

Abstract
Arterial drug concentrations determine local toxicity. As such the emergent safety concerns surrounding drug-eluting stents mandate an investigation of the factors contributing to fluctuations in arterial drug uptake. Drug-eluting stents were implanted into porcine coronary arteries, arterial drug uptake was followed and modeled using 2-dimensional computational drug transport. Arterial drug uptake in vivo occurred faster than predicted by free drug diffusion, thus an alternate, mechanism for rapid transport has been proposed involving carrier-mediated transport. Though there was minimal variation in vivo in release kinetics from stent to stent, arterial drug deposition varied by up to 114% two weeks after stent implantation. The extent of adherent mural thrombus also fluctuated by 113% within 3 days after implantation. The computational drug transport model predicted that focal and diffuse thrombi elevate arterial drug deposition in proportion to the thrombus size by reducing drug washout subsequently increasing local drug availability. Fluctuations in arterial drug uptake are commonly reported. We now explain that variable peristrut thrombus can explain such observations even in the face of a narrow range of drug release from the stent. The mural thrombus effects on arterial drug deposition may be circumvented by forcing slow, rate limiting arterial transport that cannot be further hindered by mural thrombus.
Keywords: stents, drug delivery, restenosis, thrombus
II. Introduction
There is increasing concern for the risk of thrombosis in drug-eluting stents late after implantation. It was reported that drug-eluting stents clot at 0.6% each year after implantation for up to 3 years1 and remain an issue even years after implantation2, 3. Documentation of delayed healing and re-endothelialization suggest that the unhealed tissue state might provide a nidus for thrombosis4–6. Drug-eluting stents may delay recovery of endothelial function compared to their bare metal counterparts even 6 months after implantation7, 8. Heightened local arterial drug levels may be responsible for these observations and may be an especially potent problem given the narrow therapeutic window for paclitaxel and sirolimus and their selective effects on smooth muscle and endothelial cells9, 10. Complete appreciation of the role and limitations of drug eluting stents requires understanding and control of factors governing local arterial drug delivery. As we characterize the dynamics and improve control over local drug delivery11–15, unwarranted fluctuations in tissue drug levels might be minimized and an acceptable balance achieved between efficacy and toxicity.
We hypothesized that local thrombus formation is responsible for the observed variation in local arterial drug uptake. This hypothesis is supported by previous studies showing that specific compositions and distributions of mural thrombus act as a transport barrier and capacitive reservoir for locally administered drug. Mural thrombus interposed between the arterial wall and the drug source decreased arterial drug deposition in animal models of vascular injury16. Because mural thrombus forms as a natural consequence of percutaneous intervention17 and is affected by local vascular architecture and biology, its formation is highly variable within the heterogeneous patient populations receiving drug-eluting stents. For example atherosclerotic plaque disruption18, 19, induced by patient-specific lesion morphology and device implantation strategy, increases tissue factor exposure to the luminal blood which can lead to mural thrombus formation. In addition, differences in anti-platelet regimens3 or even patient insensitivity to anti-platelet therapies20 promotes variability in the risk of clot formation. Drug-eluting stents may perpetuate mural thrombus as arterial levels of sirolimus and paclitaxel8, 21 upregulate tissue factor expression. Finally, different stent designs may well induce variable degrees of local injury and overlying thrombosis, which implies that we may well need to consider stent design in choosing a device, which is an issue in discriminating choice of bare metal stents22, 23. For all these reasons mechanistic insight into the role that thrombus may play in altering drug release kinetics from a stent and arterial drug deposition is essential.
We investigated the interrelated roles of peri-strut thrombus formation, drug release kinetics, and drug physicochemical properties in contributing to fluctuating local arterial drug deposition. We used a combination of in vivo experiments and computational modeling to demonstrate that variability in mural thrombus formation may significantly alter the time course and magnitude of arterial drug uptake and retention. The findings from this work support the existence of multiple pathways for arterial drug transport and reveal mechanisms by which mural thrombus alters drug release kinetics and arterial drug uptake.
III. Methods
Mathematical Model
Stent implantation effaces the endothelium 22, 24 and re-endothelialization is prolonged over months within stented human atherosclerotic coronary arteries 25. As such the computational domain (Fig 1) represents a longitudinal segment of blood flowing over a stent strut embedded within an artery whose endothelial layer has been denuded. Drug transport was modeled using a 2-dimensional transient mass transfer model (Fig 1) 11, 26. The luminal diameter, 3 mm, was 3 times the arterial wall thickness; and, the axial distance along the artery was determined by the fluid mechanic entry length required to restore fully developed flow 27. The coated strut measured 150 μm in height and width, based upon representative dimensions of clinically available drug-eluting stents. Blood was assumed Newtonian and non-pulsatile, and its fluid mechanics were described with continuity and steady state Navier-Stokes equations (Fig 2, Eq 1–3). The fluid mechanical blood properties and volumetric flow rate were obtained from standard reference values28. Drug transport within the stent and arterial wall were modeled using the simple transient diffusion equation (Fig 2, Eq 7,9), while drug movement through the blood was modeled with the transient diffusion-convection equation (Fig 2, Eq 8). The fluid mechanics and mass transfer boundary conditions (Fig 2, Eqs 4–6, 11–20) employed have been previously described 11.
Figure 1.

Schematic representation of the computational domain. (A) The stent strut (white box) is surrounded by a drug-laden coating (black). It resides within the intravascular blood flow field (white) and contacts the arterial wall (gray). Blood flows in the + z-direction from the proximal to distal arterial segments. Representations of mural thrombus (gray striped pattern) in (B) focal and (C) diffuse distributions. Thrombus was applied around the coated stent strut and between the blood and arterial wall.
Figure 2.

