

Abstract
The cardiovascular benefits of menopausal hormone therapy (MHT) remain controversial. The earlier clinical observations that cardiovascular disease (CVD) was less common in MHT users compared to non-users suggested cardiovascular benefits of MHT. Also, experimental studies have identified estrogen receptors ERα, ERβ and GPR30, which mediate genomic or non-genomic effects in vascular endothelium, smooth muscle, and extracellular matrix (ECM). However, data from randomized clinical trials (RCTs), most notably the Women's Health Initiative (WHI) study, have challenged the cardiovascular benefits and highlighted adverse cardiovascular events with MHT. The discrepancies have been attributed to the design of RCTs, the subjects' advanced age and preexisting CVD, and the form of estrogen used. The discrepancies may also stem from age-related changes in vascular ER amount, distribution, integrity, and post-receptor signaling pathways as well as structural changes in the vasculature. Age-related changes in other sex hormones such as testosterone may also alter the hormonal environment and influence the cardiovascular effects of estrogen. Investigating the chemical properties, structure-activity relationship and pharmacology of natural and synthetic estrogens should improve the effectiveness of conventional MHT. Further characterization of phytoestrogens, selective estrogen-receptor modulators (SERMs), and specific ER agonists may provide substitutes to conventional MHT. Conditions with excess or low estrogen levels such as polycystic ovary syndrome (PCOS) and Turner syndrome may provide insight into the development and regulation of ER and the mechanisms of aberrant estrogen-ER interactions. The lessons learned from previous RCTs have led to more directed studies such as the Kronos Early Estrogen Prevention Study (KEEPS). Careful design of experimental models and RCTs, coupled with the development of specific ER modulators, hold the promise of improving the actions of estrogen in the aging blood vessels and thereby enhancing the efficacy and safety of MHT in postmenopausal CVD.
Keywords: sex hormones, estrogen, progesterone, testosterone, endothelium, vascular smooth muscle, extracellular matrix, cardiovascular disease, hypertension
INTRODUCTION
Estrogen is the predominant sex hormone in women, affecting the development and function of the female reproductive system, including the endometrium, uterus and mammary glands. Estrogen is commonly used as a contraceptive, and is a major component of menopausal hormone therapy (MHT). Premarin, a conjugared equine estrogen (CEE) composed of estrone sulfate and the mare estrogens equilin and equilenin, was the first preparation approved by the FDA in 1942 for treatment of menopausal symptoms such as hot flashes, night sweats, vaginal dryness and atrophy. Estrogens are also used in postmenopausal women (Post-MW) to treat urinary stress incontinence, dizziness, fatigue, irritability, and to prevent osteoporosis [1]. Estrogen promotes the growth and differentiation of other tissues, including adipose, nerve, musculoskeletal and cardiovascular system [2]. Epidemiological studies have shown that CVD, such as coronary artery disease (CAD) and hypertension (HTN), is less common in pre-menopausal women (Pre-MW) than in men of the same age, suggesting cardiovascular benefits of estrogen [3,4]. Also, the risk of CVD is greater in Post-MW than Pre-MW, and has been related to the decline in plasma levels of estrogen during menopause [5]. In effect, CVD is the leading cause of death in women 65 years and older. Earlier observational studies, such as the Nurses' Health Study (NHS) in the mid 1970s, suggested that estrogen therapy in Post-MW reduced the risk of CVD by 35 to 50% [3,6]. Also, a meta-analysis of observational studies has shown that MHT is associated with ~33% less fatal CVD among MHT users compared to nonusers [1].
Studies in humans and in animals as well as in tissues and cells also suggested beneficial effects of estrogen on the vasculature. Acute administration of estrogen in female or male patients improved vasodilatory responses and ameliorated myocardial ischemia [7,8]. Also, studies in dogs in vivo and on isolated rat and rabbit hearts have shown that acute treatment with estrogen lowers coronary vascular resistance and enhances coronary blood flow [9,10]. Estrogen modulates vascular tone by targeting endothelial cells (EC), vascular smooth muscle (VSM) and extracellular matrix (ECM) [3,4]. Such beneficial effects include up-regulation of the vasodilatory substances nitric oxide (NO) and prostacyclin (PGI2), and downregulation of the vasoconstrictor molecules endothelin (ET-1) and angiotensin II (AngII) [3,4]. Thus, both in addition to the initial observational studies, experimental evidence predicted vascular protective effects of estrogen in Post-MW.
Paradoxically, RCTs, most notably Heart and Estrogen/progestin Replacement Study (HERS) and WHI, showed increased CVD and cerebrovascular events with MHT. The WHI study was halted early because the overall health risks of MHT exceeded its benefits [11,12]. The discordant findings between observational and experimental studies vs. RCTs have mitigated the need to examine the changes in the effects of estrogen on the cardiovascular system during menopause. Studies have analyzed the potential factors in the design of RCTs which may have contributed to the unanticipated data, including the age at starting MHT, pre-existing CVD, and the estrogen form used [3]. Age-related changes in vascular ER amount, subtypes, tissue distribution, integrity, and post-receptor signaling pathways as well as the blood vessel structure may have also contributed to the unanticipated results of RCTs (Fig. 1).
Fig. 1.
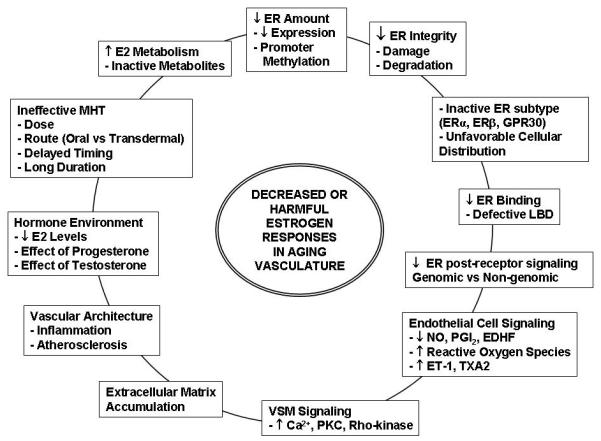
Causes of decreased estrogen responses in the aging vasculature. Age-related changes include the estrogen levels and metabolism, ER amount, distribution, integrity, and post-receptor signaling pathways, as well as structural changes in the vessel architecture. Age-related changes of other sex hormones such as progesterone and testosterone alter the hormonal environment and potentially influence the cardiovascular effects of estrogens.
The purpose of this review is to examine reports in PubMed for possible explanation of why the beneficial vascular effects of estrogen in observational and experimental studies did not translate in the RCTs. We will first describe currently known estrogenic compounds, their chemical properties and structure-activity relationship (SAR). We will then describe the vascular ERs and post-receptor signaling pathways in ECs, VSM and ECM, and the age-related changes in ERs, vascular remodeling and vessel architecture. The interaction of estrogen with other sex hormones such as progesterone and testosterone will be described. We will examine conditions with excess or low estrogen levels such as PCOS and Turner syndrome to highlight potential mechanisms of aberrant estrogen-ER interactions. We will also discuss how the lessons learned from previous RCTs, and the factors in the study design that may have affected the outcome, could help in the design of more directed RCTs. This discussion should provide insight into maximizing the effects of conventional MHT and identifying alternative MHT with greater efficacy and specificity on the aging vasculature.
CLASSIFICATION OF ESTROGENS
The first ovarian hormone estrone (E1) was isolated and its estrogenic activity in biological samples were described in the early 1920s [13]. The term “estrogen” now refers to a group of female gonadal hormones with diverse physiological functions in various tissues and cell types [14]. Many steroidal and non-steroidal compounds bind to ER and include natural and synthetic estrogens, phytoestrogens, selective estrogen receptor modulators (SERMs), and specific ER agonists and antagonists.
Natural Estrogens
Endogenous natural estrogens are C18 steroids and include estrone (E1), estradiol (E2), and estriol (E3). They have 4 rings A, B, C, D, a hydroxyl group at C3, and either a hydroxyl or a ketone group at C17 (Fig. 2) [15]. The phenolic A ring is responsible for selective high-affinity binding to ER [16]. Most alkyl substitutions in the A ring impair ER binding, but substitutions in C or D ring may be tolerated. Biosynthesis of natural estrogens starts with cholesterol binding to lipoprotein receptors on steroidogenic cells, and taken up and transferred to the steroid synthesis sites with the aid of sterol carrier protein-2 (SCP-2) [16,17]. All steroid hormones are derived from cholesterol, and hence the similar chemical structures (Fig. 2). Therefore, other steroids could affect E2 binding and action on vascular ERs. Also, since androstenedione is a main precursor of E2, it has garnered great attention in the world of sports for its body-building properties, but little is known about its role in MHT.
Fig. 2.
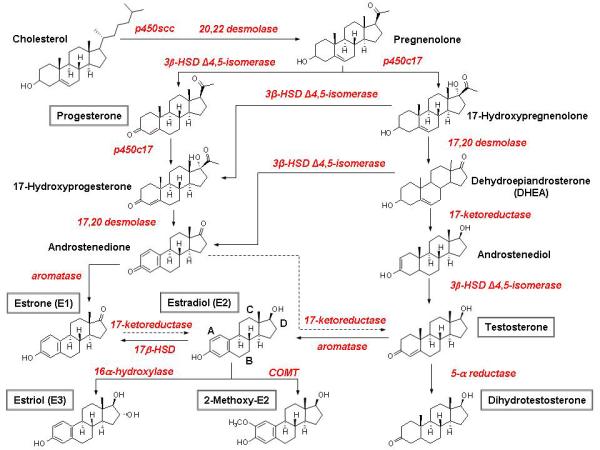
Ovarian Steroid Hormone Synthesis. In premenopausal women, the ovaries are the principal source of E2. The first, rate-limiting step in is the transfer of cholesterol from the cytosol to the inner membrane of the mitochondrion with the aid of StAR, and the conversion of cholesterol to pregnenolone. After side chain cleavage and utilizing the delta-5 or delta-4 pathway, androstenedione is the key intermediary. A fraction of androstenedione is directly “aromatized” to E1, which is subsequently converted to E2. Alternatively, androstenedione is converted to testosterone, which in turn undergoes conversion to E2 by aromatase. In postmenopause, the circulating steroids androstenedione, testosterone, and E1 become the major precursor substrates of E2 production in peripheral and estrogen target tissues.
The ovaries are the principal source of circulating E2 in Pre-MW. Most E1 and E3 are formed in the liver from E2 or in peripheral tissues from androstenedione [16]. In Post-MW, circulating androstenedione, testosterone and E1 are the major precursors of estrogen production in peripheral tissues [18]. Steroidogenic enzymes have been localized in the endoplasmic reticulum of testicular Sertoli and Leydig cells, adipose stroma, placental syncytiotrophoblasts, preimplantation blastocysts, bone, brain, and other tissues [19]. Polymorphisms in the genes coding for these steroidogenic enzymes may influence estrogen production, and careful evaluation of these polymorphisms may help to design a more individualized MHT approach in Post-MW [20].
Most of circulating E2 and other natural estrogens are bound strongly but reversibly to sex hormone-binding globulin (SHBG), and with less affinity to albumin, in a non-saturable and non-stoichiometric manner. About 2-3% of natural estrogens are unbound free physiologically active molecules that distribute rapidly and extensively due to their smaller size and lipophilic nature. E2 half-life is ~3 hours and exists in a dynamic equilibrium of metabolic inter-conversions with E1 and E3, leading to differential activation of ER subtypes and signaling pathways under different conditions [15].
Estrogens are transformed into less active metabolites that are excreted in urine and feces. Estrogen metabolism includes oxidation (hydroxylation) by cytochrome P450s (CYPs), glucuronidation by UDP-glucuronosyltransferase, sulfation by sulfotransferase, and O-methylation by catechol O-methyltransferase (COMT). Estrogen metabolism occurs mainly in the liver, where most CYPs are expressed. First, estrogens undergo hydroxylation by CYPs to many hydroxylated metabolites [18]. In the liver, ~80% of E2 is biotransformed into 2-(OH)E2 and 20% into 4-(OH)E2, which are then inactivated by COMT. Smaller amounts of E2 may be converted by CYP- or peroxidase-catalyzed reactions to semiquinones or quinones that form DNA adducts capable of removing purines from DNA or generating (via redox cycling) reactive oxygen species that oxidize DNA bases and lead to misrepair of important genes [18]. E2 metabolism varies depending on the stage of menstrual cycle, menopausal status, ethnic background and genetic polymorphisms [21]. Also, drugs and environmental factors such as cigarette smoke may induce or inhibit the enzymes that metabolize estrogens, and thus alter their clearance [16,18]. Changes in E2 metabolism with aging may also alter the effects of administered estrogen on the ERs in vasculature (Fig. 1).
