and C-PER, nuclear and cytosolic extraction reagents (Pierce) respectively.
Transfection procedure and assay of luciferase and β-galactosidase activity
HBE1 (50–60% confluence) cells were plated into each well of 12-well plates and cultured for 24 h. The cells were then transfected with 50 nM c-Myc-siRNA for an additional 24 h followed by transfection with 0.1 μg gclc (EpRE4) or gclm (EpRE) -pGL3 promoter encoding luciferase plasmid by using FuGENE 6 transfection reagent (Roche), and β-galactosidase plasmid (1/10 of total amount of plasmids) was cotransfected as an internal control. Twelve hours after transfection, the medium was replaced; 12 h later, the cells were untreated or treated with 15 μM HNE in 0.05% ethanol, or 0.05% ethanol as a control for 24 h. The cell pellet was lysed with M-PER mammalian protein extraction reagent (Pierce). The supernatant was then used for determination of the activity of luciferase and β-galactosidase.
To determine the β-galactosidase activity, 25 μl of supernatant was added to a reaction mixture containing 300 μM 4-methyllumbelliferyl β-D-galactoside. After incubation at room temperature for 20 min with shaking, β -galactosidase activity was determined in a fluorescence microplate reader (Molecular Device Corp., Sunnyvale, CA) at an excitation wavelength of 360 nm and an emission wavelength of 450 nm.
For the luciferase assay, a luciferase assay kit (Promega) was used. Briefly, 20 μl of cell lysate was added to the reaction mixture provided in the kit, and luciferase activity was determined in a luminometer (Berthold Detection Systems, Pforzheim, Germany). The final luciferase activity was normalized with the activity of co-transfected β-galactosidase.
The primer sequences used were the following: GCLC (EpRE4), 30bp (-3154:-3125) sense 5′- CCGCACAAAGCGC TGAGTCACGGGGAGGCG-3′, antisense 5′- GGCGTGTTTCGCGAC TCAGTGCCCCTCCGCG-3′; GCLM (EpRE), 30bp (-312:-283) sense 5′- CGCTACGATTTCTGCTTAGTCATTGTCTTCC-3′, antisense 5′- GCGATGCTAAAGACGAATCAGTAACAGAAG-3′.
Real time PCR Assay of mRNA levels
HBE1 (70% confluence) cells were pre-cultured in 60 mm dishes for 24 h and then treated with 10 μM HNE in 0.05% ethanol, or with 0.05% ethanol alone as control, for 18 h. Total RNA was extracted using TRIzol reagent and treated with DNA-free reagent according to the manufacturer's protocol (Ambion). RNA samples were reverse-transcribed using the TaqMan random hexamers (Applied Biosystems) and the contents of GCLM, GCLC, NQO1 and NQO2 mRNAs were measured by real-time PCR polymerase chain reaction (RT-PCR) with a Cephedi 1.2 real time PCR machine. In brief, 5 μl of reverse transcription reaction product was added to a reaction tube containing 12.5 μl SYBR green PCR Master Mix and primers specific for GCLC or GCLM mRNA. The total PCR sample reaction was 25 μl. GAPDH was used as an internal control. The primers sequences used were the following: GCLC, sense 5′-ATGGAGGTGCAATTAACAGAC-3′, antisense 5′-ACTGCATTGC CACCTTTGCA-3′; GCLM, sense 5′ –GCTGTATCAGTGGCACAG-3′, antisense 5′ –CGCTTGAATGTCAGG AATGC-3′; NQO1, sense 5′–TCTCGGCTCACTGCAACCTCT- 3′, antisense 5′–GCACTTTGGGAGGCTGAGGTA-3′; NQO2, sense 5′– CGCTCCTTTCCGTAACCAC-3′, antisense 5′ – CACAGAGTTATTGCC CGAAGT-3′; GAPDH, sense 5′-TGGGTGTGAACCATGAGAAG-3′, antisense 5′-CCATCACGACACAGT TTCC-3′.
Statistical Analysis
Results are reported as mean ± standard error for n=3-6, independent experiments, as indicated in the legends. Statistical significance was accepted when p < 0.05. Comparison of variants between experimental groups was performed with ANOVA and Tukey's test.
Results
Expression of c-Myc in HBE1 cells
c-Myc is a nuclear protein and is expressed in the nucleus of HBE1 cells under basal conditions. Following 3 h stimulation with either 10 μM 4-hydroxy-2-nonenal (HNE) or 50 μM tertiary butylhydroquinone (tBHQ), two established inducers of phase II genes through their EpREs (20,22-24), c-Myc protein decreased by 32% and 44%, respectively (Fig. 1). Treatment of cells with 10 μM HNE did not result in significant changes in phosphorylation of c-Myc after either 15 and 30 minutes (Fig. 2) or after 3 h (data not shown). Thus, a decrease in c-Myc without its phosphorylation occurred under conditions previously demonstrated to induce phase II genes.
Fig. 1.