Governing Equations and Boundary Conditions for Local Fluid Mechanics and Drug Transport within the Drug Laden Coating (c), Arterial Tissue (t), Lumenal Blood (b), and Peristrut Thrombus (th). Tissue, blood, thrombus, and coating domains are ΩTissue, ΩBlood, ΩThrombus, ΩCoat. Radial and axial axes, and time are r, z, t. Thickness of arterial tissue and coating are WTissue, LCoat. Arterial lumen radius and diameter are R, 2R. Lengths of vessel upstream and downstream from the strut are LProximal, LDistal. Duration of drug release is trelease. Maximum centerline velocity of blood flow is VC. Radial and axial component velocities are vr, vz. Dynamic pressure is P. Blood properties: dynamic viscosity and density are μ, ρ. Drug concentrations within tissue, blood, thrombus, and coating are Ct, Cb, Cth, Cc. Drug diffusivities within tissue, blood, thrombus and coating are Dt, Db, Dth, Dc.
Arterial drug diffusivities were obtained from two independent assays. Drug transport parameters in the tissue were obtained from experimentally measured values for anisotropic radial and axial free drug diffusivities29, 30. Arterial drug diffusivity was also fit to the experimental data of arterial drug uptake in stented porcine coronary arteries. Drug diffusivity within the coating was approximated from the duration of drug elution observed from the retrieved porcine implanted devices 27.
Peri-strut thrombus was simulated by placing a focal, solid mass ranging in area from 0.003 to 0.1 mm2 around the stent strut, or by distributing the mass diffusely over a 200 mm2 area around the strut and the arterial lumen (Fig 1b). The size of the mass was based upon the average peri-strut thrombus size and distribution observed histologically in porcine stented coronary arteries at 3, 14, and 30 days post-implantation. Drug diffusivity through the thrombus was based on experimentally measured values for whole blood clot16. Drug transport through the peri-strut mass occurred via transient diffusion (Fig 2, Eq 10). Drug transport continued from the coating through thrombus to the arterial wall or to lumenal blood via a continuity of flux boundary condition (Fig 2, Eq 16, 18).
Using commercially available geometry/mesh generation and computational fluid dynamics software (GAMBIT v2.3.16, FLUENT v6.3.17, Fluent Inc., Hanover, NH), previously utilized numerical schemes were employed to solve the coupled fluid mechanical and mass transfer equations 11. The computational data are reported as fractional drug release and arterial drug concentration over time.
Experimental Methods
All animal studies were performed at Cordis contract laboratory facilities. The study protocols were approved by the Animal Care and Use Committee at the respective contract facilities under the protocol numbers ETP-12-002344, ETP-12-002345, and ETP-12-002346. All studies were performed in compliance with GLP regulations. Under general anesthesia, Yucatan miniswine received CYPHER(R) Sirolimus-eluting Coronary Stent (Cordis Corporation) within the LAD, LCX and RCA coronary arteries. Devices were deployed to a target balloon-to-artery ratio of 1.1–1.3:1. Twenty-four hours prior to the procedure, the pigs received oral 650 mg aspirin, 300 mg clopidogrel and 30 mg Procardia XL. At the time of stent implantation, animals received 150–300 IU/Kg intraprocedural heparin, I.V., to achieve a target activated clotting time of >300 sec. Post-implantation, animals received 325 mg oral aspirin daily for the remainder of the study and 75 mg oral clopidogrel daily for 2 months.
A subset of stented arteries (n = 6 per time point) was harvested at 1, 8, 14, 30, 60, and 90 days post-implantation to quantify the amount of drug remaining on the stent and delivered to the artery. Separation of the tissue and stent was performed by first excising the stented arterial segment from the porcine heart, cutting longitudinally down the cylindrical segment to expose the luminal surface. The tissue was directly peeled away from the stent taking care not to disrupt the polymeric coating on the stent. Sirolimus was extracted from the stents in 3 ml of HPLC grade methanol. The fresh arterial tissue segments were frozen on dry ice and maintained at −70 °C until analysis. Tissues were homogenized in PBS and the aqueous phase extracted into methyl-t-butyl ether/n-butyl chloride (1:1), evaporated to dryness and reconstituted in 0.5 ml HPLC grade methanol. Sirolimus extracted from the stent and arterial wall samples was quantified using a validated LC/MS/MS method. Subsequently, the amount of drug released was calculated by subtracting the amount of drug extracted from the stent at the time of vessel harvest from the amount of drug on the stent prior to implantation. The data are plotted as the amount of drug released from the stent as a fraction of the original drug load on the device. It is important to note that the sirolimus extracted from the tissue represents that within the arterial wall as well as that within any peristrut thrombus and neointima, because separating arterial tissue from peristrut mass was not easily achieved. However, it is safe to assume that especially at earlier times the measurement of tissue drug levels is primarily arterial drug levels, as the arterial tissue comprises the larger volume by mass relative to the peristrut mass and hydrophobic agents such as sirolimus are known to bind significantly to the arterial wall 30 and less to adherent thrombus16.
A second subset of arteries was harvested at 3, 14, and 30 days post-implantation for histological analysis. Hearts were harvested and immediately perfusion fixed with 10% neutral buffered formalin. Vessels were carefully dissected from the hearts and embedded with methyl methacrylate after dehydration in graded ethanol. Arterial cross-sections were cut and stained with a metachromatic stain for histological analysis.
Histological Quantification of Mural Thrombus and Neointima
Histomorphometry measurements were made on the stained porcine coronary sections, using Adobe Photoshop version 5.0 to trace the perimeter of the peri-strut thrombus. The outlined thrombus area was subsequently measured and averaged over multiple stenting cross sections of a single coronary artery, enabling quantification of the local thrombus at 3, 14, and 30 days.
Statistical Analysis Methods
The amount of drug detected from the stent, porcine coronary arteries, and peri-strut thrombus area were calculated for each time point using the arithmetic mean and the corresponding standard deviation. Statistical comparison of thrombus at different time points was determined using a two-tailed Student’s t-Test assuming two samples with unequal variance. The time course of stent based drug release was compared to computational predictions of drug elution using a root mean square analysis.