Conjugated and semi-synthetic Estrogens
The natural animal-derived and semi-synthetic estrogens used in MHT have evolved since the debut of Premarin in the 1940s. While conjugated equine estrogens (CEE) are a common MHT regimen, other estrogens are available for oral, transdermal, parenteral or topical administration (Table 1). Ideally, MHT should be used at the lowest dose and shortest duration that achieve the therapeutic goal. The bioavailability of oral estrogen is usually low due to first-pass metabolism. Chemical alterations in natural estrogens produce semisynthetic estrogens and may increase their oral effectiveness. For example, ethinyl substitution at C17 position produces ethinyl E2 and minimizes the first-pass hepatic metabolism. Synthetic estrogens also include diethylstilbestrol chlorotrianisene, dienestrol, fosfestrol, mestranol, polyestradiol phosphate and quinestrol. Also, the transdermal route bypasses the liver and portal circulation, and minimizes hepatic effects of estrogens on protein synthesis, lipoprotein profiles, and triglyceride levels. Transdermal estrogen may also be safer than oral estrogen with respect to thrombotic risk and gynecologic cancers [22]. The type of estrogen administered (CEE vs E2) and the route of administration (oral vs transdermal) may partly explain the unanticipated results of HERS and WHI RCTs.
Table 1.
Estrogen and estrogen-progestin products approved by US FDA for MHT
| Route | Compound | Commercial name |
|---|---|---|
| ORAL | Estradiol - drospirenone | Angeliq® Tablets |
| Synthetic conjugated estrogens | Enjuvia™ | |
| Norethindrone acetate/ ethinyl estradiol |
Femhrt® | |
| Estradiol/norgestimate | Prefest™ | |
| CEE | Premarin® | |
| CEE/MPA | Prempro™ | |
| TRANSDERMAL | Estradiol | Alora® |
| Estradiol/Levonorgestrel | Climara Pro™ | |
| Estradiol/norethindrone acetate (NETA) |
CombiPatch® | |
| Estradiol | Estraderm® | |
| Estradiol | Menostar® | |
| Estradiol | Vivelle®, Vivelle-Dot® | |
|
TOPICAL EMULSION |
Estradiol | Estrasorb™ |
| INJECTION | Estradiol valerate | Delestrogen® |
| CEE | Premarin® | |
| VAGINAL | Estradiol acetate | Femring® |
| CEE | Premarin® |
Phytoestrogens
The realization that CEE may not be as safe as previously thought has increased the interest in phytoestrogens (Fig. 3). Phytoestrogens are polyphenolic non-steroidal compounds synthesized by plants and posses estrogenic activity. Phytoestrogens are found in a variety of foods and plants particularly soybeans, red clover, and wheat grains [23]. Interestingly, food containing phytoestrogens is consumed in significant quantities in cultures with lower rate of menopausal symptoms, osteoporosis, cancer and CVD [24]. While the classification of phytoestrogens has not been consistent in the literature, based on their chemical structure the major phytoestogens are classified into flavonoids, stillbenoids, and lignans (Fig. 3). The flavonoids are further divided into major flavonoids (flavones, flavonols and flavanones), and isoflavonoids (isoflavones and coumestanes) [25]. Stillbenoids include stillbenes such as resveratrol, which is found in red wine and peanuts, but only its trans form has estrogenic activity [23]. Plant lignans include secoisolariciresinol and matairesinol, which are not estrogenic by themselves. Secoisolariciresino is converted to enterodiol and subsequently to enterolactone by intestinal microflora. Matairesinol is converted directly to enterolactone. Enterodiol and enterolactone, also called enterolignans, are phytoestrogens with structural similarity to endogenous estrogens. Dietary sources of plant lignans include flaxseed, whole grain bread, vegetables, and tea [23]. Resorcyclic acid lactone compounds such as zearalenone, α-zearalenol and their derivatives also have estrogenic effects. They are not found in food plants but are secondary mold metabolites of fungal species, and therefore considered mycoestrogens [26], although some studies classify them as phytoestrogens [27].
Fig. 3.

Phytoestrogens are polyphenolic non-steroidal compounds synthesized by plants and posses estrogenic activity. Based on their chemical structure, the major phytoestogens are classified into: flavonoids (isoflavones, coumestans, flavones, flavanols, and flavanones), stilbenes, and lignans. Note the similarity of their chemical structures to E2.
The key structural elements crucial for the phytoestrogenic effects include the phenolic ring(s), low molecular weight, and optimal hydroxylation patterns. Although phytoestrogens could produce appreciable estrogenic effects, their efficacy at relevant doses has not been established in RCTs. Soybean foods, soybean protein extract and red-clover extract have limited effects on menopausal symptoms. On the other hand, soybean isoflavone extracts reduce hot flashes, suggesting that the benefits are subtle and not in all individuals, thus highlighting the need of adequately-powered RCTs that can definitively establish the benefits [28]. Also, while the affinity of phytoestrogens to ERs is relatively weaker than E2, their stability and therefore longer duration in the body raises concerns of potential toxicity in human.
Studies have examined the effects of dietary phytoestrogens on vasculature. Isoflavones stimulate the activity of endothelial NO synthase and induce vasodilation via NO. Isoflavones are also antithrombotic and antiatherogenic. A meta-analysis of 38 controlled human studies of soy consumption suggested positive effect on lipid profiles, including reduction in levels of LDL and triglycerides and increases in HDL [29]. On the other hand, a 12-month double-blind RCT in 202 Post-MW aged 60 to 75 years compared the effects of soy protein containing 99 mg of isoflavones per day with placebo, and found no effect on blood pressure and EC function. Also, no RCTs have examined the effects of phytoestrogens on clinical end points of CVD such as myocardial infarction. Therefore, phytoestrogens may not be recommended as a primary preventive intervention to reduce the risk of CVD. However, women may be advised that phytoestrogens used for treatment of menopausal symptoms may have additional cardiovascular benefits by reducing risk factors of atherosclerosis such as hyperlipidemia and HTN [29]. Further analysis of structure and activity of phytoestrogens should clarify their potential effects on vascular ERs and their usefulness as alternative MHT.
Specific Estrogen Receptor Modulators (SERMs)
SERMs are non-steroidal molecules that bind with high affinity to ERs, but have distinct effects depending on the drug's structure and specific tissue [30]. SERMs include raloxifene, tamoxifen, toremifene, and idoxifene (Fig. 4). SERMs have a wide range of activity from purely estrogenic, purely anti-estrogenic, to combined partial estrogenic in some tissues and anti-estrogenic or no activity in other tissues [31]. For some SERMs, the agonist/antagonist activity and tissue selectivity may be related to the ratio of co-activator/co-repressor proteins in different cell types, and the ER conformation induced by drug binding. This in turn determines how strongly the drug/receptor complex recruits co-activators, resulting in agonsim, relative to co-repressors, resulting in antagonism. For example, raloxifene exhibits ER agonism in bone and serum lipids, but ER antagonism in endometrial and breast tissue [32,33].
Fig. 4.

SERMs and specific ER agonists and antagonists. SERMs are non-steroidal molecules that bind with high affinity to ERs, but have a wide range of activity from purely estrogenic, purely anti-estrogenic, to combined partial estrogenic and anti-estrogenic effects. Selective ERα agonist propylpyrazole triol (PPT) and ERβ agonist diarylpropionitrile (DPN) have been developed. Selective ERα antagonists such as methyl-piperidino-pyrazole (MPP) have also been developed. Note the similarity of their chemical structures to E2.
The Raloxifene Use for The Heart (RUTH) RCT in Post-MW with CVD or at increased risk for CVD demonstrated that treatment with raloxifene for a median of 5.6 years reduced the risk of invasive breast cancer but did not change the incidence of coronary events [34]. Also, in post-MW women (mean age 67.5 years) raloxifene had no effect on the risk of primary coronary events, but was associated with increased risk of fatal stroke and venous thromboembolism [35]. However, as in the WHI study, the advanced age of the participants could have affected the estrogenic response. Ex vivo studies on rat renal and pulmonary arterial rings and porcine arterial rings have shown that raloxifene induces endothelium-independent relaxation through inhibition of Ca2+ influx via voltage-gated channels [36,37]. Also, in overiectomized (OVX) rats, tamoxifen demonstrated protective effects and antioxidant properties in myocardial ischemia-reperfusion injury [38]. Additionally, tamoxifen causes rapid relaxation in isolated rabbit and porcine coronary artery [39,40]. Although currently available SERMs show limited vascular benefits, as new SERMs are developed and more studies are done on existing SERMs, they may be used in MHT to decrease the risk of CVD in Post-MW.
Selective ER Agonists and Antagonists
Significant progress has been made to develop compounds that act selectively on various ER subtypes. Selective ER agonists such as propylpyrazole triol (PPT) and diarylpropionitrile (DPN) have been developed (Fig. 4). DPN is a potent ERβ agonist with a 30- to 70-fold selectivity over ER [41]. Members of the triarylpyrazole class such as propylpyrazole trisphenol (PPT) are 400-fold more potent on ERα than ERβ [42,43]. PPT increases flow-mediated relaxation in small mesenteric arteries from female but not male mice [44]. Studies with selective ER agonists suggest that ERα mediates most of the vascular actions of estrogen [45]. However, DPN, a selective ERβ agonist, also induces acute NO-dependent vasodilation [46].
ER antagonists bind to the ER ligand-binding pocket, thereby occluding agonist access and inducing a conformational change in ER that prevents efficient interaction with transcriptional coactivators. Blocking ER function can also be achieved downstream of ligand binding through direct interference with ER-coactivator interactions [47]. The search for agents that block ER signaling have led to the synthesis of a series of steroidal 7-alkylamide analogues of estradiol. ICI 164,384, the first estrogen antagonist described, blocks the uterotrophic action of E2 or tamoxifen in rats [48]. ICI 182,780 (fulvestrant) is a more potent estrogen antagonist approved for treatment of advanced breast cancer in Post-MW [49]. Both ICI 164,384 and ICI 182,780 are classical “pure” antiestrogens (full antagonists) that completely block the activity of E2 via inhibition of both AF-1 and AF-2 action [50]. ICI 182,780 is non-selective and does not distinguish between ER subtypes. By introducing basic side chain substituents such as those found in tamoxifen, ERα selective antagonists, such as methyl-piperidino-pyrazole (MPP) have been developed [51].
ESTROGEN RECEPTORS (ER)
Estrogenic ligands bind to ERs with high affinity and specificity. The effects of estrogens were thought to be mediated solely via nuclear ERs. Two nuclear ERs have been cloned: ERα (NR3A1) and ERβ (NR3A2) [52]. Recently, a novel 7-transmembrane G protein-coupled receptor, termed GPR30 has been identified [53]. A less-defined ER-X has been found in brain but not in vasculature [54].
ERα and ERβ
Studies on ERα knock-out (KO), ERβ KO, and double ERα ERβ KO mice revealed that life is possible without either or both ERs, although the reproductive functions are severely impaired [55,56]. ERα is expressed abundantly in the uterus, vagina, ovaries, mammary gland, and hypothalamus [57]. ERβ is expressed highly in the prostate and ovaries, with smaller amount in lung, brain, and bone [58]. Also, ERα and ERβ have distinct roles in the immune, skeletal, central nervous system and cardiovascular system [59,60]. In many cell types, ERα and ERβ form either homodimers or heterodimers. ERα and ERβ have been localized in ECs and VSM [61]. The expression of vascular ERs appears to vary with gender and the vascular bed studied. For example, both ERα and ERβ mRNA are expressed in human VSM from the coronary artery, iliac artery, aorta, and saphenous vein, and the expression of ERβ is greater in females [62]. In rats, ERα is found mainly in the uterine vasculature, whereas ERβ immunoreactivity is more abundant in ECs and VSM from the aorta, tail and uterine arteries [63].
ERα and ERβ are members of the nuclear receptor superfamily, which include the thyroid hormone, retinoic acid, and vitamin D receptors [64]. Like other members of this superfamily, one gene may result in the expression of multiple receptor proteins and diverse responses [65]. The mechanisms of this diversity include epigenetic changes and methylation of the genes encoding these receptors, alternative RNA splicing leading to multiple mRNA isoforms of each receptor, and multiple sites for initiation of translation of receptor mRNA [66].
The two nuclear ER genes are located on separate chromosomes: ESR1 is located on chromosome 6q(25.1) and encodes ERα, while ESR2 is located on chromosome 14q(23-24.1) and encodes ERβ [15]. ESR1 and ESR2 comprise 8 exons separated by seven intronic regions and span more than 140 kilobases and approximately 40 kilobases, respectively [67]. The wild-type full-length human ERα mRNA encodes 595 amino acids, while the wild-type full-length human ERβ mRNA encodes 530 amino acids [57].