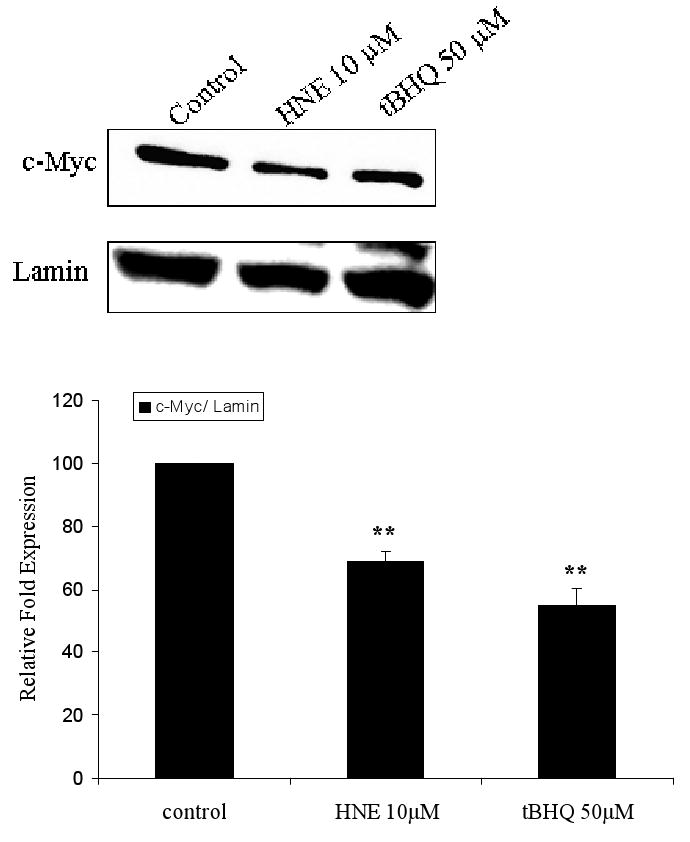
Expression of c-Myc protein in the nucleus of HBE1 cells under basal and induced conditions. Cells were grown to confluence and treated with 10 μM HNE, 50 μM tBHQ, or vehicle control for 3 h. Nuclear extracts were collected and 30 μg protein aliquots were subjected to western blot analyses with anti c-Myc antibody. Lamin was used as loading control and identified with anti-lamin antibody. The band intensity of c-Myc and lamin was quantified, and the c-Myc protein level was normalized to the lamin intensity. The error bars indicate standard deviations from 3 experiments.
Fig. 2.
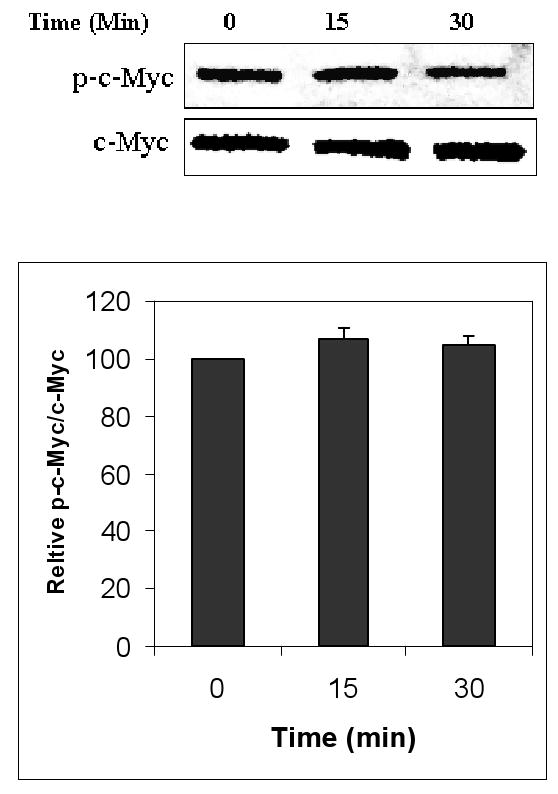
Effect of HNE on the phosphorylation of c-Myc. HBE1 cells were treated with 10 μM HNE for the indicated time points. Whole cell extracts were collected and subjected to immunobloting with anti p-c-Myc. c-Myc was used as loading control and identified with anti-c-Myc antibody. The figure shows a representative experiment and graph reported as mean ± S.E. out of three independent experiments.
Interactions of c-Myc with Nrf2 and p-c-Jun
We performed reciprocal immuno-precipitation assays using nuclear extracts from HBE1 cells. Immunoprecipitation of c-Myc followed by immunoblotting with anti-Nrf2 antibody, showed the presence of Nrf2 in the same complex with c-Myc (Fig. 3A). Immunoprecipitation of Nrf2 followed by immunoblotting with anti-c-Myc antibody also demonstrated the presence of both proteins in the same complex (Fig. 3B). When p-c-Jun was immunoprecipitated and then immunoblotted with either anti-c-Myc or anti-Nrf2 antibodies, the results revealed that all three proteins could appear in the same complex (Fig. 3C). The interaction existed under both basal and induced conditions (stimulation with HNE or tBHQ), however, it is clear that protein intensities were changed following cell stimulation. Following stimulation with HNE, c-Myc decreased while Nrf2 increased in the complex as shown by immunoprecipitation of c-Myc or Nrf2 followed by immunoblotting for the other protein (Figure 3, A and B). We have previously shown the interaction of Nrf2 with p-c-Jun in HBE1 cells (20). No association of c-Myc with unphosphorylated c-Jun was observed (Fig. 3A) nor was association of Nrf2 with p-c-Myc found (Fig. 3B). Endogenous expression of the proteins in total lysates is also shown in Figure 3 as positive controls for detection of the proteins.
Fig. 3.