IV. Results
In Vivo Variability in Arterial Drug Deposition and Mural Thrombus Formation
Sirolimus eluting stents implanted into porcine coronary arteries released 93% of their drug load within 90 days. The variability in drug release was defined as the standard deviation of the mean amount of drug released normalized by the arithmetic mean. Drug release from the stent strut reached its maximum variability of 30% after 1 day of drug elution. Variability diminished to 5% of the mean two weeks after implantation (Fig 3a). In contrast, arterial drug uptake reached its peak level one day after implantation and decayed 75% after 30 days. Arterial drug levels fluctuated more substantially, such that maximum variability in arterial drug uptake was 114% two weeks after implantation (Fig 3b).
Figure 3.

Experimental drug release and arterial drug uptake data obtained from drug-eluting stent implants within porcine coronary arteries. (A) Fractional Drug Release from the Stent, (B) Average Arterial Drug Concentrations. Quantification of drug released from the stent and absorbed by the arterial wall was obtained for 1, 8, 14, 30, 60, 90 days post-implantation. (Note: all error bars represent the standard deviation from the mean.)
Histological analysis of explanted porcine coronary arteries revealed that foci of mural thrombus formed around stent struts as early as 3 days after implantation. Thrombus size ranged from 0.3% to 70% of the 0.02 mm2 strut area (Fig 4a,d). The peri-strut mass grew over time to become on average 5 times larger in area by 14 days post-implantation, ranging in size from 70%–250% of the strut area (Fig 4b,d). By 30 days, the peri-strut region was surrounded by a diffuse neointimal covering that was on average more than 25-fold larger than the 3 day thrombotic mass (Fig 4c–d). Interestingly, the variability in thrombus area reached its maximum of 120% 3 days after stent implantation, and declined over time reaching 60% at 14 days (Fig 4d). By 30 days, the extent of variability in neointimal area was reduced to 45%. The overlapping duration of maximal variability in the peristrut mass (thrombus and neointima) and arterial drug deposition (Fig 3b, 4d) suggests that they are related.
Figure 4.
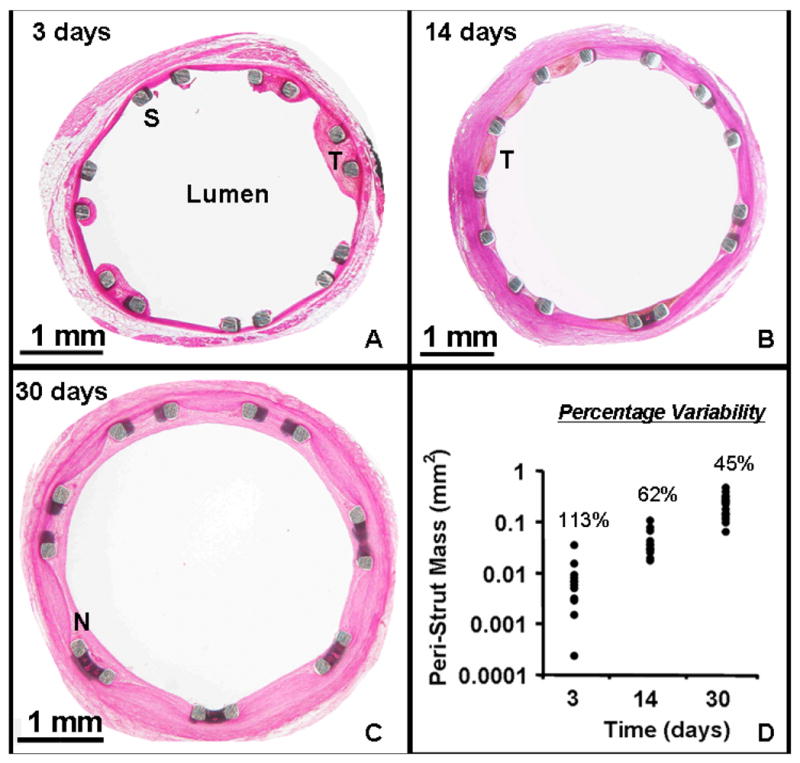
Histological evidence of mural thrombus and neointima around the drug-eluting stent strut at various times after implantation. Images obtained from stented porcine coronary artery explants at (A) 3 days, (B) 14 days, and (C) 30 days post-implantation. (D) Quantification of peristrut mass area (mm2) at 3, 14, and 30 days post-implantation. Percentage variability is defined as the standard deviation of the peristrut mass area as a percentage of the arithmetic mean of the peristrut mass area. Note: the y-axis is plotted on a log scale.
Computational Predictions of Thrombus Formation
Transient, 2-dimensional computational simulations were performed to examine drug transport from the coated strut through the surrounding thrombus and finally to the underlying arterial wall. The goal was to test how variations in mural thrombus of physiologic size and distribution contribute to fluctuating arterial drug levels. The simulations indicate that the slow rate of drug release from the stent cannot be altered by either diffuse or focal, peri-strut thrombi (Fig 5a), because of the resistance to drug transport within the polymeric coating of the stent is significantly greater than resistance imposed by peri-strut masses such as thrombi or neointima. This prediction is consistent with the experimental drug release in vivo that appears well-controlled with little variability in fractional release (Fig 3a). The simulated drug release from the stent matches in vivo drug release with a root mean square value of <0.1.
Figure 5.