Human ERα and ERβ have 44% homology in overall amino acid sequence and share the domain structure common to members of the nuclear superfamily: A/B, C, D, E and F domains (Fig. 5) [52]. The A/B region is involved in protein-protein interactions and transcriptional activation of target-gene expression. This region harbors an activation function 1 (AF-1), located toward the N-terminal end, that is ligand-independent and shows promoter and cell-specific activity [68]. Human ERα and ERβ share <20% amino acid identity in the AF-1 region, suggesting that it may contribute to ER-specific actions on target genes. AF-1 in ERα is very active on a variety of estrogen-sensitive promoters, but under identical conditions, the activity of AF-1 in ERβ is minimal [52]. The central C-domain represents the DNA-binding domain (DBD), which contains 4 cysteines arranged in 2 zinc fingers, and is involved in specific DNA binding and receptor dimerization. It is highly conserved between ERα and ERβ and shares 95% amino acid identity, suggesting that both ERs recognize similar DNA sequences and hence regulate many of the same target genes [68]. The D-domain, or hinge domain, is not well-conserved between ERα and ERβ (30%), and works as a flexible hinge between the DBD and ligand-binding domain (LBD). D domain also functions in promoting the association of ER with heat shock protein 90 (HSP90) and nuclear localization of ER [52]. The E-domain is the LBD, and ERα and ERβ share ~55% amino acid identity in this region. The LBD contains a ligand-dependent AF-2, and is important for ligand binding and receptor dimerization. The F-domain has <20% amino acid homology between the two ERs, and the function of this domain is unclear. The two ERs have similar affinities for E2 and bind the same DNA response elements [52,57]. Full transcriptional activation by ERs is mediated by synergism between AF-1 and AF-2 [2,52]. AF-1 and AF-2 are also required for ligand-independent receptor functions, including growth factor activation by AF-1 and cAMP activation by AF-2 [47].
Fig. 5.
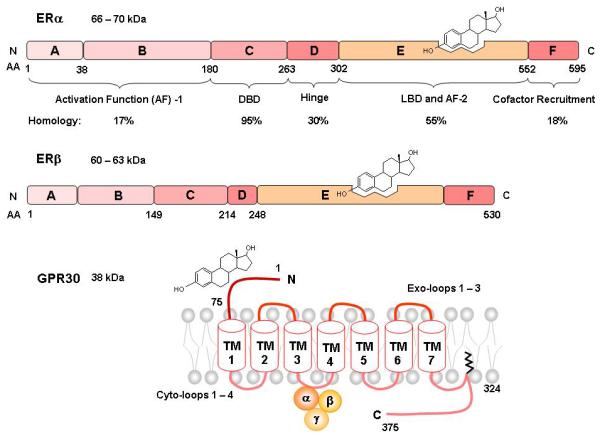
Primary Structure of ERα, ERβ, and GPR30. Similar to other members of the nuclear receptor superfamily, ERα and ERβ share a common structure with five functional domains termed A/B, C, D, E and F. Domain A/B contains the activation function-1 1 (AF-1). Domain C is the DNA binding domain. Domain D is the hinge domain, linking domain C and E. The ligand binding cavity and activation function 2 (AF-2) are located in domain E. Domain F includes co-factor recruitment regions. GPR30 is a G-coupled protein receptor that has estrogenic activity and shares little homology with the classical ERs. GPR30 consists of an N-terminal domain, a 7- transmembrane domain and a C-terminal domain.
Polymorphisms in ER genes may account for differences in ER function and may modify the outcome of MHT. Two common polymorphisms of ERα at positions c.454–397 T>C (PvuII) and c.454–351 A>G (XbaI) may be associated with the severity of CAD in Post-MW undergoing angiography for suspected CAD [69]. Post-MW with the ERα IVS1-397 polymorphism C/C genotype (recessive) have more prominent increases in HDL and apolipoprotein A-1 levels compared to those with the dominant phenotype, as well as a greater forearm blood flow, brachial artery diameter, and endothelium-dependent dilation [70]. In Post-MW with established CAD and being treated with estrogen alone or estrogen+progesterone, a 2 times greater increase in HDL levels was seen in women with the ERα IVSI-401 polymorphism C/C genotype as compared to those without the polymorphism [71].
Several splice variants or isoforms of ER subtypes have been described. Most ERα variants differ in their 5'-untranslated region, not in the coding sequence. ERα isoforms have not been identified in tissues, but their biological function and role in mediating the effects of estrogen in vivo are unclear. However, they are important research tools as they heterodimerize with the full-length ERα and repress AF-1-mediated activity. They may also localize in plasma membrane and thereby help to elucidate the mechanisms of rapid non-genomic estrogen signaling [72]. Multiple ERβ isoforms exist as a result of either alternative splicing of the last coding exon 8, deletion of one or more coding exons, or alternative usage of untranslated exons in the 5' region [73]. Among them, 5 full-length transcripts ERβ1-5 have been reported in human [60]. Whether ER isoforms are distributed differently in vasculature and whether they change with aging is an important area for further investigation.
A major mechanism for downregulation of gene expression is methylation of the CpG island, a cytosine and guanine rich area in the promoter region of the gene, resulting in an epigenetic change and permanent inactivation of gene transcription without associated genetic abnormality [74,75]. ERβ expression is regulated through methylation of CpG islands in the ERβ promoter. An increase in promoter methylation may be a possible epigenetic mechanism contributing to age-related diseases. Methylation of ER genes and several others may be associated with physiologic aging of human colonic mucosa. Also, ERβ promoter methylation may represent an epigenetic change in atherosclerosis and vascular aging [75]. Methylation of ER genes also increases with passage of cultured ECs and VSM and may be implicated in the decreased ER responsiveness and vascular genomic effects of estrogen with aging [75,76].
Although estrogen levels and release patterns change with aging, little is known about the corresponding changes in ERs. Studies have shown no significant changes in ER expression in aorta of aging compared with adult OVX spontaneously hypertensive rats (SHR) [77]. However, gender and age-related differences in ER mRNA expression have been demonstrated in other tissues from human and experimental animals. ERα protein was detected in the retina and retinal pigment epithelium of young female eyes, but not in eye tissues dissected from men and Post-MW [78]. Studies have demonstrated a decrease in ERβ mRNA positive cells in specific regions of the brain of old compared with young and middle-aged female rats, and this expression is not altered by estrogen replacement [79]. Aging may also affect the subcellular distribution of ERα in cholinergic neurons of transgenic and wild-type mice, and is associated with translocation of ERα from the nucleus to cytoplasm [80].
Estrogenic Structure Activity Relationship (SAR)
Compounds with similar estrogenic activity appear to share common structural features. Studies have examined a large database from the National Center for Toxicological Research (NCTR) on the ER relative binding affinity of 230 chemicals, of which 130 were active [27]. These SAR studies revealed 5 structural features that are important for binding to ER; the more key features a chemical contains, the more active it is (Fig. 6). These 5 structural features are present in E2 and include: 1) H-bonding ability of a phenolic ring, mimicking the C3-OH group of the A ring of E2, 2) H-bond donor mimicking the C17β-OH, and a certain O-O distance equivalent to that between C3-OH and C17β-OH, 3) hydrophobicity, 4) precise steric hydrophobic centers mimicking steric C7α and C11β substituents, and 5) a ring structure [81].
Fig. 6.
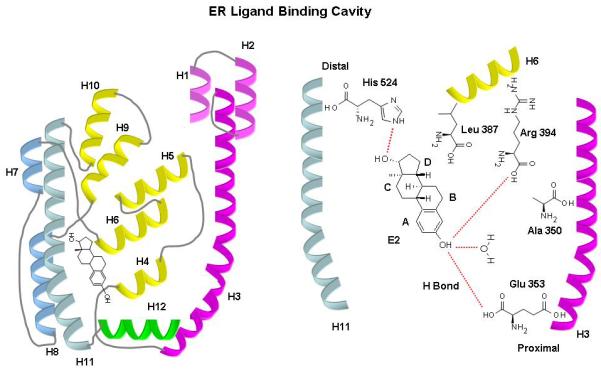
Schematic 3D structure of ERα and ERβ ligand binding domain (LBD). The LBD has twelve helices (H1-H12), folded into a three-layered anti-parallel α-helical sandwich comprising a central core (H5–6, H9, and H10) sandwiched between two additional layers of α-helices (H1-4) and (H7, H8, H11). This helical arrangement creates a ‘wedge-shaped’ molecular scaffold that maintains a sizeable ligand-binding cavity at the narrower end of the domain, into which the hormone binds. Amino acid residues that interact with E2 are shown in their approximate positions. The molecules that contribute to direct hydrogen bonds are water and the imidazole group of His524 on H11, the guanidium group of Arg394 on H6, and the carboxylate group of Glu353 on H3. H12 sits snugly over the ligand binding cavity packed against H3, H5–6, and H11. Although, H12 makes no direct contact with E2, it forms the lid of the binding cavity and projects its inner hydrophobic surface towards the bound hormone.
The 3D structures of ERα and ERβ LBD are very similar and reflect their high sequence identity. The LBD has twelve helices (H1-H12), folded into a three-layered anti-parallel α-helical sandwich comprising a central core layer of three α-helices (H5–6, H9, and H10) sandwiched between two additional layers of α-helices (H1-4) and (H7, H8, and H11). This helical arrangement creates a ‘wedge-shaped’ molecular scaffold that maintains a sizeable ligand-binding cavity at the narrower end of the domain, into which the hormone binds. The cavity is formed by residues from H3, H6, the loop region between H7 and H8, H8, H11, and H12; and is completely buried within the core of the LBD and flanked by a small two-stranded anti-parallel β-sheet (S1 and S2) and the C-terminal helix (H12) [57,82]. All the ligands bind across the cavity between H3 and H11. The ligand binding sites of ERα and ERβ are nearly identical; only two residues are different: Leu384/Met421 in ERα correspond to Met336/Ile373 in ERβ. Also, the ligand binding cavity of ERβ is smaller (<20%) than that of ERα and this may have implications for the selective affinity and pharmacology of ligands and ERs [52,57,83].
The binding of E2 with ER typically involves non-covalent H-bonding and hydrophobic interactions. In terms of H-bonding, the contribution of the phenolic ring to binding is more important than any other structural feature. 108 of the 130 active chemicals in the NCTR data contain a phenolic ring. Also, of the 130 active chemicals, 125 chemicals contain an O atom and 3 chemicals contain a Cl atom that serves as a weak H-bond acceptor. The phenolic -OH is a better H-bond donor than an acceptor [27]. Replacing -OH with the weak H-bond donor -NH2 reduces the activity of E2 [84]. Strong estrogens have an additional OH group within a certain distance from the phenolic ring. The H-bonding residues at the A-ring binding region in the proximal end of LBD (Glu353 on helix H3, Arg394 on H6, and a structurally conserved buried water molecule in ERα) and at the D-ring binding region in the distal end (His524 on H11) form a polar pocket between H3, H6, and H11 (Fig. 6). Access to this polar pocket is somewhat restricted by a ‘pincer-like’ arrangement of the side chains of Ala350 on H3, Leu387 on H6, and Phe404 on S1 in ERα. The planar moieties of ER ligands, such as the A-ring of E2, bind at this part of the cavity [50]. Mutational studies also indicated that Cys residues Cys381, 417, 447 and 530 in ERα LBD, and Cys334, 369, 399, and 481 in ERβ LBD play a role in ligand binding. In particular, Cys530 builds up the primary site for E2 binding, whereas Cys381 and Cys417 are involved in the recognition of non-steroidal anti-estrogens [57,85]. Interestingly, E2 binds ‘upside-down’ to ERβ compared to ERα, as observed in the crystal structure of the ERβ-ICI 164,384 complex [50,57]. A secondary E2 binding site runs from the A-ring -OH and the buried water bond to the exterior of the LBD between H3, H5 and H6 [86,87]. Ligands might exhibit preference for one or the other binding site; however, there seems to be no obvious function for the secondary E2 binding site [86,87]
With regard to hydrophobicity in the ER-E2 complex, H12 sits snugly over the ligand binding cavity packed against H3, H5–6 and H11. Although H12 makes no direct contact with E2, it forms the lid of the binding cavity and projects its inner hydrophobic surface towards the bound hormone. While the overall conformation of the ER LBD is markedly similar in various ligand complexes, the orientation of the C-terminal transactivation H12 is highly sensitive to the nature of ligand. H12 affects the LBD surface and contains a sequence critical to the AF-2 transcription activating function where most of the co-activators/co-repressors bind in a ligand-dependent manner. For example, partial-agonists such as genistein and raloxifene, and antagonists such as ICI 164,384, bind across the ligand-binding cavity in a similar manner to the natural and synthetic agonists E2 and diethylstilbesterol, respectively. However, their large side chain substituents are not accommodated within the confines of the binding cavity, resulting in non-allosteric displacement of H12 and prevention of its alignment over the cavity. Also, in ERβ-genistein, ERα-4-hydroxytamoxifen, and ERα-raloxifene complexes, H12 lies in a groove formed by H3, H4, and H5. As a whole, the partial agonist and antagonist capabilities are mainly dependent on their ability to prevent the formation of a transcriptionally competent AF-2 conformation, thus favoring or impairing the recruitment of co-activators [50,57].