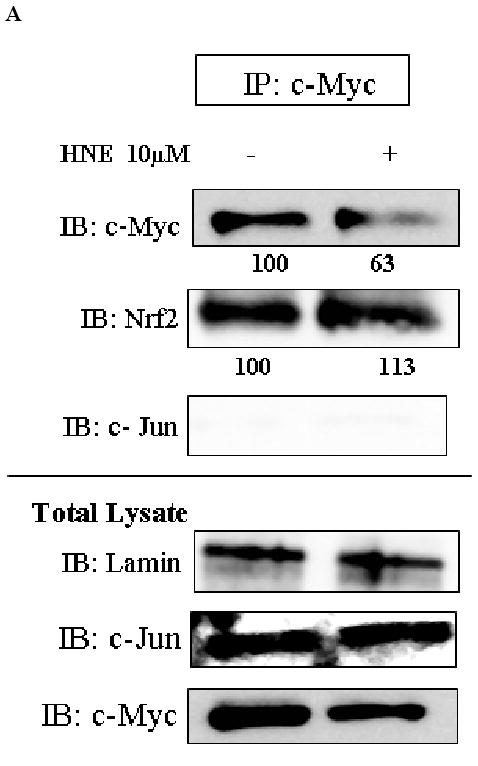
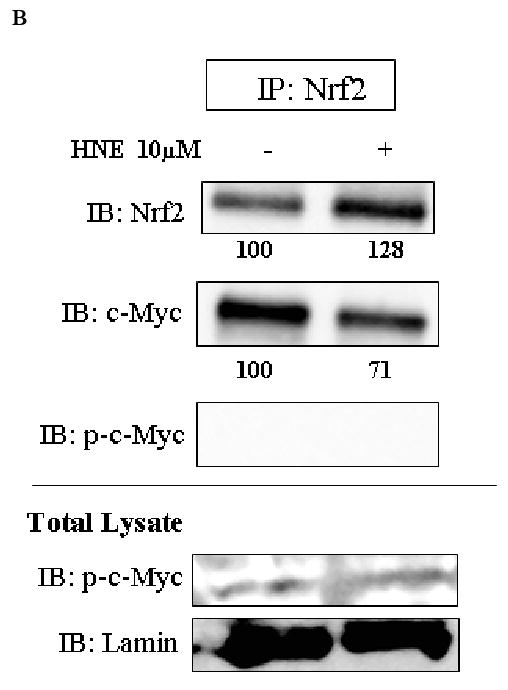
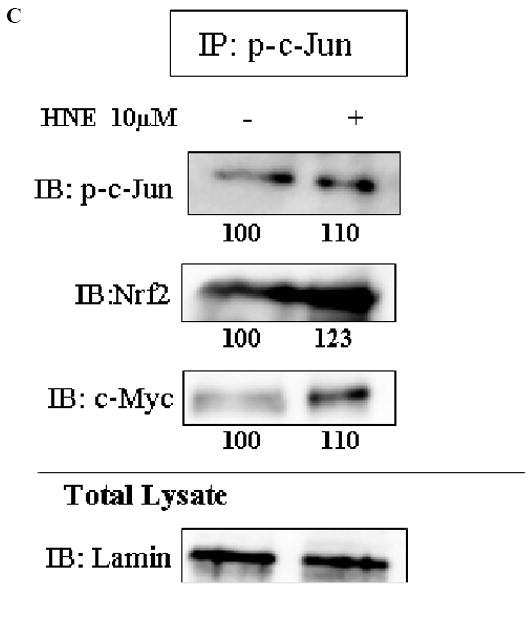
Interaction of c-Myc with either Nrf2 or p-c-Jun at both basal and induced conditions. HBE1 cells were treated with 10 μM HNE or vehicle control for 3 hours. Nuclear proteins were immunoprecipitated with the indicated antibodies: A. c-Myc. B. Nrf2. C. p-c-Jun, and visualized by Western blot analysis with anti Nrf2, anti c-Myc, anti p-c-Jun, anti p-c-Myc or anti c-Jun antibodies. Lamin was used as loading control and identified with anti-lamin antibody. Expression levels of the indicated proteins in the whole cell extract are shown in total lysate (lower panels).
c-Myc knockdown incresases transcription of phase II genes
As c-Myc bound to Nrf2 we further investigated the effects of c-Myc on the transcription of Nrf2-dependent genes using RNA interference (siRNA). In order to verify that c-Myc-siRNA knockdown was effective, we transfected cells with 50 nM c-Myc-siRNA for 48 h and subjected the proteins to western blot analyses. c-Myc protein level were reduced by 48% compared to control siRNA (Fig. 4A). Nrf2 protein level was increased by 37% compared to control siRNA in c-Myc knockdown cells (Fig. 4A).
Fig. 4.