Thrombus size, distribution, and drug transport characteristics impact arterial drug deposition and distribution without altering drug release kinetics. (A) Fractional drug release from the stent (Note: all curves with and without thrombus overlap) (B) Average arterial drug concentration resulting from focal and diffuse thrombus (C) Axial arterial drug distribution in the presence of focal and diffuse thrombus 1 day after implantation at a depth of 10 um into the arterial wall, (D) Average arterial drug concentration resulting from two extremes in thrombus drug transport properties, which represent different thrombi compositions. (Note: For all these cases, the arterial drug diffusivity in the transmural direction (Dr) was 10 um2/s and in the axial direction (Dz) was 1 um2/s; the drug diffusivity within the stent coating (Dc) was 1e-5 um2/s. Strut width was 150 um.)
In contrast, modeling predicts that the size and distribution of peri-strut thrombi determines the extent of variability in arterial drug uptake and retention (Fig 5). Focal, peri-strut thrombi of 0.1 mm2 (Fig 1b) increase the peak average drug concentration and cumulative drug exposure within the arterial wall by 80% even without protecting drug from washout from the arterial wall (Fig 5b). Focal thrombus increases the residence time and surface contact of drug eluted from the stent within the local arterial milieu. As a result, more drug is available at arterial interface where it is absorbed by the artery from the thrombus (Fig 5c). In this way, focal thrombus acts as an extension of the stent strut, increasing the footprint of the strut-thrombus complex along the arterial wall. When the drug travels even more slowly through the thrombus, which is approximated using drug diffusion characteristics within a heterogeneous clot16, simulations predict that thrombus elevates arterial drug levels to a greater extent (Fig 5d). Diffusely distributed thrombus (Fig 1c) primarily shields drug from washout from the arterial wall (Fig 5c). When drug transport is simulated in the presence of a diffuse thrombus, arterial drug uptake is predicted to increase 3.5-fold. Thus, thrombus induced elevations in arterial drug uptake and exposure are predicted as a consequence of better local arterial drug availability and reduced drug washout into the luminal blood.
Drug Physicochemical Properties Govern Thrombus Induced Arterial Drug Deposition
It is predicted that the extent to which thrombus influences arterial drug deposition depends upon the relative rates of drug transport within the arterial wall and thrombus, determined by drug physicochemical properties within each domain. Drugs that are rapidly transported in the arterial tissue relative to thrombus, with isotropic arterial drug diffusivity of 50 um2/s compared to thrombus drug diffusivity of 20 um2/s, should exhibit thrombus-induced increases in drug deposition and distribution by more than 2-fold (Fig 6b). This occurs because thrombus locally sequesters drug to improve availability for arterial drug transport, an event which occurs relatively rapidly. In contrast, drugs that move exceedingly slowly through the arterial wall, with an arterial drug diffusion coefficient of 0.02 um2/s compared to thrombus drug diffusivity of 20 um2/s, should not cause thrombus-induced fluctuations in arterial drug deposition. In these cases, the slow nature of drug entry and exit into the tissue is rate limiting, thus thrombus does not retain drug long enough to increase arterial drug uptake (Fig 6b).
Figure 6.

The extent to which thrombus impacts arterial drug levels depends on the rate of drug transport within the arterial wall, which is determined by the arterial drug diffusivity. (A) Fractional release of drug from the stent (Note: pink and black curves representing cases with and without thrombus overlap) and (B) Average arterial drug concentration achieved with drugs possessing different diffusivities in the arterial wall, where the arterial drug diffusivity is either isotropic (D) or anisotropic (Dr, Dz). (Note: black curve = without thrombus, pink curve= with focal thrombus 0.1 mm2; Strut width and height = 150 um.)
In reality, most drugs interact with the layered arterial ultrastructure so that they travel faster axially and circumferentially than transmurally. Preferential axial transport enables spreading along the arterial wall but minimizes radial penetration. Two different anisotropic drug transport diffusivities were simulated. In one instance, a previously measured diffusion coefficient for anisotropic free paclitaxel30 (Fig 6b, Dr = 0.02 um2/s, Dz = 50 um2/s) was used. Thrombus increases average arterial drug levels by only 20% when free paclitaxel transport is simulated, which is small compared to the thrombus effect on faster moving agents (Fig 6b). In the other case, the transport value was obtained from fitting the computations with the experimental arterial drug uptake data. Hydrophobic drugs diffuse faster through the arterial wall than expected from free drug diffusion alone (Fig 4b, 5b). The rate of arterial drug diffusion matched the arterial diffusion properties of carrier proteins, such as albumin (Fig 5b, Dr = 1 um2/s, Dz = 10 um2/s). The faster rate of arterial drug transport predicted from computational fitting of experimental data suggests an alternative arterial paclitaxel transport modality: carrier-protein mediated transport. Focal thrombus in the setting of faster anisotropic arterial drug diffusion leads to 80% increase in the average arterial drug concentration (Fig 5b).
Though potentially a rare event, a compound may traverse preferentially in the transmural penetrating direction compared to axial transport, for instance due to transmural pressure driven flow from porous catheter drug delivery. Such anisotropically transported compounds spread minimally (Dr = 50 um2/s, Dz = 0.02 um2/s), so the available surface area for washout is also minimal (Fig 6b). As a result any mural thrombus only minimally impacted arterial drug uptake.
Role of Thrombus Induced Variation in Arterial Drug Uptake in the Setting of Rapid Drug Release Kinetics
Peristrut thrombus should slow drug release kinetics only when release is sufficiently rapid compared to drug transport through the thrombus. For example, when the entire drug load is released within minutes or hours, then mural thrombus imposes a transport barrier to drug diffusion from the coated stent strut and through the surrounding thrombus, which subsequently slows the overall rate of drug release (Fig 7a). The thrombus induced deceleration in drug release increases local drug availability for arterial uptake, leading to elevated arterial drug deposition (Fig 7b). Thus, when drug transport through thrombus and not drug release from the stent is rate limiting, the extent of arterial drug deposition is governed by the mass, distribution and transport properties of peristrut thrombus. Though stent release of drug occurs predominantly by a slow mechanism over weeks to months, the earliest period of drug release may be rapid, known as an early burst release phase. Within this period, thrombus may slow drug release from the stent resulting in increased local drug availability and subsequent elevation in arterial drug levels. By contrast, when the stent strut eluting drug over several weeks is surrounded by a thrombus, the majority of drug release from the stent is so slow that the thrombus is predicted to impose a negligible barrier for drug transport. As a result, mural thrombus around a slowly eluting stent is predicted to have no effect on drug release, because it is the drug release from the stent and not transport through thrombus that is rate limiting (Fig 5a).