The positive effect of hydrophobicity on binding is more apparent for chemicals containing C3α and C17β -OH, compared to those with only a C3 -OH. Hydrophobicity is quantitatively expressed as the ratio of solubility in octanol vs water, or log P (Partition Coefficient). Interestingly, the mean log P value of chemicals with one OH group is larger than that of chemicals containing two OH groups mimicking the C3α and C17 -OH of E2. This indicates that chemicals lacking the effective O-O distance require greater hydrophobicity to reach binding activities similar to those of chemicals with the C3α and C17 groups [27].
In terms of precise steric hydrophobic centers mimicking steric C7α and C11β substituents, the volume of the ER binding pocket is about twice that of the E2 molecule [88]. Therefore, there are large unoccupied cavities opposite the C7α and C11β positions of E2. These cavities allow steric groups of certain sizes to fit, and are important for estrogenic compounds including diethylstilbesterol and other chemicals such as biphenyls. While the rigid protein architecture around the A-ring pocket imposes an absolute requirement for effective ER ligands to contain a planar ring group, the remainder of the binding cavity is quite accommodating. Particularly, the distal end or D-ring binding site of the cavity is quite flexible and permits a variety of ligand-binding modalities. The discrepancy between the volume of the ER binding cavity and the size of its cognate ligand suggests that the LBD have evolved sufficiently so that it can discriminate between E2 and other endogenous steroids whilst retaining some remnants of its ancestral character. In addition, the sub-optimal architecture in certain regions of the LBD suggests that unknown endogenous ER modulators remain to be identified [50,88].
A ring structure in ER ligands increases the rigidity of both the structure and the steric center, which favors ER binding. The flat aromatic ring, which fits better to the narrow “tunnel-like” arrangement in ER for the phenolic A ring, is more favorable than other ring structures. Only 5 chemicals without aromatic rings were found to be active, of which four are steroids in addition to kepone. These 5 chemicals possess H-bond capability with a rigid hydrophobic backbone that matches the A, B and C rings of E2, suggesting that non-aromatic estrogens require several key structures to exhibit ER binding activity, including a strong electronegative atom such as O for H-bonding and a rigid hydrophobic backbone [27].
The binding affinity of other estrogenic compounds and metabolites to ERs has been studied. Tamoxifen has almost identical binding affinities for human ERα and ERβ, although its binding affinity is only 3–4% of those of E2 and 7–10% of ICI-182,780. Raloxifene has a similar binding affinity for ERβ as tamoxifen, 16-fold higher binding affinity for ERα than tamoxifen, and comparable binding affinity as ICI-182,780. Therefore, raloxifene has a preferential binding affinity for human ERα over ERβ. The phytoestrogen genistein has high affinity for ERβ, almost identical to that of E2, although its affinity for ERα is only 6% of that. Coumestrol has very high binding affinity for human ERα and slightly higher affinity for ERβ. Daidzein has very weak binding affinity for both ERα and ERβ, but its relative affinity for ERβ is higher than that for ERα. Metabolites such as C-3 sulfated estrogens (E1-3-sulfate and E2-3-sulfate) have little affinity for ERα and ERβ, while 16α-OH-E1 has a higher affinity than E1 [15]. Further elucidation of the estrogen-ER interactions should permit the rational design of drugs with selective estrogenic activity particularly in aging women.
GPR30
GPR30 is a structurally unrelated protein to ERα or ERβ that binds E2 with high affinity, and may mediate some of its non-genomic effects [89]. The existence of this receptor was suggested because of evidence of G-protein mediated signaling by estrogen and localization of estrogen binding sites to membranes [90]. GPR30 was cloned in 1998 and is located on chromosome 7p22 [91]. GPR30 was identified as a novel E2 binding receptor by two groups, independently [89,92]. GPR30 has an extracellular N terminal, seven transmembrane α helices, 3 exo-loops involved in ligand binding, 3 or 4 cyto-loops involved in G protein subunit binding, and a C terminal linked to the membrane through lipid addition, and also involved in binding G protein subunits. GPR30 is widely distributed in the brain and peripheral tissues and may play a functional role in vasculature [58]. GPR30 is expressed in human mammary artery and saphenous vein, and its expression decreases after exposure to E2 in mammary artery, but not in saphenous vein [93]. The cellular localization of GPR30 appears to vary depending on the cell type and the GPR30-tag used for analysis. One group reported localization of GPR30 in the endoplasmic reticulum [89], while other groups found GPR30 to be expressed in the plasma membrane [92, 94].
The activation of GPR30 by estrogen results in intracellular Ca2+ mobilization and synthesis of phosphatidylinositol 3,4,5-trisphosphate in the nucleus [92]. GPR30 may also function in conjunction with ERα, to assemble a signal complex essential for rapid estrogen signaling [95]. The ER antagonists tamoxifen and ICI 187,280 may act as GPR30 agonists [92]. Apart from the unknown physiological significance of GPR30, there is controversy regarding its role as an ER. In contrast to previously published data [92], some studies could not demonstrate cAMP or ERK activation in GPR30-positive, ER-negative breast cancer cells [96]. Also, in ER-positive, GPR30-positive MCF-7 cells, nongenomic E2 responses were blocked by ICI 187,280 and were dependent on ER. Silencing of GPR30 function in these cells had no effect on E2-induced cAMP elevation and ERK activation [97]. Thus, although GPR30 has been found in the vasculature, its functional role needs further investigation. It is also important to note that a decrease in the vascular effects of estrogen with aging could be related to downregulation of not only vascular ERs, but also postreceptor signaling mechanisms.
ER POST-RECEPTOR SIGNALING PATHWAYS
Estrogen has multiple vascular effects including alteration of serum lipid concentrations, coagulation and fibrinolysis, antioxidant properties, and the production of vasoactive molecules, such as NO and prostaglandins. Estrogen also causes vasodilation and inhibits the blood vessel response to injury and the development of atherosclerosis [61]. These biological processes are regulated via both genomic and non-genomic pathways [58].
Genomic Effects of Estrogen
Estrogen via ERs regulates transcriptional processes that involve nuclear translocation, binding to specific estrogen response elements (ERE) and regulation of target gene expression [98]. Estrogen affects genes regulating vascular tone, as well as the response to vascular injury and atherosclerosis. Estrogen increases the expression of genes for vasodilatory enzymes such as NOS and prostacyclin synthase. In the absence of estrogen, ER exists as an inactivated monomer bound with HSP90. Upon binding to estrogen ER undergoes conformational changes that result in dissociation of HSP90 and formation of a homo- or heterodimer with high affinity for estrogen and DNA [99]. Ligand-bound ER can bind directly to ERE in the promoters of target genes or interact with other transcription factor complexes like Fos/Jun (AP-1-responsive elements) or SP-1 (GC-rich SP-1 motifs) and influence transcription of genes whose promoters do not harbor ERE [100,101]. ER can also affect the transcription of genes through preventing the recruitment of other transcription factors [99]. Many of the estrogen-regulated genes also encode transcription factors [102]. Ligand-dependent activation triggers recruitment of a variety of coregulators to ER in a complex that alters chromatin structure and facilitates recruitment of the RNA polymerase II transcriptional machinery. Ligand-independent pathways may also activate ERs [60]. Growth factor signaling leads to activation of kinases that may phosphorylate and activate ERs or associated coregulators in the absence of ligand [103]. The growth factor activated kinase pathway may contribute to hormone-independent growth in some tumors [104].
Studies on the aorta of wild-type and ER KO mice have suggested that ERα primarily mediates estrogen-induced gene expression, while ERβ may mediate a decrease in gene expression. While most plasma membrane ERα and ERβ form homodimers in the presence of E2, a small portion of the ER pool forms ERα/ERβ heterodimers [105].
Nongenomic effects of Estrogen
Non-genomic effects are rapid responses that occur too quickly to be mediated by gene transcription, are independent of protein synthesis, and typically involve modulation of membrane bound and cytoplasmic regulatory proteins [98]. Rapid vasodilation occurs within seconds or minutes of adding E2, and involves activation of kinases and phosphatases and changes in ion fluxes across membranes [95,98]. Whether the nongenomic effects involve the nuclear ERs or distinct membrane associated receptors is unclear [58,60]. In several cell types, ERs associate with caveolae and other signaling molecules to trigger G protein-coupled receptor-mediated second messengers and intracellular pathways including MAPK and PI3K/Akt, and activation of ion channel fluxes [95,106].
Much of the current investigation of a distinct membrane receptor has focused on GPR30, although this association remains unclear [58]. GPR30 is thought to promote rapid estrogen actions [89,107]. Estrogen binding to GPR30 results in the dissociation of Gα-GTPase from the heterotrimeric Gαβγ complex. Dissociated Gβγ-subunit activates membrane associated matrix metalloproteinases (MMPs) with subsequent transactivation of epidermal growth factor receptor (EGFR) through the cleavage and release of pro-heparan-bound epidermal growth factor (ProHB-EGF) from the cell surface and transient activation of MAPK [108,109]. This is supported by reports that E2 induces the phosphorylation of p38 and p42/44 MAPK (ERK-1/2) as well as proliferation and migration of porcine aortic endothelial cells [110]. Gα-GTPase acts on membrane-associated adenylyl cyclase, generating the second messenger cAMP which in turn inhibits ERK-1/2 activity via a cAMP-dependent protein kinase mechanism [111].
Estrogen and the Endothelium
Estrogen affects vascular reactivity through direct effects on endothelial cells (ECs) [112]. Vascular ECs are involved in regulation of many processes including vascular tone and angiogenesis, and EC dysfunction can lead to CVD including HTN, CAD, stroke, and atherosclerosis [113]. Human umbilical vein ECs (HUVECs) express both ERα and ERβ, and estrogen modulates EC function in human, experimental animals and cultured cells [61].
Endothelium-dependent vascular relaxation is greater in female than male in SHR [114]. Selective ERα agonists also improve EC dysfunction in blood vessels of OVX female SHR [115]. Also, E2-induced vascular relaxation and NO production are greater in mice expressing only functional ERα [116,117]. E2 administration in OVX female mice in vivo causes rapid non-genomic arterial dilation of elastic and muscular arteries, as a result of ER-mediated NO production. Rapid activation of both MAPK/ERK and PI3K activity was found in the E2-exposed arteries, and inhibiting either kinase prevented E2-induced vasodilatation. The kinase activation and vasodilator responses to E2 were absent in both ERα and ERβ knockout mice. These results indicate that E2 modulation of arterial tone through plasma membrane ER and rapid signaling could underlie many of the observed actions of estrogen in vivo [118]. In support of these findings, studies on small arteries isolated from healthy Post-MW not receiving MHT have shown that the morphology and function of the endothelium are impaired, and these impairments are improved upon treating the isolated vessels with E2 [113]. Also, E2 induces the phosphorylation and activation of MAPK, and proliferation of ECs via cytosolic and nuclear ERs [119]. The mechanisms of estrogen-induced endothelium-dependent vasodilation include modifications of the synthesis, release and bioactivity of relaxing factors such as NO, PGI2 and endothelium-derived hyperpolarizing factor (EDHF), as well as contracting factors such as endothelin-1 (ET-1) and thromboxane A2 (TXA2) [120].
Estrogen and Endothelium-Derived Vasodilators
Estrogen and NO
NO is a powerful vasodilator produced from the transformation of L-arginine to L-citrulline by NO synthase (NOS) [121]. E2-mediated vasodilation has been attributed to increased eNOS transcription via genomic pathways as well as increased eNOS activity and NO production via non-genomic EC activation [122]. Increased eNOS expression has been demonstrated in the uterine vasculature during E2 treatment in the follicular phase and during pregnancy, suggesting that endothelial-derived NO is involved in the vasorelaxant actions of E2 [121]. Also, endothelial NO release is greater in arteries of females compared with males, and estrogen may mediate the gender differences in NO production [123].
In vitro studies have demonstrated gender differences in NO production. The inhibitory effect of the NOS inhibitor L-NAME on acetylcholine (Ach)-induced relaxation is more pronounced in mesenteric artery of female than male rats [124]. Also, in isolated basilar arteries from OVX female rabbits, E2 treatment increases the response to NO in VSM cells [125]. Other studies have examined the effects of E2, PPT (ERα agonist) and DPN (ERβ agonist) on aortic rings isolated from wild type mice with trauma-induced hemorrhage. It was found that trauma hemorrhage increased ET-1 induced vasoconstriction, and treatment with E2 and DPN counteracted the vasoconstriction, while PPT had no effect [126]. These data suggest that ERβ contributes to attenuation of ET-1 mediated vasoconstriction, particularly in trauma hemorrhages, and that specific ERs may be involved in NO production. Studies in cultured ECs have also shown that estrogen increases eNOS mRNA expression [122].
Estrogen has antioxidant properties, decreases the expression of NADPH oxidase and the generation of superoxide and peroxynitrite, and increases NO bioavailability [127]. Estrogen deficiency decreases NO bioavailability for vascular relaxation and reduction in blood pressure. In OVX female rats, the increase in blood pressure is associated with lower plasma antioxidant levels and increased plasma lipoperoxides and vascular free radicals, and E2 replacement prevents these effects [127]. Also, measurement of vascular superoxide has shown greater amounts in blood vessels of male compared with female rats [120].