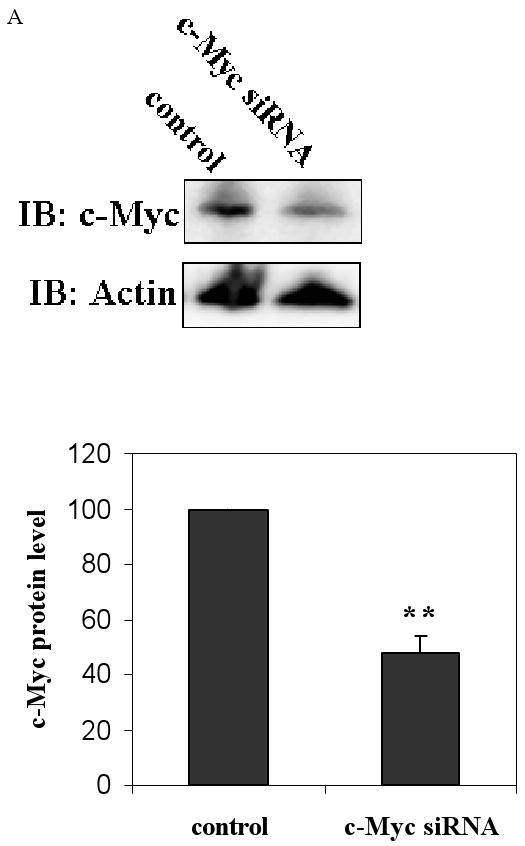
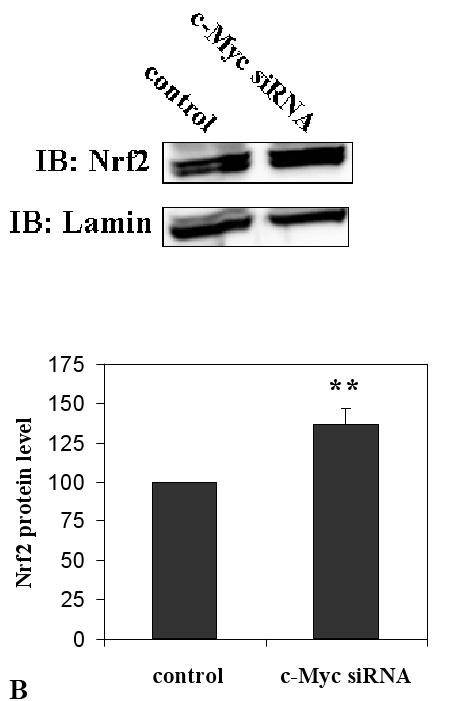


Knockdown of c-Myc up- regulates transcription of self defense genes. A. c-Myc knockdown decreased the protein level of c-Myc. Cells were transfected with 50 nM c-Myc-siRNA or control siRNA for 48 h. c-Myc and Nrf2 proteins were then detected by western blot analyses. B. c-Myc knockdown increases the expression of GCLC (black columns) and GCLM (net columns) mRNA. HBE1 cells were transfected with control si-RNA or 50 nM c-Myc-siRNA. Following 24 h of transfection cells were treated or untreated with HNE for 18 h and mRNA expression was measured. Cells were also treated with 10 μM HNE alone. C. c-Myc knockdown increases the expression of NQO1 (black columns) and NQO2 (net columns) mRNA. HBE1 cells were transfected with control siRNA or 50 nM c-Myc–siRNA. Following 24 h transfection cells were treated or untreated with HNE for 18 h. Cells were treated with 10 μM HNE alone and mRNA expression were analyzed by RT-PCR. C. Results are reported as mean ± S.E for four independent experiments. * p<0.05, significantly different from control. ** p<0.01, significantly different from its control.
HBE1 cells were transfected with 50 nM c-Myc-siRNA for 24 h in order to silence c-Myc expression and subjected to RT-PCR. Both GCLC and GCLM mRNAs were increased by 5.3 and 6.1 fold, respectively following knock down of c-Myc (Fig. 4B). When cells were exposed to 10 μM HNE, which causes maximal induction, the expression of GCLC and GCLM increased by 3.3 and 2.6 fold, respectively. When a combined treatment of HNE and c-Myc-siRNA was applied to cells, the expression of both GCLC or GCLM was also significantly greater than the control (Fig. 4B). Similar increments, but with lower magnitude, were observed for the expression of NQO1 and NQO2 mRNAs following silencing of c-Myc (Fig. 4C). Again, the combined effect of HNE and c-Myc-siRNA was also significantly greater than the control (Fig. 4C).
c-Myc down-regulates EpRE promoter activity of GCLC and GCLM
We next determined whether c-Myc regulation of GCL transcription is through the EpRE sequences using EpRE-driven luciferase reporter constructs. We have used short constructs, for both GCLC and GCLM that contain only EpRE4 and EpRE sequence, as described in material and methods. Transfection of cells with 50 nM c-Myc siRNA increases EpRE4 (GCLC) promoter activity by 2 fold, compared to control (Fig. 5A). Treatment with 10 μM HNE, alone, increased promoter activity by 2.3 fold. When transfected cells were treated with 10 μM HNE and c-Myc siRNA, the combination resulted in 3.8 fold increase in promoter activity (Fig. 5A). Similar sets of experiments were applied using EpRE (GCLM) promoter-reporter constructs. Knocking down c-Myc up-regulated promoter activity by 15 % and HNE increased activity by 2 fold. But here, the combined effect was higher than the effect of c-Myc siRNA alone but lower than that of HNE alone (Fig. 5B). Again, in both basal and induced condition, silencing c-Myc increased the promoter activity of both genes.
Fig. 5.


Silencing of c-Myc up-regulated the EpRE-reporter gene activity of GCLC and GCLM. HBE1 cells were transfected with control siRNA or c-Myc-si-RNA. Expression vectors contained: A. EpRE4 (GCLC) or B. EpRE (GCLM). Luciferase reporter constructs were transfected 24 h post si-RNA transfection. The transfected cells were treated with 10μM HNE or vehicle control for 24 h prior to the measurement of luciferase activities. A & B. Cells were transfected with 50 nM c-Myc-siRNA or non-specific si-RNA followed by transfection with either EpRE4 (GCLC) or EpRE (GCLM) plasmid, respectively. Then the cells were treated or untreated with HNE 10μM for additional 24 h. Results are reported as mean ± S.E. for at least three determinations. ** p<0.01, significantly different from control. # p<0.05, ## p<0.01, significantly different from control siRNA.
c-Myc interacts with EpREs in the promoter regions of phase II genes
Since c-Myc silencing increased EpRE promoter activity of both GCLC and GCLM, we next investigated whether c-Myc associates with EpRE4 (GCLC) and EpRE (GCLM) promoters. We used ChIP assays to determine binding sites in vivo. We have found that c-Myc associate with the functional EpRE sequences from nqo1, gclc and gclm genes with and without 10 μM HNE as a stimulant (Fig. 6, A, B and C). It was observed that c-Myc-siRNA eliminated the recruitment of c-Myc to the EpRE-containing promoter sequences. We have also shown that knocking down c-Myc enhanced recruitment of Nrf2 to the promoter region of gclc (Fig. 6C). This suggested an inhibitory role of c-Myc in recruitment of Nrf2.