Figure 7.

Thrombus slows drug release kinetics when drug release occurs rapidly over minutes to hours, which is faster than the typical release rate achieved by intravascular stents. Subsequently, thrombus increases local drug availability leading to elevated arterial drug levels. (A) Fractional release of drug from the stent, (B) Average arterial drug concentration. The arterial drug diffusivity in the transmural direction (Dr) was 10 um2/s and in the axial direction (Dz) was 1 um2/s; the drug diffusivity within the stent coating was 10 um2/s; drug diffusivity within the mural thrombus was 20 um2/s. (Note: black curve = without thrombus, pink curve= with focal thrombus 0.1 mm2; Strut width and height = 150 um. (A) and (B) are plotted on different time scales on the x-axis)
V. Discussion
The role of thrombus in local vascular therapy continues to emerge as an increasingly important and critical phenomenon. Recent clinical trial results demonstrate a 6-fold increase in subsequent thrombosis when a drug-eluting stent was placed in an artery with substantial pre-existing thrombosis31. While intimal hyperplasia is controlled only by a threshold dose, above which there is an effect and below which there is not, thrombosis is significantly dose responsive. Since efficacy and toxicity are governed by the amount of drug absorbed by the artery not simply the amount released from the stent and presented to the vessel, it is incumbent upon us to define the parameters that determine arterial uptake.
Within this study, we found that the experimental time course for in vivo arterial uptake of sirolimus was faster than that predicted with free drug diffusion alone, which suggests that drug transport within the arterial wall must occur via a faster alternate mechanism. Additionally, variations in peri-strut mural thrombi were found to cause fluctuations in arterial drug levels. Methods of controlling thrombus induced variation in arterial drug levels include altering the relative drug transport characteristics within the stent, arterial wall, and mural thrombus.
Mechanisms for Arterial Drug Transport
Hydrophobic drugs such as sirolimus and paclitaxel diffuse exceedingly slowly through the arterial wall, which permits their high arterial retention and subsequent superior biologic efficacy over biologically potent hydrophilic agents30, 32. Arterial drug transport is dictated by drug interactions with the local arterial ultrastructure, a composite structure of smooth muscle cell layers residing between sheets of elastin. This laminate tissue structure hinders drug diffusion transmurally but does not impose much resistance to circumferential or axial diffusion29, 30. Thus, computations predicted that hydrophobic drugs penetrate through the arterial wall poorly, but spread axially and circumferentially with greater ease, achieving the highest drug concentrations near the arterial-lumenal interface.
However, free drug diffusion alone predicts a significantly slower rate of arterial drug deposition compared to that seen in vivo (Fig 3b, 6b), which implies that there is likely an additional, expedited mechanism of arterial drug transport. In addition to free diffusion, drug may travel rapidly within the arterial wall by associating with carrier proteins that diffuse faster than free drug alone33. When drug diffusion coefficients were calculated by fitting the experimental data to the computational model (Fig 5b), the resultant diffusion coefficient corresponded with previously measured transport coefficients for carrier proteins within the arterial wall 29. This suggests a carrier mediated mechanism of arterial drug transport, which is supported by prior experimental data showing that the carrier protein, albumin, penetrates the arterial wall, especially when the endothelium is denuded 34, which occurs during stent implantation24. Endothelium remains at least partially denuded for many months after implantation in atherosclerotic human coronary arteries 25, Hydrophobic drugs such as paclitaxel bind albumin, with potentially strong affinity35. Thus, hydrophobic drugs, which move slowly in their free state, may actually associate with carrier proteins in the arterial wall and subsequently diffuse faster leading to higher arterial drug levels. In healthy porcine coronary arteries, which re-endothelialize more rapidly than diseased human vessels 24, rapid drug delivery after vessel re-endothelialization could still occur via carrier mediated transport. The re-endothelialized vessel would be particularly subject to carrier protein penetration especially if the endothelium is not yet confluent or has not regained complete barrier function, though the magnitude of carrier mediated transport is likely to be reduced. Additionally, the endothelial denudation may lead to vessel edema, which could increase the proportion of drug transported via convective forces even after re-endothelialization. Though not applied in this computational model, transmural hydrophobic drug convection through a porous arterial wall could also dominate local transport, analogously to hydrophilic drug convective transport36.
Mural Thrombus Accounts for Variability in Arterial Drug Deposition
Thrombus was first proven to influence arterial drug levels when arterial drug deposition was found to decrease in response to mural thrombus interposed between tissue and drug source 16. On this basis, variable mural thrombus formation was investigated as an initiator of fluctuating arterial drug deposition. Healthy pig coronary arteries implanted with drug-eluting stents had significant variability in arterial drug deposition (Fig 3b). A component of variability is always attributable to experimental error, which can arise from the technical difficulty of harvesting tissue and separating it from the stent, as well as from drug quantification methods and detection instruments. However, if random errors were the only source of variability, one would expect deviations to remain either constant over time or at a fixed percentage of the value at specific points in time. Yet, the variability in arterial drug deposition was greatest within the first two weeks post-implantation after which it declined. A similar trend of maximal variability in arterial drug deposition occurring 2 weeks after stent implantation was reported in another experimental study, corroborating our observations 37. The variation in arterial drug deposition was hypothesized to arise from an identifiable and predictable source. If drug release from the stent fluctuated substantially, one might attribute varying arterial drug levels to release kinetics. However, drug elution was relatively constant (Fig 3a).