The role of mitochondria in the effects of E2 in the cardiovascular system is increasingly recognized [58]. ER has been localized to mitochondria and E2 may decrease mitochondrial superoxide production and mitochondrial oxidative stress. Besides nuclear genes, ER may mediate effects on mitochondrial-encoded genes [58].
Estrogen and Prostacyclin (PGI2)
The effects of estrogen on ECs may also involve the release of PGI2, a potent endogenous inhibitor of platelet aggregation and a strong vasodilator. PGI2 is produced from free arachidonic acid by cyclooxygenase-1 (COX-1) and COX-2. Estrogen may modulate cross-talk between NOS and COX pathways where estrogen-induced NO-dependent vasodilation may be associated with a decrease in PGI2-dependent vascular relaxation pathway [128].
In Post-MW, the COX-2 pathway plays a specific role in the rapid E2-induced potentiation of cholinergic vasodilation [129]. Also, in arteries from OVX female monkeys with induced atherosclerosis, the amount of PGI2 is inversely related to plaque size, and arteries treated with E2 show increased PGI2 production [130]. E2 stimulates urinary excretion of COX-2-derived PGI2 metabolites in ERβ but not ERα deficient mice, supporting a role for ERα in PGI2 production [131]. COX inhibitors such as indomethacin inhibit a significant portion of endothelium-dependent vascular relaxation, and gender differences in indomethacin-sensitive vascular relaxation have been attributed to differences in COX products [132]. Other studies have shown that indomethacin does not affect E2-induced relaxation in endothelium-intact coronary artery [133]. In OVX female rats, administration of E2 is associated with COX-2 upregulation in the uterus, but its down-regulation in the vena cava [134]. Also, In the presence of E2 COX-2 is upregulated in human uterine microvascular ECs, but not in dermal microvascular ECs [135], suggesting that COX modulation by E2 may be tissue specific.
Studies in cultured ECs demonstrated a more positive relation between estrogen, COX and PGI2. E2 causes upregulation of COX-1 expression and PGI2 synthesis in ECs [4]. E2 also causes rapid ER mediated stimulation of PGI2 synthesis in ovine fetal pulmonary artery ECs via a Ca2+-dependent, but MAPK-independent pathway [136]. Interestingly, PGI2 production by HUVECs is stimulated after exposure to serum from Post-MW treated with a mixture of phytoestrogens [137].In cultured HUVECs treated with raloxifene, PGI2 synthesis is increased possibly by modification of activity or expression of COX-1 and -2 [138].
Estrogen and EDHF
In some blood vessels treated with NOS and COX inhibitors, ECs still release EDHFs that cause hyperpolarization and relaxation of VSM. Estrogen-induced vascular relaxation could involve the release of EDHF. ER stimulation increases the production of EDHF, which activates K+ channels, causes hyperpolarization, and in turn inhibits Ca2+ influx via Ca2+ channels and leads to VSM relaxation [4]. Ach-induced hyperpolarization and relaxation of mesenteric arteries are reduced in intact male and OVX female compared with intact female rats, and the differences are eliminated by K+ channel blockers. The vascular hyperpolarizing and relaxation response to Ach is improved in E2-replaced OVX female rats, confirming that estrogen-deficient states attenuate vascular relaxation by EDHF [139]. Phytoestrogens may induce vascular relaxation through production of EDHF [140].
Estrogen and Endothelium-Derived Vasoconstrictors
Estrogen and ET-1
ECs release other vascular modulators such as ET-1. ET-1 activates endothelial ETB1 receptors, which mediate the release of relaxing factors and cause vasodilation. ET-1 also stimulates ETA and ETB2 receptors in VSM to cause vasoconstriction. ET-1 induces greater contraction in mesenteric arteries of male DOCA-salt hypertensive rats than those of females. Ovariectomy in females is associated with increased ET-1 and ETB2 receptor mRNA in mesenteric arteries, and E2 replacement reverses these changes. The ETB agonist IRL-1620 induces less vasoconstriction in mesenteric arteries of intact compared with OVX females, and E2 replacement decreases IRL-1620-induced vasoconstriction in OVX females. These data suggest that estrogen attenuates ET-1/ETB receptor expression and their vascular responses in DOCA-salt hypertensive rats [141]. E2 modulates the expression and release of ET-1 in human ECs [142]. Prolonged E2 treatment of ECs inhibits ET-1 production in response to serum, tumor necrosis factor-α, transforming growth factor β1, and AngII [143]. Also, ET-1 release is less in ECs of female than male SHR.
Other endothelium-derived vasoconstrictors include AngII and TXA2. The Angiotensin Converting Enzyme (ACE) Insertion/Deletion(I/D) polymorphism appears to be involved in EC dysfunction in Post-MW [144]. Also, basal release of TXA2 from platelets is greater in raloxifene- compared to E2-treated OVX pigs. Raloxifene treatment, compared to E2, increases the production of contractile and proaggregatory prostanoids from venous ECs and platelets, suggesting a greater thrombotic risk with SERMs compared to natural estrogen [145].
Although no significant changes in vascular ER expression were observed in aging compared with adult OVX SHR, possible age-related reduction in the estrogen/ER binding or the post-ER signaling mechanisms of vasodilation may occur [77]. The findings in the aging OVX SHR model of postmenopausal HTN highlight the importance of timing of HRT in relation to the onset of menopause and patient's age.
Estrogen and Mechanisms of VSM Contraction
Estrogen causes relaxation of endothelium-denuded vascular strips, suggesting direct effects on VSM contraction mechanisms [4]. VSM contraction is triggered by Ca2+ release from the sarcoplasmic reticulum and Ca2+ entry from the extracellular space. Activation of myosin light chain (MLC) kinase, Rho-kinase and MAPK as well as inhibition of MLC phosphatase also contribute to VSM contraction. PKC activation also increases the myofilament force sensitivity to [Ca2+]i and thereby maintains vascular contraction (Fig. 8) [146].
Fig. 8.
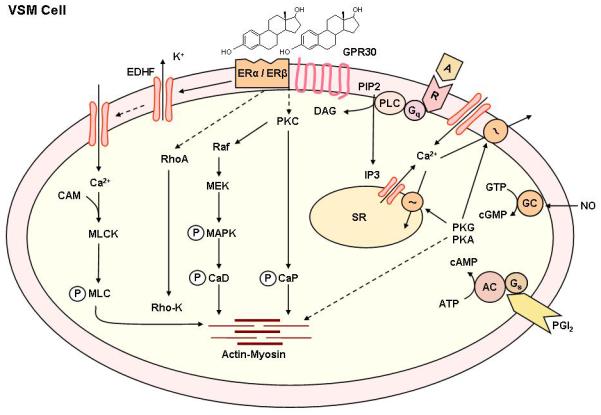
Non-genomic estrogen mediated mechanisms of VSM contraction. Agonists activate specific VSM receptors and stimulates PLC, and increases the production of IP3 and diacylglycerol (DAG). IP3 stimulates Ca2+ release from the sarcoplasmic reticulum (SR). Agonists also stimulate Ca2+ entry through Ca2+ channels. Ca2+ binds CAM, activates myosin light chain (MLC) kinase, causes MLC phosphorylation, and initiates VSM contraction. DAG causes activation of PKC, and PKC then phosphorylates calponin (CaP) and/or activate a protein kinase cascade involving Raf, MAPK kinase (MEK), and MAPK, leading to phosphorylation of caldesmon (CaD) and an increase in the myofilament force sensitivity to Ca2+. Estrogen binds to plasma membrane ER, leading to inhibition of agonist-activated mechanisms of VSM contraction. Possible nongenomic effects of estrogen include activation of K+ channels, leading to membrane hyperpolarization, inhibition of Ca2+ entry through Ca2+ channels, and thereby inhibition of Ca2+-dependent MLC phosphorylation and VSM contraction. Estrogen may also inhibit PKC and/or the MAPK pathway through activation of plasma membrane ERs and thereby further inhibit VSM contraction. Estrogen-induced NO release from the endothelium activates guanylate cyclase in VSM leading to increased cGMP and stimulation of cGMP-dependent protein kinase (PKG). PKG decreases [Ca2+]i by stimulating Ca2+ extrusion pumps in the plasma membrane and Ca2+ uptake pumps in SR and/or decrease the sensitivity of the contractile myofilaments to [Ca2+]i and thereby promote VSM relaxation. Estrogen also induces the release of PGI2 from the endothelium to activate the PGI2-cAMP pathway, or EDHF to activate Ca2+-activated K+ channels and to cause hyperpolarization and relaxation of VSM.
Estrogen and VSM [Ca2+]i
The contraction to the α-adrenergic agonist phenylephrine (Phe) is greater in aortic strips from intact male compared to female Sprague-Dawley rats. There is no difference in Phe-induced contraction between intact and castrated males, suggesting that the enhanced vascular contraction may not be related to endogenous androgens. In contrast, Phe-induced contraction is greater in OVX compared with intact female rats, suggesting a role of estrogen in the reduced vascular response in females [147].
In isolated aortic strips incubated in Ca2+-free solution Phe or caffeine induces a transient contraction that is not different between intact and gonadectomized male and female rats, suggesting no sex differences in the Ca2+ release mechanism. Membrane depolarization by high KCl solution stimulates Ca2+ entry from the extracellular space through voltage-gated Ca2+ channels. KCl-induced contraction is reduced in vascular strips from intact female compared to male rats or OVX female rats. Also, both Phe and high KCl cause stimulation of 45Ca2+ influx that is reduced in intact female compared to male rats, suggesting that the reduced vasoconstriction in females is likely due to an effect of estrogen on the expression/permeability of voltage-gated Ca2+ channels [147].
The gender differences in vascular contraction appear to be related to direct effects of E2 on the vasculature. In endothelium-denuded porcine coronary artery strips and isolated coronary VSM cells, E2 (10−10 to 10−5 M) causes concentration-dependent relaxation of PGF2α-induced contraction. E2 does not affect caffeine-induced coronary contraction in Ca2+-free solution, suggesting that it does not affect Ca2+ release from the intracellular stores. In contrast, E2 causes relaxation of high KCl-induced coronary artery contraction and inhibits PGF2α- and high KCl-induced Ca2+ influx and [Ca2+]i, suggesting inhibitory effects of E2 on Ca2+ entry mechanism of coronary VSM contraction. Topical application of progesterone or testosterone also causes reduction in the maintained [Ca2+]i, but E2 is still more effective [148,149]. E2 also attenuates voltage-dependent Ca2+ current in A7r5 VSM cell line [150].
If estrogen has protective vascular effects, one would predict that its inhibitory effects on the mechanisms of VSM contraction would be enhanced in CVD such as HTN. We have examined the sex differences in VSM contraction and [Ca2+]i in aortic VSM cells isolated from male and female WKY and SHR [151]. In VSM cells of intact male WKY Phe (10−5 M) causes an initial peak followed by maintained increase in [Ca2+]i. The maintained Phe-induced [Ca2+]i is reduced in intact female compared to male WKY and SHR. Also, the reduction in female compared to male SHR (~38%) is greater than that in female compared to male WKY (~25%). Similarly, the reduction in high KCl-induced [Ca2+]i in female compared to male SHR (~31%) is greater than that observed in female compared to male WKY (~20%). These sex differences appear to be related to endogenous estrogen because they are eliminated in OVX female rats, and restored in E2-replaced OVX females. E2 also caused greater reduction of Phe- and high KCl-induced [Ca2+]i in VSM cells of OVX female SHR compared to WKY, suggesting enhanced vascular protective effects of E2 in animal models of genetic HTN [151].
Estrogen and PKC
PKC is a ubiquitous enzyme and comprises a family of Ca2+-dependent and Ca2+-independent isoforms, expressed in different proportions in VSM of various vascular beds. During cell activation, PKC translocation to the cell surface may trigger a cascade of protein kinases that ultimately interact with the contractile myofilaments and cause VSM contraction [146]. The gender-related decrease in VSM contraction in female WKY rats compared with males is associated with reduction in the expression/activity of vascular α-, δ-, and ζ-PKC isoforms. Treatment of OVX females with E2 subcutaneous implants causes reduction in Phe and phorbol 12,13-dibutyrate induced contraction and PKC activity that is greater in SHR than WKY. These data suggest sex differences in VSM contraction and PKC activity that are possibly mediated by estrogen and are enhanced in the SHR model of genetic HTN [152].