Fig. 6.
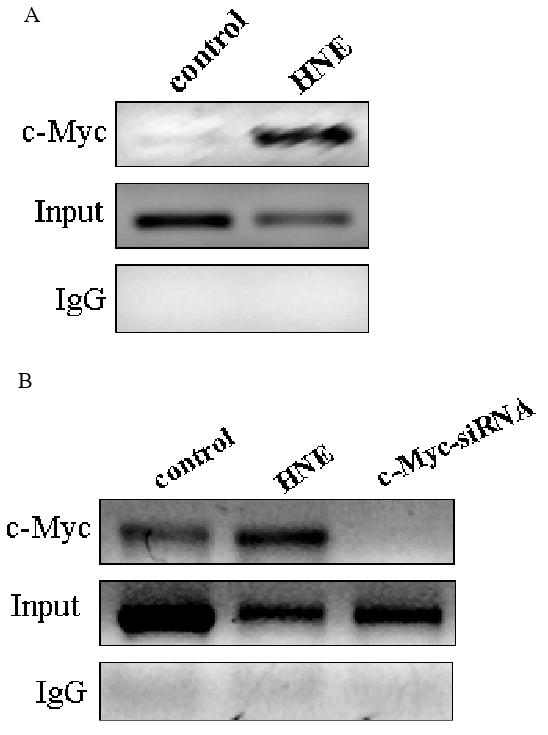
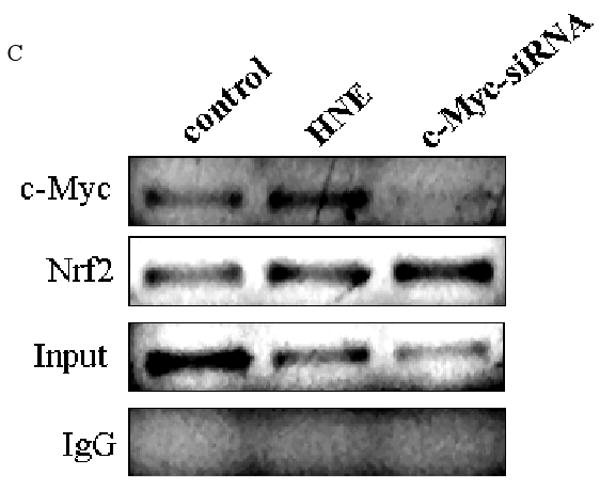
Recruitment of c-Myc to EpRE promoter. c-Myc interacts with the promoter of A. GCLM (EpRE) B. NQO1 (EpRE) and C. GCLC (EpRE4) under both basal and induced conditions in HBE1 cells. ChIP assays were performed to examine the in vivo interaction of c-Myc with EpRE that either treated or untreated with 10 μM HNE. In panel B and C cells were transfected with 50 nM c-Myc-siRNA for 24 h or treated with HNE for 3 h. Vehicle and non-specific siRNA was used in the controls. After stimulation with HNE or transfection with si-RNA, cells were fixed with 1% formaldehyde, lysed, and sonicated to shear chromatin in 0.2-0.8-kb fragments, which were then immunoprecipitated with anti c-Myc antibody or anti Nrf2 antibody. Purified DNA was then analyzed with RT-PCR with specific primers amplifying EpRE elements. Results are indicating a representative experiment out of four.
Silencing of c-Myc prolongs the half-life of Nrf2
Since we demonstrated that c-Myc interacted with Nrf2 and regulated phase II genes through the EpRE we further verified whether the regulation of EpRE by c-Myc involved changes in Nrf2 stability. We measured the half-life of Nrf2 protein in control cells and compared to that of cells which expressed lower c-Myc levels using si-RNA. Both sets of samples were exposed to 25 μM cyclohexamide (CHX) with samples from different time points subjected to immunoblot analysis. In c-Myc silenced cells, Nrf2 half-life was extended compared to that of control cells (Fig. 7, A and B). This indicated that c-Myc shortened the half-life of Nrf2 protein level.
Fig. 7.