Variability in peri-strut thrombus was hypothesized to cause fluctuating arterial drug levels because the timing of maximal fluctuations in mural thrombus overlapped with that of maximum variability in arterial drug deposition (Fig 4). Computational studies confirmed that physiologically sized thrombus could elevate arterial drug uptake in a manner comparable to experimental observation (Fig 5). Thus, uncontrolled formation of mural thrombus around the stent strut was concluded to be a significant contributor to the observed variability in arterial drug deposition.
Mechanisms of Thrombus-Induced Arterial Drug Uptake
Thrombus-induced fluctuations in arterial drug deposition may increase the risk for local toxicity due to the narrow therapeutic window of the current hydrophobic agents9, 10. To achieve uniform local therapy, one can either minimize variability in local thrombus formation, or minimize the effect of thrombus on arterial drug levels. The former can be achieved by minimizing local thrombus formation either pharmacologically or via improvements in procedural and device related risk factors for thrombus. Still, fine control over focal thrombi as small as 0.1 mm2 may prove difficult, considering that local tissue levels of sirolimus and paclitaxel may promote thrombus formation21, 38. Alternatively, some degree of mural thrombus may be essential for efficacious drug delivery, and may be problematic only when it is excessive and uncontrolled. Thus, it is useful to understand the mechanisms by which thrombus induces fluctuations in arterial drug levels and possibly alter these pathways.
The pattern of distribution of thrombus around the stent determines the mechanism by which it modulates arterial drug levels. Local, focal thrombi formed adjacent to the stent strut and arterial wall effectively increase the footprint of the stent strut along the arterial wall, enhancing arterial drug deposition. These thrombi prolong the drug residence time and retention at the arterial interface long enough to increase tissue absorption (Fig 5b). Such local thrombi are ubiquitous even 3 days after stent implantation (Fig 4a), and even small thrombi 0.1 mm2 were predicted to increase tissue drug deposition (Fig 5b). By contrast, diffusely distributed thrombi/neointima, which are observed at 14 and 30 days post-implantation, can shield arterial drug from washout into the lumen (Fig 4b–c, 5b).
The extent to which thrombi impact arterial drug deposition depends on how drug transport through the thrombus alters the relative kinetics of drug washout and retention in the local milieu (Fig 5–7). Thrombus was predicted to increase arterial drug deposition if it imposes a diffusion barrier to drug washout (Fig 5) and/or drug release (Fig 7). Thrombus prevents drug washout when drug moves exceeding slowly through the thrombus relative to that in the artery, which occurs with carrier-mediated arterial drug diffusion (Fig 5). Similarly, when the rate of drug elution is fast, which may be expected when drug delivery occurs from a coated angioplasty balloon or during the early burst release phase from stents, mural thrombus slows the overall drug release rate (Fig 7). Subsequently, the drug residence time in the local milieu increases due to impeded drug transport from the stent and through the thrombus, which enables better arterial drug absorption.
Conversely, when transport in the arterial wall and stent coating are exceedingly slow relative to that in thrombus, thrombi of any size or distribution are not able to further impede drug movement. Stent based drug delivery is an example of a predominantly thrombus-independent drug elution process, where the slow rate of drug elution from the stent cannot be further decelerated by the thrombus. This prediction is supported by the experimental and computational observations that stent release kinetics did not vary despite mural thrombus formation (Fig 5a). To further control thrombus induced fluctuations in arterial drug deposition, arterial drug transport could be slowed by steric modification of the drug. However, this approach has the drawback that slow drug transport results in poor distribution of drug through the arterial tissue. This approach might also alter the pharmacological effectiveness of the drug so modification of drug structure must be carefully considered.
VI. Conclusions
Arterial drug diffusion was found to be faster in vivo than would be predicted by free drug diffusion alone, thus computations support the existence of a carrier-mediated drug transport mechanism. A significant component of arterial drug variability was found to be caused by locally varying thrombus formation. Small, focal thrombi may be virtually impossible to prevent yet they should substantially increase local drug deposition, which may subsequently impact local toxicity. The impact of thrombus induced variability can be minimized if drug release from the stent and uptake into the artery are rate limiting relative to drug transport through mural thrombus. Alternatively, the variability in thrombus induced drug delivery would be less significant if locally delivered agents had greater selective activity against smooth muscle cells with less effect on the local thrombotic cascade and endothelialization.