Estrogen and Rho-Kinase
Rho-kinase is known to inhibit MLC phosphatase and to enhance the VSM myofilament sensitivity to [Ca2+]i. Rho-kinase is upregulated in CVD and may play a role in the pathogenesis of coronary arteriosclerosis and vasospasm [153]. Estrogen may inhibit Rho-Kinase expression and activity. For instance, the expression of Rho-kinase may involve a PKC/NF-κB pathway that is inhibited by estrogen [154]. Also, the vasodilator response to the Rho-kinase inhibitor Y-27632 is similar in OVX female and male rats, and E2 treatment of OVX rats normalizes the vasodilator effects of Y-27632 to those observed in intact females [155]. In vivo studies have shown that long-term inhibition of Rho-kinase causes regression of coronary arteriosclerosis, and in cultured human coronary VSM cells, treatment with E2 causes a decrease in Rho-kinase mRNA expression [153].
Estrogen and Extracellular Matrix (ECM)
The actin cytoskeleton forms the backbone of the cell, and its spatial organization is crucial for cell migration. Modification of the form and positioning of actin fibers in relation to membrane anchoring integrins and focal adhesion complexes allow cell movement in the extracellular environment. ERα interacts with Gα13 to induce activation of the RhoA/Rho-kinase pathway and phosphorylation of the actin-regulatory protein moesin, leading to remodeling of the actin cytoskeleton and EC migration [156].
Vascular remodeling occurs during all stages of atherosclerotic progression, and MMPs, a family of zinc-binding proteolytic enzymes, are involved in these processes [157]. Increased MMP activity occurs in cancer, arthritis and CVD. Also, MMP-induced ECM degradation within the atherosclerotic plaque may be involved in plaque instability and cardiovascular events. Studies suggest that changes in the levels of MMP-2, -9 and -10 in women receiving MHT may contribute to the potential risk of cardiovascular events and cancer [158].
Aging and the Vascular Architecture
The reduced vascular effects of estrogen after menopause could also be due to age- related structural changes in the blood vessel architecture. Vascular aging involves changes in the mechanical and structural properties of the vascular wall. Arterial compliance is defined as the change in area, diameter, or volume of an artery for a given change in pressure [159]. Elastin fibers play an important role in determining the mechanical strength of the vessels at lower pressures and collagen fibers bear most of the strength at higher pressures [160]. Aging has a major effect on arterial stiffness, and thereby compliance. During vascular aging, there is a progressive arterial stiffening and arteriosclerosis, due to quantitatively less elastin and more collagen, qualitative changes in the content of the vessel wall, and impaired endothelial-mediated vasodilation [161]. In larger elastic arteries, aging is associated with an increase in collagen content, covalent cross-linking of collagen, elastin fracture, reduction in the elastin content, and calcification. There are also changes in wall thickness, media to lumen ratio and EC function. With age, NO production decreases and ET-1 production increases. This favors a procoagulant state and promotes VSM growth. Also, atherosclerosis, an inflammatory process involving EC dysfunction and excess deposition of oxidized lipids, progresses through lifetime and is enhanced by smoking, dyslipidemia, diabetes, and HTN. Atherosclerosis leads to thick stiff arterial wall, calcification and plaque formation, which changes the mechanical properties of the arteries and increase the risk of cardiovascular events [160]. Arteries of Post-MW have some degree of atherosclerosis that could mechanically impede the vasodilator effects of estrogen on vasculature.
The Sex Hormone Environment
Other sex hormones may modify the vascular actions of estrogen, and could also have direct effects on the vasculature. Studies have suggested that the relationship between circulating levels of free E2, free testosterone, and SHBG may be more predictive of changes in carotid intimal thickening than the levels of any of these hormones alone [162]. Also, conversion of testosterone to E2 may contribute to the regulation of the peripheral circulation in men, and administration of an aromatase inhibitor to young men results in a decrease in endothelial vasodilator function [163]. Additionally, the circulating levels of estrogen and androgens may not reflect those at the tissue level, as both aromatase and 5-α-reductase are found in a number of tissues including blood vessels [164]. In addition, the gonadal steroid hormone metabolism is so complex and allows extensive inter-conversion of sex hormones precursors and metabolites (Fig. 2). Furthermore, with the growing therapeutic use of steroid metabolism inhibitors, including aromatase inhibitors and 5-α-reductase inhibitors, particularly in women with a history of breast cancer, cardiovascular side effects are predicted [165].
Role of Progesterone
Progesterone is a steroid hormone produced by the adrenal cortex and gonads, and by the placenta during pregnancy. Progesterone receptors have been identified in ECs and VSM in human, mice, rat, rabbit and primates [166]. Similar to E2, progesterone is anti-atherosclerotic, decreases LDL, and increases HDL. Progesterone causes pulmonary vasodilation by activating both endothelium-dependant and -independent pathways. It stimulates eNOS expression, NO production and NO-mediated relaxation in rat aorta and ovine uterine artery. It also causes direct non-genomic COX activation and increases vascular PGI2 production. It inhibits VSM proliferation/migration and facilitates the inhibitory effects of estrogen. It may also alter the vascular effects of estrogen in MHT, and modify the effects of E2 on vascular contraction. Progesterone also causes rapid relaxation of agonist- or KCl-induced contraction in endothelium-denuded porcine coronary artery [148].
However, the combined endothelium-dependent and -independent vasorelaxation effects of progesterone are less than those of estrogen, and some studies have shown antagonistic effects of progesterone. For example, progesterone counteracts the stimulatory effects of estrogen on NO production and vascular relaxation in canine coronary artery. In porcine coronary artery rings, estrogen blocks progesterone-induced EC dysfunction and reduction in NO production [167]. Also, progesterone antagonizes the antioxidant effect of estrogen, and enhances NADPH oxidase activity and the production of reactive oxygen species in OVX mice [168]. Progesterone also promotes vasoconstriction by upregulating vascular AT1R mRNA and protein [169,170]. Progestins may also diminish estrogen-mediated the anti-inflammatory effects and attenuation of ischemic brain injury [171]. Also, treatment of surgically postmenopausal OVX female monkeys with CEE alone, but not in combination with MPA, inhibits aortic connective tissue remodeling after plasma lipid lowering [172]. Furthermore, MPA antagonizes the inhibitory effects of CEE on coronary artery atherosclerosis [173]. These complex interactions of estrogen and progesterone on the vasculature highlight the need to determine the benefits vs. risk of combined estrogen/progestins in postmenpausal CVD.
Androgens and CVD
Androgens are sex steroids that control the development and maintenance of masculine secondary sexual characteristics, and are produced in the testis, adrenal glands, and ovaries. In women, androgens are important for maintaining bone mass, secondary sex characteristics and libido. The adrenal steroid dehydroepiandrosterone (DHEA) and its sulfate ester (DHEAS) are the most abundant steroids in the human circulation [174]. DHEA and androstenedione do not have significant biological activity, but they are converted to testosterone which binds to and activates the androgen receptor. Testosterone is converted to dihydrotestosterone (DHT), which has 5 times higher binding affinity to the androgen receptor. Testosterone is also aromatized peripherally, mainly in the adipose tissue, to E2.
Because men have higher BP than women throughout most of their lives, and CVD develops at an earlier age in men than in women, it has been suggested that androgens may promote CVD in men [175]. However, serum testosterone levels are lower in men with chronic CVD such as HTN than in healthy age-matched men [175,176], suggesting that androgens may not be responsible for mediating CVD. However, downregulation of androgen synthesis may be a protective compensatory mechanism that occurs once the CVD is initiated [175].
Experimental studies suggest that androgens may mediate CVD in males. Adult male SHR have higher BP than females, and castration of male rats is associated with reduction in BP to the levels found in females. Also, testosterone treatment of OVX female rats increases their BP in a dose-dependent manner [177]. Sex differences in BP have also been found in Dahl salt-sensitive (DS) rats on a high-salt diet and DOCA-salt treated rats [175,178].
While it is classically thought that testosterone may exert unfavorable cardiovascular effects, some clinical and epidemiological studies suggest beneficial effects. For example, low circulating testosterone levels in men are positively correlated with numerous risk factors for CAD [179]. Also, testosterone may have beneficial effects on the blood lipid profile and against atheroma formation [180]. Furthermore, acute intra-coronary or intravenous infusion of testosterone in men rapidly improves myocardial ischemia [181,182].
Androgens may play a role in determining the cardiovascular risk in Post-MW. Serum testosterone levels are markedly lower in women than in men, but whether serum testosterone levels decrease after menopause is not clear [175,177]. The conventional wisdom is that androgen levels, like estrogen, do decrease with age. However, serial measurements of sex hormones in Post-MW for 10 years after cessation of cycling showed increases in serum testosterone and androstenedione and decreases in serum E2 and dihydrotestosterone with age [183]. In cross-sectional studies of women in the Rancho Bernardo cohort, serum testosterone decreased immediately following menopause, but thereafter increased with age, reaching Pre-MW levels at 70-79 years of age. Also, in women who had undergone surgical menopause serum testosterone levels did not increase with age, but were 40–50% lower than in women with natural menopause [184]. These data support the contention that the natural postmenopausal period is a relatively hyperandrogenic state [175,177,185].
Disease Models with Disturbed Sex Steroids Environment
Conditions with excess or deficient estrogen levels such as PCOS, Turner syndrome and obesity may serve as models to identify potential mechanisms of aberrant estrogen-ER interactions. PCOS is a heterogeneous disorder characterized by obesity, menstrual abnormalities, hirsutism, anovulatory infertility, elevated androgens and estrogens, insulin resistance, and lipid abnormalities. In PCOS, both insulin and leutinizing hormone stimulate androgen production from ovarian theca cell, leading to elevated levels of testosterone and androstenedione [186]. In turn, elevated androgen levels contribute to increased E1 levels through peripheral aromatization. E2 levels also increase in PCOS due to increased production of E2 from E1 in peripheral tissues, increased biologically available circulating E2 because of decreased SHBG, and local conversion of E1 to E2 at target tissues. Most women with PCOS are severely overweight or obese, which generally aggravates the endocrine disturbances [187]. In both normoandrogenic Pre-MW and Post-MW without PCOS, excess weight and chronic hyperinsulinemia are associated with changes in total and bioavailable plasma sex steroid levels. Overweight results in increased estrogen concentrations from peripheral conversion of androgens to E1 in adipose tissue by aromatase enzyme [187]. Since Post-MW and women with PCOS have similar sex hormone profiles, namely the lack of progesterone, and E1 being the major estrogen, investigating ERs in vasculature of women with PCOS may give an insight into ER regulation and evolution. Further, the excess stimulation by androgens may give an insight into the role of other sex hormones in affecting the actions of estrogen on ERs.
Estrogen is known to regulate fat mass, adipose deposition and differentiation, and adipocyte metabolism [187]. Estrogen deficiency results in increases in adipose tissue and visceral fat, and ss the epidemic of obesity increases, Post-MW are especially at higher risk [188]. Adipose tissue has P450 aromatase activity, which converts androstenedione into E1, the main estrogen in Post-MW. The conversion rate of androstenedione into E1 increases with age and obesity [189]. Also, adipose tissue secretes inflammatory substances such as the cytokines interleukin-6 (IL-6) and tumor necrosis factor-α (TNFα), complement factors C3, factor B, and Adipsin [190]. Since obesity is prevalent in Post-MW, the increased inflammatory factors from excess adipose tissue could affect vascular ER or their responses in vasculature.
Turner syndrome is characterized by the absence of a part of or the entire X chromosome, with the typical karyotype 45,X; although most women have several different variants. Turner syndrome is characterized by short stature, estrogen deficiency due to ovarian dysgenesis, primary amenorrhoea, and cardiovascular malformations, and is often complicated by HTN, ischemic heart disease, and aortic dilation with or without aortic aneurysm [191]. Assuming potential vascular benefits of estrogen, the increased cardiovascular morbidity in Turner syndrome could be partly explained by lack of estrogens. Estrogen supplementation would likely have positive effects on aortic ‘stiffness’ in the long term [192]. Investigating the long-term effects of estrogen supplementation on cardiovascular morbidity in Turner syndrome may give better insight into the regulation of ERs, when the levels of estrogen are low.
MHT RCTs: Lessons Learned and Future Directions
The results of HERS and WHI RCTs challenged the concept that MHT protects the cardiovascular system and suggested that such therapy may actually increase the risk of CVD. Many factors in the design of RCTs may have led to the unexpected results including the participants’ age and timing of MHT, preexisting CVD, type of estrogen used, the combination of progestins in MHT, and socioeconomic status. In the NHS, which showed protective effects of MHT, ~80% of women initiated MHT within 2 years of menopause [193]. In contrast, the women in WHI and HERS were on average 63 and 67 years of age, and had been postmenopausal for ~10 years at the time of enrollment. Even younger healthy participants (aged 50 to 59) in the WHI study probably had been menopausal ~6 years before MHT. Also, there was no information on the subjects’ age at menopause, which may be important in defining the cardiovascular status as it changes with age and more rapidly after menopause [3].