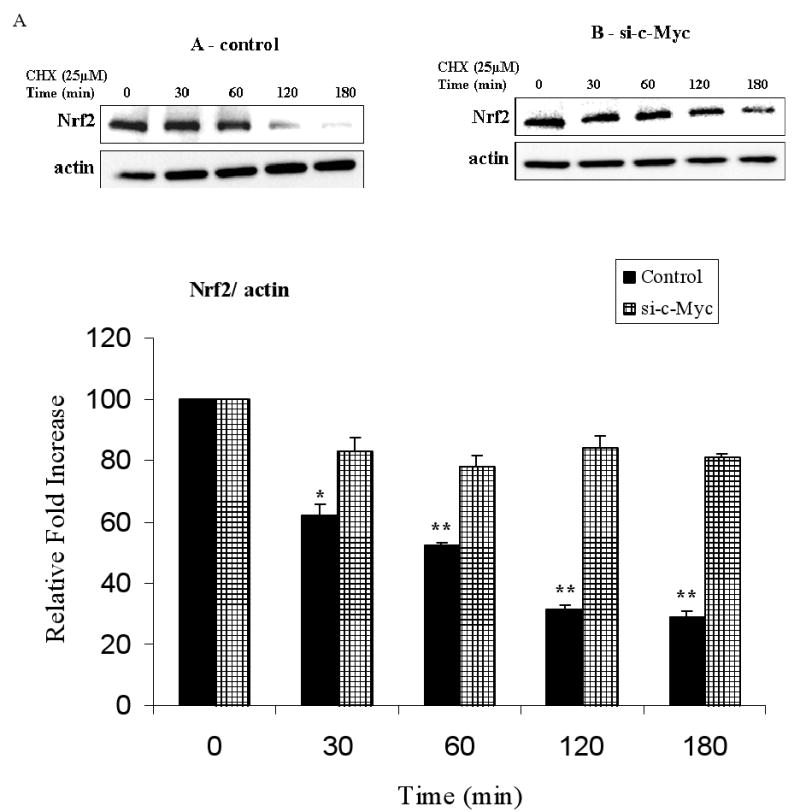
c-Myc knock down stabilized Nrf2 by increasing its half-life. HBE1 control cells A. or transfected with 50 nM c-Myc-siRNA B. were treated with 25 μM cyclohexamide (CHX) at 48 h post transfection and incubated for the time periods indicated in figure. Total proteins were collected and endogenous Nrf2 was detected by immunoblot analysis, and the intensity of the Nrf2 bands was quantified compared to actin. The amount of Nrf2 before addition of CHX was set as 1. Blot shown is a representative experiment. Graph shows the results as mean ± S.E. of 3 determination. * p<0.05, ** p<0.01, significantly different from its corresponding c-Myc-siRNA.
Discussion
In this report, we present our finding that c-Myc is a Nrf2-interacting protein that negatively regulates phase II genes in the HBE1 human bronchial epithelial cell line under both basal and induced conditions.
c-Myc has multiple functions that occur mainly through transcriptional regulation of target genes. It binds to several hundred genomic loci that contain consensus c-Myc binding sites, termed E-boxes, resulting in transcriptional activation of those genes (25). Additionally, a large number of genes such as cell cycle/growth arrest genesgas1, p21 and p27 are down-regulated by c-Myc (26,27). This dual role of c-Myc as a transcriptional factor involved in signaling depends upon the absence or presence of growth factors, over-expression of c-Myc (as in MEF cells), and differences in cell models. c-Myc has been reported to both increase ROS generation and participate in the induction of antioxidant enzymes (12-14). These pleiotropic effects of c-Myc are due to the large number of genes under its control (5).
It has been previously shown that c-Myc regulates GCL genes through the ERK/ c-Myc phosphorylation pathway (13,28). We have previously shown that induction of both GCL genes in HBE1 cells by HNE is however, through activation of the JNK pathway rather than either ERK or p38 (10,29,30). In addition, we detect only minor and insignificant phosphorylation of c-Myc following stimulation with HNE (Fig. 2). Moreover, it became evident that the basic phosphorylation state of c-Myc is low in HBE1 cells under both basal and induced conditions (Fig. 3B). Thus, differences in cell type and stimulant appear to alter the involvement of c-Myc in gclc and gclm regulation.
It is well established that c-Myc regulates genes through binding to the canonical E-box sequence CACGTG, as well as to non-canonical sequences including CATGTG (31). Here we demonstrated that c-Myc also affects gene expression through EpRE sequences. We have used constructs containing either the GCLC EpRE4 or GCLM EpRE sequence. The two constructs included only those EpRE sequences and none of the other cis elements in the promoters. While this does not eliminate the possibility that another element could theoretically interact with c-Myc, work done by Mulcahy's laboratory showed that GCLC EpRE4 and GCLM EpRE were the only EpRE sequences that responded to electrophiles and that mutation of them decreased the response (32,33). Our luciferase reporter assays indicated that c-Myc causes down-regulation of EpRE-driven genes (Fig. 5) while ChIP assays demonstrated recruitment of c-Myc to EpRE-containing DNA sequence in vivo (Fig. 6). Thus, the data suggested that c-Myc associate with EpRE sequences and suppressed induction of phase II genes (gclm, gclc and nqo1). In addition, silencing of c-Myc increased the recruitment of Nrf2 to the EpRE promoter (Fig. 6C). This effect of knocking down c-Myc may be due to a decrease in c-Myc replacing Nrf2 in the EpRE complex (Fig. 6c) and/or a decrease in c-Myc-induced degradation of Nrf2 (Fig. 7). Regardless of the mechanism, the results demonstrated that c-Myc inhibited Nrf2 dependent gene expression.
Both HNE and tBHQ are well known phase II inducers (30,34). We have shown that stimulation of HBE1 cells with either HNE or tBHQ decreased the expression of c-Myc protein (Fig. 1). This is in agreement with others findings, which showed that HNE inhibits c-Myc protein expression in three human leukemic cell lines (HL-60, U937, ML-1), and that the DNA binding activity of c-Myc is inhibited by HNE in HL-60 cells (35).
We have shown that under both basal and induced conditions knocking down c-Myc significantly increased mRNA transcription. HNE decreased c-Myc protein expression (Fig. 1) but the combined treatment of HNE and silencing of c-Myc did not enhance the transcription of any of the three phase II genes examined more than the sum of each of the two treatments alone (Fig. 4, A and B). While this is consistent with the possibility that HNE regulates transcription partially through its decrease of c-Myc expression, HNE also participates in the activation of Nrf2 by inactivating Keap1 (36). Furthermore, HNE may also affect the phosphorylation of Nrf2 that is needed for its translocation to the nucleus as was demonstrated with acrolein, another α,β-unsaturated aldehyde in HBE1 cells (37). Thus, multiple effects on signaling involving HNE, c-Myc and Nrf2 combine to affect gene expression through the EpRE cis element in phase II genes. The results also suggest the existence of an additional, HNE-independent, c-Myc-sensitive pathway.