Acknowledgments
The study was supported in part by grants from the NIH (R01 HL-49309) and an unrestricted gift from Cordis Corporation (a Johnson and Johnson Company).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
VIII. References
- 1.Daemen J, Wenaweser P, Tsuchida K, Abrecht L, Vaina S, Morger C, Kukreja N, Juni P, Sianos G, Hellige G, van Domburg RT, Hess OM, Boersma E, Meier B, Windecker S, Serruys PW. Early and late coronary stent thrombosis of sirolimus-eluting and paclitaxel-eluting stents in routine clinical practice: data from a large two-institutional cohort study. Lancet. 2007;369(9562):667–678. doi: 10.1016/S0140-6736(07)60314-6. [DOI] [PubMed] [Google Scholar]
- 2.Carlsson J, von Wagenheim B, Linder R, Anwari TM, Qvist J, Petersson I, Magounakis T, Lagerqvist B. Is late stent thrombosis in drug-eluting stents a real clinical issue? A single-center experience and review of the literature. Clin Res Cardiol. 2006 doi: 10.1007/s00392-007-0464-x. [DOI] [PubMed] [Google Scholar]
- 3.Pfisterer M, Brunner-La Rocca HP, Buser PT, Rickenbacher P, Hunziker P, Mueller C, Jeger R, Bader F, Osswald S, Kaiser C. Late clinical events after clopidogrel discontinuation may limit the benefit of drug-eluting stents: an observational study of drug-eluting versus bare-metal stents. J Am Coll Cardiol. 2006;48(12):2584–2591. doi: 10.1016/j.jacc.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Drachman DE, Edelman ER, Seifert P, Groothuis AR, Bornstein DA, Kamath KR, Palasis M, Yang D, Nott SH, Rogers C. Neointimal thickening after stent delivery of paclitaxel: change in composition and arrest of growth over six months. J Am Coll Cardiol. 2000;36(7):2325–2332. doi: 10.1016/s0735-1097(00)01020-2. [DOI] [PubMed] [Google Scholar]
- 5.Farb A, Heller PF, Shroff S, Cheng L, Kolodgie FD, Carter AJ, Scott DS, Froehlich J, Virmani R. Pathological analysis of local delivery of paclitaxel via a polymer-coated stent. Circulation. 2001;104(4):473–479. doi: 10.1161/hc3001.092037. [DOI] [PubMed] [Google Scholar]
- 6.Sousa JE, Costa MA, Farb A, Abizaid A, Sousa A, Seixas AC, da Silva LM, Feres F, Pinto I, Mattos LA, Virmani R. Images in cardiovascular medicine. Vascular healing 4 years after the implantation of sirolimus-eluting stent in humans: a histopathological examination. Circulation. 2004;110(1):e5–6. doi: 10.1161/01.CIR.0000134307.00204.B3. [DOI] [PubMed] [Google Scholar]
- 7.Hofma SH, van der Giessen WJ, van Dalen BM, Lemos PA, McFadden EP, Sianos G, Ligthart JM, van Essen D, de Feyter PJ, Serruys PW. Indication of long-term endothelial dysfunction after sirolimus-eluting stent implantation. Eur Heart J. 2006;27(2):166–170. doi: 10.1093/eurheartj/ehi571. [DOI] [PubMed] [Google Scholar]
- 8.Togni M, Windecker S, Cocchia R, Wenaweser P, Cook S, Billinger M, Meier B, Hess OM. Sirolimus-eluting stents associated with paradoxic coronary vasoconstriction. J Am Coll Cardiol. 2005;46(2):231–236. doi: 10.1016/j.jacc.2005.01.062. [DOI] [PubMed] [Google Scholar]
- 9.Axel DI, Kunert W, Goggelmann C, Oberhoff M, Herdeg C, Kuttner A, Wild DH, Brehm BR, Riessen R, Koveker G, Karsch KR. Paclitaxel inhibits arterial smooth muscle cell proliferation and migration in vitro and in vivo using local drug delivery. Circulation. 1997;96(2):636–645. doi: 10.1161/01.cir.96.2.636. [DOI] [PubMed] [Google Scholar]
- 10.Matter CM, Rozenberg I, Jaschko A, Greutert H, Kurz DJ, Wnendt S, Kuttler B, Joch H, Grunenfelder J, Zund G, Tanner FC, Luscher TF. Effects of tacrolimus or sirolimus on proliferation of vascular smooth muscle and endothelial cells. J Cardiovasc Pharmacol. 2006;48(6):286–292. doi: 10.1097/01.fjc.0000248233.22570.8b. [DOI] [PubMed] [Google Scholar]
- 11.Balakrishnan B, Dooley JF, Kopia G, Edelman ER. Intravascular drug release kinetics dictate arterial drug deposition, retention, and distribution. J Control Release. 2007;123(2):100–108. doi: 10.1016/j.jconrel.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westedt U, Kalinowski M, Wittmar M, Merdan T, Unger F, Fuchs J, Schaller S, Bakowsky U, Kissel T. Poly(vinyl alcohol)-graft-poly(lactide-co-glycolide) nanoparticles for local delivery of paclitaxel for restenosis treatment. J Control Release. 2007;119(1):41–51. doi: 10.1016/j.jconrel.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Westedt U, Wittmar M, Hellwig M, Hanefeld P, Greiner A, Schaper AK, Kissel T. Paclitaxel releasing films consisting of poly(vinyl alcohol)-graft-poly(lactide-co-glycolide) and their potential as biodegradable stent coatings. J Control Release. 2006;111(1–2):235–246. doi: 10.1016/j.jconrel.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Zweers ML, Engbers GH, Grijpma DW, Feijen J. Release of anti-restenosis drugs from poly(ethylene oxide)-poly(DL-lactic-co-glycolic acid) nanoparticles. J Control Release. 2006;114(3):317–324. doi: 10.1016/j.jconrel.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 15.Huh KM, Lee SC, Cho YW, Lee J, Jeong JH, Park K. Hydrotropic polymer micelle system for delivery of paclitaxel. J Control Release. 2005;101(1–3):59–68. doi: 10.1016/j.jconrel.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Hwang CW, Levin AD, Jonas M, Li PH, Edelman ER. Thrombosis modulates arterial drug distribution for drug-eluting stents. Circulation. 2005;111(13):1619–1626. doi: 10.1161/01.CIR.0000160363.30639.37. [DOI] [PubMed] [Google Scholar]