One criticism of HERS was that the participants had CVD at baseline. Also, while WHI was designed as a primary prevention RCT in “healthy” women, of the women assigned MHT 36% had HTN, 49% were current or past smokers, and 34% were obese, suggesting an active atherosclerosis process in the participants. Because atherosclerosis and vascular remodeling are age-dependent processes, a delay in MHT by even a few years may influence outcome [3]. Studies conducted in nonhuman primates demonstrated that the vascular protective effects of MHT were greater if MHT was started before the onset of atherosclerosis, suggesting that MHT may only afford primary prevention. Early MHT administration caused a 70% protection in OVX primates on an atherosclerotic diet, whereas delay in therapy until after development of moderate atherosclerosis resulted in only 50% protection. In primates that had received an atherosclerotic diet for 2 years before initiating MHT, MHT did not protect against atherosclerosis [194]. Administration of E2 before and during, but not 7 days after, balloon injury resulted in inhibition of neointima formation in rats [195]. Furthermore, delayed administration of E2 failed to prevent neointima formation in rabbits [196]. Thus, a 6-year delay in MHT may be sufficient to reduce their protective actions [3].
Another factor that may have affected the RCTs’ outcome is socioeconomic status. In general, women who take MHT are more educated, wealthier, have healthier lifestyle, and fewer cardiovascular risk factors. A meta-analysis showed that the previously observed reduced risk for CAD among MHT users was lost when the statistical analysis included socioeconomic status [3].
The type of estrogen used in RCTs has also been debated. HERS and WHI used CEE, a mixture extracted from pregnant equine urine, and its active ingredients include sodium estrone sulfate, sodium equilin sulfate, and sodium 17α-dihydroequilinenin. After menopause, women lose E2, whereas the levels of E1 remain unchanged. The nomenclature seems to obscure the important distinction that CEE does not contain E2. Chemically, CEE and E2 are different molecular entities, and therefore their pharmacological properties vary considerably and may influence the outcome of RCTs [3]. Compared to E2, estrogens in CEE have different metabolic products and distinct binding affinity, selectivity, and agonistic properties to ER subtypes [143]. Because both ER-dependent and ER-independent mechanisms play a role in mediating the cardiovascular actions of E2, CEE may not mimic the cardiovascular protective effects of E2. Thus non-E2 estrogens may be less effective in counteracting CVD and may even induce deleterious effects. For example, a Swedish study found a reduced risk of myocardial infarction for E2 compared with oral E3 or vaginal estriol/dienestrol [193].
Studies on human aortic VSMs demonstrated that estrogens present in CEE were less potent than E2 in inhibiting mitogen-induced VSM growth and MAPK activity [197], which may the negative outcomes of HERS and WHI. In a primate model, administration of CEE had no effect on intimal hyperplasia after balloon injury [198]. Importantly, in the Estrogen in the Prevention of Atherosclerosis Trial (EPAT), administration of E2 to Post-MW without CVD reduced the progression of intimal thickening [199]. These findings provide evidence that use of estrogens other than E2 may contribute to the lack of protective actions of MHT.
In women with intact uterus, estrogens are given in combination with a progestin in order to reduce the risk of endometrial cancer. The negative findings of HERS and one arm of WHI may have been caused by concomitant use of MPA. For instance, there was an increase in stroke risk in women taking CEE plus MPA versus women never using MHT [200]. Also, in Postmenopausal Estrogen/Progestin Interventions (PEPI) trial, CEE caused beneficial effects on LDL and HDL levels that were attenuated by MPA [3]. However, other studies did not find any attenuation of CEE-induced dilatation by MPA or micronized progesterone [201,202]. Also, in one arm of WHI in women taking estrogen alone, no protective effects were observed even though lipids were favorably changed. Moreover, the NHS demonstrated a similar risk reduction for CHD among women taking CEE alone and those taking CEE plus MPA.
In cynomolgus monkeys, chronic E2 or E2+progesterone had similar anti-atherosclerotic effects [3]. In contrast, loss of protective effects were observed in monkeys administered CEE+MPA as compared to CEE alone (72% reduction in coronary artery atherosclerosis) [203]. Also, studies on postmenopausal cynomolgus monkeys suggested that MPA abrogates the vascular benefits of estrogen [204]. In these studies Ach caused vasoconstrictor responses in estrogen-deprived monkeys not receiving MHT; however, a vasodilatory response was observed in monkeys treated with estrogen alone, and the beneficial effect of estrogen was reduced by 50% by co-administration of MPA [204]. In a rat model, MPA abrogated the ability of E2 to attenuate balloon injury-induced intimal thickening, a process independent of lipids [205]. On the other hand, studies in rabbits indicated that the protective actions of CEE or E2 on atherosclerosis were not prevented by MPA, or other progestins [3]. This suggests that MPA may block the protective actions of E2 that are mediated via its direct interaction with vascular cells. Whether progestins attenuate the anti-atherosclerotic effects of estrogen is still unclear. Also, progesterone and MPA inhibit mitogen-induced proliferation of VSM cells in culture. Thus, whether MPA is responsible for the lack of vascular protective actions of MHT in RCTs needs to be further examined. The termination of the estrogen alone arm of the WHI study suggest that factors other than MPA may be involved [3].
Another important distinction between E2 and CEE is the route of administration. Oral, but not transdermal estrogen increases C-reactive protein and IL-6 levels [206,207]. Because CEE is given orally, whereas E2 is often administered transdermaly to humans and subcutaneously to animals, it is possible that the differences in outcome between CEE and E2 are caused in part by the route of administration.
New clinical trial data and re-analyses of the WHI data suggest that MHT started within a few years of menopause reduces atherosclerosis progression, clinical CHD events and total mortality. Also, basic science studies indicate that estrogen could reduce early atherogenesis but augment its later stages [208]. These observations have prompted two new RCTs: The Kronos Early Estrogen Prevention Study (KEEPS) and Early Versus Late Intervention Trial With Estradiol (ELITE). KEEPS investigates the effects of MHT on carotid intima–media thickness (CIMT, a measure of atherosclerosis) and the accrual of coronary calcium in 720 early menopausal women (aged 42-58 years) women randomized at 6 to 36 months postmenopause, with women post-hysterectomy excluded. Participants are given low dose oral CEE and weekly transdermal E2 (both in combination with cyclic oral, micronized progesterone) or placebo 12 days per month [209]. On the other hand, ELITE will randomize 504 women, either < 6 years or > 10 years postmenopausal, to receive oral E2 for 2–5 years, and as in KEEPS, the primary endpoint is change in CIMT. ELITE will test for differences between early and late initiation of MHT, whereas KEEPS will examine differences between low-dose oral CEE and low-dose transdermal E2. The results of these two studies might help clarify the benefits of MHT timing, and the possible differences in atherosclerosis effects between oral E2 and oral CEE, and between oral and transdermal E2 [58,208].
Perspective
While MHT may still be useful to treat vasomotor menopausal symptoms such as hot flashes and night sweats, its use to prevent CVD remains to be elucidated. Although RCTs have documented increased rates of myocardial infarction and stroke in women receiving MHT, the final chapter on estrogen therapy may have not yet been written. Understanding the structural basis and molecular mechanisms by which ER transduce E2 signals in target cells will help develop new therapies for CVD. Vascular ERα, ERβ, and GPR30 mediate estrogen-induced genomic or non-genomic effects. Also, estrogen promotes endothelium-dependent vascular relaxation, inhibits VSM contraction, modulates the cytoskeleton, and the vascular inflammatory response. The failure of the beneficial effects of estrogen on vasculature to materialize into RCTs is likely related to age-related changes in ER amount, distribution, integrity, and downstream signaling mechanisms as well as structural changes in the vasculature. These changes may be attributed to age-related gene mutations or ER gene methylation. Investigation of the SARs of estrogenic compounds would enhance our understanding of the molecular interaction of estrogen and ER. Further characterization of currently available estrogens and the exploration of novel estrogenic compounds would provide more efficient and specific ER modulators. Study of diseases associated with abnormal estrogen production or metabolism such as PCOS and Turner syndrome may provide further insights into ER regulation and evolution in Post-MW. Also, the potential benefits/risks of combined therapy of multiple sex hormones and modulators of progesterone and androgen receptors should be examined. As newer forms of MHT become available, and if used at the right dose, route of administration and timing depending on the subjects’ age and preexisting cardiovascular condition, the beneficial vascular effects of estrogen could be translated into the outcome of MHT in post-menopausal CVD.
Fig. 7.
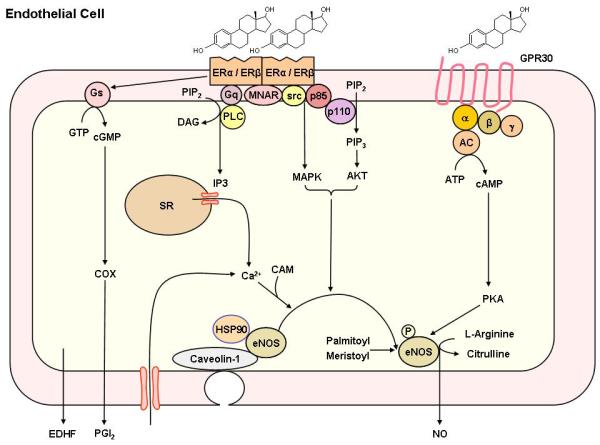
Non-genomic estrogen mediated endothelial pathways of vascular relaxation. Estrogen binds to endothelial surface membrane ER and activates phospholipase C (PLC), leading to the generation of inositol 1,4,5- triphosphate (IP3) and DAG. IP3 causes the release of stored Ca2+ from the endoplasmic reticulum, and influx of extracellular Ca2+. Ca2+ forms a complex with calmodulin (CAM), which causes initial activation of eNOS, its dissociation from caveolin-1, and its translocation to intracellular sites. Estrogen also activates phosphatidylinositol 3-kinase (PI3K), which transforms phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3), which activates Akt. ER-mediated activation of Akt or MAPK pathway causes phosphorylation of cytosolic eNOS and its second translocation back to the cell membrane where it undergoes myristoylation and palmitoylation to become fully activated. Fully activated eNOS promotes the transformation of L-arginine to L-citrulline and the production of NO, which diffuses through the endothelial cell and causes VSM relaxation. Estrogen also activates COX to produce prostacyclin (PGI2), and induces the release of EDHF.
ACKNOWLEDGEMENTS
This work was supported by grants from National Heart, Lung, and Blood Institute (HL-65998 and HL-70659.