The interaction of c-Myc with Nrf2 was complex. We previously demonstrated that HNE increased the nuclear content of Nrf2 in HBE1 cells (20). Here we have shown, using reciprocal immunoprecipitation, that c-Myc bound to Nrf2 and was altered upon stimulation with HNE (Fig. 3, A and B). We have previously shown that Nrf2 interacts with p-c-Jun and binds to EpRE sequences in HBE1 cells (20). Here, we have shown that c-Myc also interacted with p-c-Jun and Nrf2 simultaneously to form a ternary complex (Fig. 3). This suggests that c-Myc is a new partner in the complex binding that activates and regulates phase II genes through EpRE. We have previously shown in HBE1 cells, using electrophoretic mobility assays and an EpRE consensus sequence, that HNE caused changes in the mobility and intensity of bands suggesting alteration in the composition of EpRE binding complexes (38). The alterations in binding of c-Myc and Nrf2 observed here explain, in part, those previous observations.
In looking at the interactions among the transcription factors studied here, the finding that c-Myc did not bind to c-Jun was not surprising, as we have shown in our previous study that Nrf2 bound to p-c-Jun but not c-Jun in HBE1 cells (20). As c-Myc appeared to be a partner in the complex with Nrf2, the result here was in agreement with our previous data. Further examination regarding the interaction between c-Myc and Nrf2 revealed that c-Myc shortened the half-life of Nrf2 (Fig. 7; however, how this occurred is beyond the scope of the current investigation).
In summary, we have shown that c-Myc regulated both the mRNA transcription and the promoter activity of phase II genes in human bronchial endothelial cells. Silencing of c-Myc increased EpRE-regulated gene expression with and without induction by HNE. The effects of c-Myc were achieved by a combination of effects including recruitment of c-Myc to the EpRE promoter while forming ternary complex with Nrf2 and p-c-Jun and decreased stability of Nrf2. The effects of HNE were therefore explained, at least in part, by decreased c-Myc and increased Nrf2 both of which contributed to HNE-induction of phase II genes.
Significance
c-Myc is a transcription factor that is over-expressed in most human cancers (2). Knowing that c-Myc down-regulates EpRE will help to better understand the regulation of the genes involved in the metabolism of xenobiotics, cancer chemopreventive agents and multi-drug resistance proteins and provide potential new avenues for exploration in chemotherapy.
Acknowledgments
This work was supported by the National Institutes of Health grant ES 05511.
Abbreviations
- Nrf2
nuclear factor-erythroid 2 (NF-E2) related factor 2
- EpRE
electrophile responsive elements
- HBE1
human bronchial epithelial
- HNE
4-hydroxy-2-nonenal
- GCLC
catalytic subunit of GCL
- GCLM
modulator subunit of GCL
- NQO2
NAD(P)H:quinone oxidoreductase 2
- NQO1
NAD(P)H:quinone oxidoreductase-1
- ChIP
chromatin immunoprecipitation
References
- 1.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22(56):9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 2.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18(19):3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 3.Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, Weintraub H. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol. 1993;13(9):5216–5224. doi: 10.1128/mcb.13.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang CV, Resar LM, Emison E, Kim S, Li Q, Prescott JE, Wonsey D, Zeller K. Function of the c-Myc oncogenic transcription factor. Exp Cell Res. 1999;253(1):63–77. doi: 10.1006/excr.1999.4686. [DOI] [PubMed] [Google Scholar]
- 5.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19(1):1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deneke SM, Fanburg BL. Regulation of cellular glutathione. Am J Physiol. 1989;257(4 Pt 1):L163–173. doi: 10.1152/ajplung.1989.257.4.L163. [DOI] [PubMed] [Google Scholar]
- 7.Forman HJ, Dickinson DA. Oxidative signaling and glutathione synthesis. BioFactors (Oxford, England) 2003;17(1-4):1–12. doi: 10.1002/biof.5520170101. [DOI] [PubMed] [Google Scholar]
- 8.Gipp JJ, Bailey HH, Mulcahy RT. Cloning and sequencing of the cDNA for the light subunit of human liver gamma-glutamylcysteine synthetase and relative mRNA levels for heavy and light subunits in human normal tissues. Biochemical and biophysical research communications. 1995;206(2):584–589. doi: 10.1006/bbrc.1995.1083. [DOI] [PubMed] [Google Scholar]
- 9.Wild AC, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression: insights into transcriptional control of antioxidant defenses. Free Radic Res. 2000;32(4):281–301. doi: 10.1080/10715760000300291. [DOI] [PubMed] [Google Scholar]
- 10.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free radical biology & medicine. 2002;33(7):974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 11.Rahman I, Bel A, Mulier B, Lawson MF, Harrison DJ, Macnee W, Smith CA. Transcriptional regulation of gamma-glutamylcysteine synthetase-heavy subunit by oxidants in human alveolar epithelial cells. Biochemical and biophysical research communications. 1996;229(3):832–837. doi: 10.1006/bbrc.1996.1888. [DOI] [PubMed] [Google Scholar]
- 12.Biroccio A, Benassi B, Filomeni G, Amodei S, Marchini S, Chiorino G, Rotilio G, Zupi G, Ciriolo MR. Glutathione influences c-Myc-induced apoptosis in M14 human melanoma cells. The Journal of biological chemistry. 2002;277(46):43763–43770. doi: 10.1074/jbc.M207684200. [DOI] [PubMed] [Google Scholar]