- 17.Diaz-Sandoval LJ, Bouma BE, Tearney GJ, Jang IK. Optical coherence tomography as a tool for percutaneous coronary interventions. Catheter Cardiovasc Interv. 2005;65(4):492–496. doi: 10.1002/ccd.20340. [DOI] [PubMed] [Google Scholar]
- 18.Toschi V, Gallo R, Lettino M, Fallon JT, Gertz SD, Fernandez-Ortiz A, Chesebro JH, Badimon L, Nemerson Y, Fuster V, Badimon JJ. Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation. 1997;95(3):594–599. doi: 10.1161/01.cir.95.3.594. [DOI] [PubMed] [Google Scholar]
- 19.Wilcox JN, Smith KM, Schwartz SM, Gordon D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci U S A. 1989;86(8):2839–2843. doi: 10.1073/pnas.86.8.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruef J, Kranzhofer R. Coronary stent thrombosis related to aspirin resistance: what are the underlying mechanisms? J Interv Cardiol. 2006;19(6):507–509. doi: 10.1111/j.1540-8183.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- 21.Steffel J, Latini RA, Akhmedov A, Zimmermann D, Zimmerling P, Luscher TF, Tanner FC. Rapamycin, but not FK-506, increases endothelial tissue factor expression: implications for drug-eluting stent design. Circulation. 2005;112(13):2002–2011. doi: 10.1161/CIRCULATIONAHA.105.569129. [DOI] [PubMed] [Google Scholar]
- 22.Rogers C, Edelman ER. Endovascular stent design dictates experimental restenosis and thrombosis. Circulation. 1995;91(12):2995–3001. doi: 10.1161/01.cir.91.12.2995. [DOI] [PubMed] [Google Scholar]
- 23.Rogers C, Tseng DY, Squire JC, Edelman ER. Balloon-artery interactions during stent placement: a finite element analysis approach to pressure, compliance, and stent design as contributors to vascular injury. Circ Res. 1999;84(4):378–383. doi: 10.1161/01.res.84.4.378. [DOI] [PubMed] [Google Scholar]
- 24.Rogers C, Parikh S, Seifert P, Edelman ER. Endogenous cell seeding. Remnant endothelium after stenting enhances vascular repair. Circulation. 1996;94(11):2909–2914. doi: 10.1161/01.cir.94.11.2909. [DOI] [PubMed] [Google Scholar]
- 25.Jeremias A, Sylvia B, Bridges J, Kirtane AJ, Bigelow B, Pinto DS, Ho KK, Cohen DJ, Garcia LA, Cutlip DE, Carrozza JP., Jr Stent thrombosis after successful sirolimus-eluting stent implantation. Circulation. 2004;109(16):1930–1932. doi: 10.1161/01.CIR.0000127105.99982.21. [DOI] [PubMed] [Google Scholar]
- 26.Balakrishnan B, Tzafriri AR, Seifert P, Groothuis A, Rogers C, Edelman ER. Strut position, blood flow, and drug deposition: implications for single and overlapping drug-eluting stents. Circulation. 2005;111(22):2958–2965. doi: 10.1161/CIRCULATIONAHA.104.512475. [DOI] [PubMed] [Google Scholar]
- 27.Deen WM. Analysis of Transport Phenomena. Oxford University Press, Inc; Oxford: 1998. [Google Scholar]
- 28.Berne R, Levy MN, editors. Physiology. 4. Mosby, Inc; St. Louis: 1998. [Google Scholar]
- 29.Hwang CW, Edelman ER. Arterial ultrastructure influences transport of locally delivered drugs. Circ Res. 2002;90(7):826–832. doi: 10.1161/01.res.0000016672.26000.9e. [DOI] [PubMed] [Google Scholar]
- 30.Levin AD, Vukmirovic N, Hwang CW, Edelman ER. Specific binding to intracellular proteins determines arterial transport properties for rapamycin and paclitaxel. Proc Natl Acad Sci U S A. 2004;101(25):9463–9467. doi: 10.1073/pnas.0400918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sianos G, Papafaklis MI, Daemen J, Vaina S, van Mieghem CA, van Domburg RT, Michalis LK, Serruys PW. Angiographic stent thrombosis after routine use of drug-eluting stents in ST-segment elevation myocardial infarction: the importance of thrombus burden. J Am Coll Cardiol. 2007;50(7):573–583. doi: 10.1016/j.jacc.2007.04.059. [DOI] [PubMed] [Google Scholar]
- 32.Hwang CW, Wu D, Edelman ER. Physiological transport forces govern drug distribution for stent-based delivery. Circulation. 2001;104(5):600–605. doi: 10.1161/hc3101.092214. [DOI] [PubMed] [Google Scholar]
- 33.Lovich MA, Creel C, Hong K, Hwang CW, Edelman ER. Carrier proteins determine local pharmacokinetics and arterial distribution of paclitaxel. J Pharm Sci. 2001;90(9):1324–1335. doi: 10.1002/jps.1085. [DOI] [PubMed] [Google Scholar]
- 34.Ramirez CA, Colton CK, Smith KA, Stemerman MB, Lees RS. Transport of 125I-albumin across normal and deendothelialized rabbit thoracic aorta in vivo. Arteriosclerosis. 1984;4(3):283–291. doi: 10.1161/01.atv.4.3.283. [DOI] [PubMed] [Google Scholar]
- 35.Paal K, Muller J, Hegedus L. High affinity binding of paclitaxel to human serum albumin. Eur J Biochem. 2001;268(7):2187–2191. doi: 10.1046/j.1432-1327.2001.02107.x. [DOI] [PubMed] [Google Scholar]
- 36.Lovich MA, Edelman ER. Mechanisms of transmural heparin transport in the rat abdominal aorta after local vascular delivery. Circ Res. 1995;77(6):1143–1150. doi: 10.1161/01.res.77.6.1143. [DOI] [PubMed] [Google Scholar]
- 37.Klugherz BD, Llanos G, Lieuallen W, Kopia GA, Papandreou G, Narayan P, Sasseen B, Adelman SJ, Falotico R, Wilensky RL. Twenty-eight-day efficacy and phamacokinetics of the sirolimus-eluting stent. Coron Artery Dis. 2002;13(3):183–188. doi: 10.1097/00019501-200205000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Stahli BE, Camici GG, Steffel J, Akhmedov A, Shojaati K, Graber M, Luscher TF, Tanner FC. Paclitaxel enhances thrombin-induced endothelial tissue factor expression via c-Jun terminal NH2 kinase activation. Circ Res. 2006;99(2):149–155. doi: 10.1161/01.RES.0000233379.92010.fd. [DOI] [PubMed] [Google Scholar]