- 13.Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, Zupi G, Biroccio A. c-Myc phosphorylation is required for cellular response to oxidative stress. Molecular cell. 2006;21(4):509–519. doi: 10.1016/j.molcel.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Benassi B, Zupi G, Biroccio A. Gamma-glutamylcysteine synthetase mediates the c-Myc-dependent response to antineoplastic agents in melanoma cells. Mol Pharmacol. 2007;72(4):1015–1023. doi: 10.1124/mol.107.038687. [DOI] [PubMed] [Google Scholar]
- 15.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. The Journal of biological chemistry. 1997;272(11):7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- 16.Dickinson DA, Iles KE, Zhang H, Blank V, Forman HJ. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. Faseb J. 2003;17(3):473–475. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- 17.Mulcahy RT, Gipp JJ. Identification of a putative antioxidant response element in the 5′-flanking region of the human gamma-glutamylcysteine synthetase heavy subunit gene. Biochemical and biophysical research communications. 1995;209(1):227–233. doi: 10.1006/bbrc.1995.1493. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Jaiswal AK. Human antioxidant-response-element-mediated regulation of type 1 NAD(P)H:quinone oxidoreductase gene expression. Effect of sulfhydryl modifying agents. European journal of biochemistry / FEBS. 1994;226(1):31–39. doi: 10.1111/j.1432-1033.1994.tb20023.x. [DOI] [PubMed] [Google Scholar]
- 19.Harada S, Fujii C, Hayashi A, Ohkoshi N. An association between idiopathic Parkinson's disease and polymorphisms of phase II detoxification enzymes: glutathione S-transferase M1 and quinone oxidoreductase 1 and 2. Biochemical and biophysical research communications. 2001;288(4):887–892. doi: 10.1006/bbrc.2001.5868. [DOI] [PubMed] [Google Scholar]
- 20.Levy S, Jaiswal AK, Forman HJ. The Role of c-Jun Phosphorylation in EpRE Activation of Phase II Genes. Free radical biology & medicine. 2009 doi: 10.1016/j.freeradbiomed.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends in biochemical sciences. 2000;25(3):99–104. doi: 10.1016/s0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- 22.Vasiliou V, Shertzer HG, Liu RM, Sainsbury M, Nebert DW. Response of [Ah] battery genes to compounds that protect against menadione toxicity. Biochemical pharmacology. 1995;50(11):1885–1891. doi: 10.1016/0006-2952(95)02083-7. [DOI] [PubMed] [Google Scholar]
- 23.Ahlgren-Beckendorf JA, Reising AM, Schander MA, Herdler JW, Johnson JA. Coordinate regulation of NAD(P)H:quinone oxidoreductase and glutathione-S-transferases in primary cultures of rat neurons and glia: role of the antioxidant/electrophile responsive element. Glia. 1999;25(2):131–142. [PubMed] [Google Scholar]
- 24.Iles KE, Liu RM. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free radical biology & medicine. 2005;38(5):547–556. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 25.Levens DL. Reconstructing MYC. Genes & development. 2003;17(9):1071–1077. doi: 10.1101/gad.1095203. [DOI] [PubMed] [Google Scholar]
- 26.Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, Tyner AL. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(8):4510–4515. doi: 10.1073/pnas.081074898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu-Mourez R, Freedman LP, Sonenshein GE. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20(14):1688–1702. doi: 10.1038/sj.onc.1204245. [DOI] [PubMed] [Google Scholar]
- 28.Burdo J, Schubert D, Maher P. Glutathione production is regulated via distinct pathways in stressed and non-stressed cortical neurons. Brain research. 2008;1189:12–22. doi: 10.1016/j.brainres.2007.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rinna A, Forman HJ. SHP-1 inhibition by 4-hydroxynonenal activates Jun N-terminal kinase and glutamate cysteine ligase. American journal of respiratory cell and molecular biology. 2008;39(1):97–104. doi: 10.1165/rcmb.2007-0371OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna A, Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Archives of biochemistry and biophysics. 2008;477(2):183–195. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome biology. 2003;4(10):R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wild AC, Gipp JJ, Mulcahy T. Overlapping antioxidant response element and PMA response element sequences mediate basal and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase catalytic subunit gene. The Biochemical journal. 1998;332(Pt 2):373–381. doi: 10.1042/bj3320373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moinova HR, Mulcahy RT. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochemical and biophysical research communications. 1999;261(3):661–668. doi: 10.1006/bbrc.1999.1109. [DOI] [PubMed] [Google Scholar]
- 34.Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Archives of pharmacal research. 2005;28(3):249–268. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- 35.Pizzimenti S, Briatore F, Laurora S, Toaldo C, Maggio M, De Grandi M, Meaglia L, Menegatti E, Giglioni B, Dianzani MU, Barrera G. 4-Hydroxynonenal inhibits telomerase activity and hTERT expression in human leukemic cell lines. Free radical biology & medicine. 2006;40(9):1578–1591. doi: 10.1016/j.freeradbiomed.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 36.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signaling: role of cystein modification in controlling antioxidant defences in response to electrophilic lipid peroxidation products. The Biochemical journal. 2004;378(Pt 2):373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, Forman HJ. Acrolein induces heme oxygenase-1 through PKC-delta and PI3K in human bronchial epithelial cells. American journal of respiratory cell and molecular biology. 2008;38(4):483–490. doi: 10.1165/rcmb.2007-0260OC. [DOI] [PubMed] [Google Scholar]
- 38.Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free radical biology & medicine. 2004;37(8):1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]