List of Abbreviations
- Akt
protein kinase B
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- CEE
conjugated equine estrogen
- CHD
coronary heart disease
- CRP
c-reactive protein
- CVD
cardiovascular disease
- E2
17β-estradiol
- EC
endothelial cell
- ECM
extracellular matrix
- ELITE
Early versus Late Intervention Trial with Estradiol
- eNOS
endothelial nitric oxide synthase
- ER
estrogen receptor
- GPR
G protein-coupled receptor
- HERS
Heart and Estrogen/progestin Replacement Study
- HTN
hypertension
- KEEPS
Kronos Early Estrogen Prevention Study
- MAPK
mitogen-activated protein kinase
- MHT
menopausal hormone therapy
- MMP
matrix metalloproteinase
- MPA
medroxyprogesterone acetate
- NO
nitric oxide
- NHS
Nurses' Health Study
- OVX
ovariectomized
- PCOS
polycystic ovary syndrome
- PI3K
phosphatidylinositol 3-kinase
- Pre-MW
premenopausal women
- Post-MW
postmenopausal women
- SAR
structure-activity relationship
- SHR
spontaneously hypertensive rat
- VSM
vascular smooth muscle
- WHI
Women's Health Initiative
REFERENCES
- 1.Stefanick ML. Estrogens and progestins: background and history, trends in use, and guidelines and regimens approved by the US Food and Drug Administration. Am. J. Med. 2005;118(Suppl 12B):64–73. doi: 10.1016/j.amjmed.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 2.Pettersson K, Gustafsson JA. Role of estrogen receptor beta in estrogen action. Annu. Rev. Physiol. 2001;63:165–192. doi: 10.1146/annurev.physiol.63.1.165. [DOI] [PubMed] [Google Scholar]
- 3.Dubey RK, Imthurn B, Zacharia LC, Jackson EK. Hormone replacement therapy and cardiovascular disease: what went wrong and where do we go from here? Hypertension. 2004;44(6):789–795. doi: 10.1161/01.HYP.0000145988.95551.28. [DOI] [PubMed] [Google Scholar]
- 4.Orshal JM, Khalil RA. Gender, sex hormones, and vascular tone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;286(2):R233–R249. doi: 10.1152/ajpregu.00338.2003. [DOI] [PubMed] [Google Scholar]
- 5.Hodis HN, Mack WJ, Lobo RA, Shoupe D, Sevanian A, Mahrer PR, Selzer RH, Liu CR, Liu CH, Azen SP. Estrogen in the Prevention of Atherosclerosis Trial Research Group. Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2001;135(11):939–953. doi: 10.7326/0003-4819-135-11-200112040-00005. [DOI] [PubMed] [Google Scholar]
- 6.Manson JE, Martin KA. Clinical practice. Postmenopausal hormone-replacement therapy. N. Engl. J. Med. 2001;345(1):34–40. doi: 10.1056/NEJM200107053450106. [DOI] [PubMed] [Google Scholar]
- 7.Rosano GM, Sarrel PM, Poole-Wilson PA, Collins P. Beneficial effect of oestrogen on exercise-induced myocardial ischaemia in women with coronary artery disease. Lancet. 1993;342(8864):133–136. doi: 10.1016/0140-6736(93)91343-k. [DOI] [PubMed] [Google Scholar]
- 8.Blumenthal RS, Heldman AW, Brinker JA, Resar JR, Coombs VJ, Gloth ST, Gerstenblith G, Reis SE. Acute effects of conjugated estrogens on coronary blood flow response to acetylcholine in men. Am. J. Cardiol. 1997;80(8):1021–1024. doi: 10.1016/s0002-9149(97)00596-1. [DOI] [PubMed] [Google Scholar]
- 9.Eckstein N, Nadler E, Barnea O, Shavit G, Ayalon D. Acute effects of 17 beta-estradiol on the rat heart. Am. J. Obstet. Gynecol. 1994;171(3):844–848. doi: 10.1016/0002-9378(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 10.Node K, Kitakaze M, Kosaka H, Minamino T, Sato H, Kuzuya T, Hori M. Roles of NO and Ca2+-activated K+ channels in coronary vasodilation induced by 17beta-estradiol in ischemic heart failure. FASEB. J. 1997;11(10):793–799. doi: 10.1096/fasebj.11.10.9271364. [DOI] [PubMed] [Google Scholar]
- 11.Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, Hsia J, Hulley S, Herd A, Khan S, Newby LK, Waters D, Vittinghoff E, Wenger N, HERS Research Group Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II) J.A.M.A. 2002;288(1):49–57. doi: 10.1001/jama.288.1.49. [DOI] [PubMed] [Google Scholar]
- 12.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Writing Group for the Women's Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. J.A.M.A. 2002;288(3):321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 13.Lieberman S. Are the differences between estradiol and other estrogens, naturally occurring or synthetic, merely semantical? J. Clin. Endocrinol. Metab. 1996;81(2):850–851. doi: 10.1210/jcem.81.2.8636315. [DOI] [PubMed] [Google Scholar]
- 14.Hewitt SC, Harrell JC, Korach KS. Lessons in estrogen biology from knockout and transgenic animals. Annu. Rev. Physiol. 2005;67:285–308. doi: 10.1146/annurev.physiol.67.040403.115914. [DOI] [PubMed] [Google Scholar]
- 15.Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology. 2006;147(9):4132–4150. doi: 10.1210/en.2006-0113. [DOI] [PubMed] [Google Scholar]
- 16.Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. N. Engl. J. Med. 2002;346(5):340–352. doi: 10.1056/NEJMra000471. [DOI] [PubMed] [Google Scholar]
- 17.Gallegos AM, Atshaves BP, Storey SM, Starodub O, Petrescu AD, Huang H, McIntosh AL, Martin GG, Chao H, Kier AB, Schroeder F. Gene structure, intracellular localization, and functional roles of sterol carrier protein-2. Prog. Lipid. Res. 2001;40(6):498–563. doi: 10.1016/s0163-7827(01)00015-7. [DOI] [PubMed] [Google Scholar]
- 18.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227(2):115–124. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase--a brief overview. Annu. Rev. Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- 20.Feigelson HS, McKean-Cowdin R, Pike MC, Coetzee GA, Kolonel LN, Nomura AM, Le Marchand L, Henderson BE. Cytochrome P450c17alpha gene (CYP17) polymorphism predicts use of hormone replacement therapy. Cancer Res. 1999;59(16):3908–3910. [PubMed] [Google Scholar]
- 21.Herrington DM, Klein KP. Pharmacogenetics of estrogen replacement therapy. J. Appl. Physiol. 2001;91(6):2776–2784. doi: 10.1152/jappl.2001.91.6.2776. [DOI] [PubMed] [Google Scholar]
- 22.Canonico M, Bouaziz E, Carcaillon L, Verstuyft C, Guiochon-Mantel A, Becquemont L, Scarabin PY. Estrogen and Thromboembolism Risk (ESTHER) Study Group. Synergism between oral estrogen therapy and cytochrome P450 3A5*1 allele on the risk of venous thromboembolism among postmenopausal women. J. Clin. Endocrinol. Metab. 2008;93:3082–3087. doi: 10.1210/jc.2008-0450. [DOI] [PubMed] [Google Scholar]
- 23.Cornwell T, Cohick W, Raskin I. Dietary phytoestrogens and health. Phytochemistry. 2004;65(8):995–1016. doi: 10.1016/j.phytochem.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Murkies AL, Lombard C, Strauss BJ, Wilcox G, Burger HG, Morton MS. Dietary flour supplementation decreases post-menopausal hot flushes: effect of soy and wheat. Maturitas. 1995;21(3):189–195. doi: 10.1016/0378-5122(95)00899-v. [DOI] [PubMed] [Google Scholar]
- 25.Ververidis F, Trantas E, Douglas C, Vollmer G, Kretzschmar G, Panopoulos N. Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part I: Chemical diversity, impacts on plant biology and human health. Biotechnol. J. 2007;2(10):1214–1234. doi: 10.1002/biot.200700084. [DOI] [PubMed] [Google Scholar]
- 26.Ososki AL, Kennelly EJ. Phytoestrogens: a review of the present state of research. Phytother. Res. 2003;17(8):845–869. doi: 10.1002/ptr.1364. [DOI] [PubMed] [Google Scholar]
- 27.Fang H, Tong W, Shi LM, Blair R, Perkins R, Branham W, Hass BS, Xie Q, Dial SL, Moland CL, Sheehan DM. Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens. Chem. Res. Toxicol. 2001;14(3):280–294. doi: 10.1021/tx000208y. [DOI] [PubMed] [Google Scholar]
- 28.Cassidy A, Albertazzi P, Lise Nielsen I, Hall W, Williamson G, Tetens I, Atkins S, Cross H, Manios Y, Wolk A, Steiner C, Branca F. Critical review of health effects of soyabean phyto-oestrogens in post-menopausal women. Proc. Nutr. Soc. 2006;65(1):76–92. doi: 10.1079/pns2005476. [DOI] [PubMed] [Google Scholar]
- 29.Tempfer CB, Bentz EK, Leodolter S, Tscherne G, Reuss F, Cross HS, Huber JC. Phytoestrogens in clinical practice: a review of the literature. Fertil. Steril. 2007;87:1243–1249. doi: 10.1016/j.fertnstert.2007.01.120. [DOI] [PubMed] [Google Scholar]
- 30.Park WC, Jordan VC. Selective estrogen receptor modulators (SERMS) and their roles in breast cancer prevention. Trends Mol. Med. 2002;8(2):82–88. doi: 10.1016/s1471-4914(02)02282-7. [DOI] [PubMed] [Google Scholar]
- 31.Morello KC, Wurz GT, DeGregorio MW. SERMs: current status and future trends. Crit. Rev. Oncol. Hematol. 2002;43(1):63–76. doi: 10.1016/s1040-8428(02)00022-7. [DOI] [PubMed] [Google Scholar]
- 32.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295(5564):2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 33.Smith CL, O'Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004;25(1):45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 34.Mosca L, Collins P, Herrington DM, Mendelsohn ME, Pasternak RC, Robertson RM, Schenck-Gustafsson K, Smith SC, Jr., Taubert KA, Wenger NK. American Heart Association. Hormone replacement therapy and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 2001;104(4):499–503. doi: 10.1161/hc2901.092200. [DOI] [PubMed] [Google Scholar]
- 35.Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, McNabb MA, Wenger NK. Raloxifene Use for The Heart (RUTH) Trial Investigators. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N. Engl. J. Med. 2006;355(2):125–137. doi: 10.1056/NEJMoa062462. [DOI] [PubMed] [Google Scholar]
- 36.Chan YC, Leung FP, Yao X, Lau CW, Vanhoutte PM, Huang Y. Raloxifene relaxes rat pulmonary arteries and veins: roles of gender, endothelium, and antagonism of Ca2+ influx. J. Pharmacol. Exp. Ther. 2005;312(3):1266–1271. doi: 10.1124/jpet.104.077990. [DOI] [PubMed] [Google Scholar]
- 37.Leung HS, Seto SW, Kwan YW, Leung FP, Au AL, Yung LM, Yao X, Huang Y. Endothelium-independent relaxation to raloxifene in porcine coronary artery. Eur. J. Pharmacol. 2007;555(2-3):178–184. doi: 10.1016/j.ejphar.2006.10.035. [DOI] [PubMed] [Google Scholar]
- 38.Ek RO, Yildiz Y, Cecen S, Yenisey C, Kavak T. Effects of tamoxifen on myocardial ischemia-reperfusion injury model in ovariectomized rats. Mol. Cell Biochem. 2008;308(1-2):227–235. doi: 10.1007/s11010-007-9633-0. [DOI] [PubMed] [Google Scholar]
- 39.Figtree GA, Webb CM, Collins P. Tamoxifen acutely relaxes coronary arteries by an endothelium-, nitric oxide-, and estrogen receptor dependent mechanism. J. Pharmacol. Exp. Ther. 2000;295(2):519–523. [PubMed] [Google Scholar]
- 40.Hutchison SJ, Chou TM, Chatterjee K, Sudhir K. Tamoxifen is an acute, estrogen-like, coronary vasodilator of porcine coronary arteries in vitro. J. Cardiovasc. Pharmacol. 2001;38(5):657–665. doi: 10.1097/00005344-200111000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol. Cell Endocrinol. 2003;206(1-2):13–22. doi: 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 42.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J. Med. Chem. 2000;43(26):4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 43.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-alpha or estrogen receptor-beta. Endocrinology. 1999;140(2):800–804. doi: 10.1210/endo.140.2.6480. [DOI] [PubMed] [Google Scholar]
- 44.Douglas G, Cruz MN, Poston L, Gustafsson JA, Kublickiene K. Functional characterization and sex differences in small mesenteric arteries of the estrogen receptor-beta knockout mouse. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008;294(1):R112–R120. doi: 10.1152/ajpregu.00421.2007. [DOI] [PubMed] [Google Scholar]
- 45.Bolego C, Cignarella A, Sanvito P, Pelosi V, Pellegatta F, Puglisi L, Pinna C. The acute estrogenic dilation of rat aorta is mediated solely by selective estrogen receptor-alpha agonists and is abolished by estrogen deprivation. J. Pharmacol. Exp. Ther. 2005;313(3):1203–1208. doi: 10.1124/jpet.104.082867. [DOI] [PubMed] [Google Scholar]
- 46.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 47.Hall JM, McDonnell DP. Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol. Interv. 2005;5(6):343–357. doi: 10.1124/mi.5.6.7. [DOI] [PubMed] [Google Scholar]
- 48.Wakeling AE, Bowler J. Steroidal pure antioestrogens. J. Endocrinol. 1987;112(3):R7–R10. doi: 10.1677/joe.0.112r007. [DOI] [PubMed] [Google Scholar]
- 49.Howell A. Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr. Relat. Cancer. 2006;13(3):689–706. doi: 10.1677/erc.1.00846. [DOI] [PubMed] [Google Scholar]
- 50.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure. 2001;9:145–153. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 51.Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbogen BS. Antagonists selective for estrogen receptor alpha. Endocrinology. 2002;143(3):941–947. doi: 10.1210/endo.143.3.8704. [DOI] [PubMed] [Google Scholar]
- 52.Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl. Recept. Signal. 2008;6:e003. doi: 10.1621/nrs.06003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prossnitz ER, Oprea TI, Sklar LA, Arterburn JB. The ins and outs of GPR30: A transmembrane estrogen receptor. J. Steroid Biochem. Mol. Biol. 2008;109(3-5):350–353. doi: 10.1016/j.jsbmb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly ES, Jr., Nethrapalli IS, Tinnikov AA. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J. Neurosci. 2002;22(19):8391–8401. doi: 10.1523/JNEUROSCI.22-19-08391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc. Natl. Acad. Sci. U. S. A. 1998;95(26):15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr. Rev. 1999;20(3):358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 57.Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol. Aspects Med. 2006;27(4):299–402. doi: 10.1016/j.mam.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol. Rev. 2008;60(2):210–241. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris HA. Estrogen receptor-beta: recent lessons from in vivo studies. Mol. Endocrinol. 2007;21(1):1–13. doi: 10.1210/me.2005-0459. [DOI] [PubMed] [Google Scholar]
- 60.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 2007;87(3):905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 61.Mendelsohn ME. Genomic and nongenomic effects of estrogen in the vasculature. Am. J. Cardiol. 2002;90(1A):3F–6F. doi: 10.1016/s0002-9149(02)02418-9. [DOI] [PubMed] [Google Scholar]
- 62.Hodges YK, Tung L, Yan XD, Graham JD, Horwitz KB, Horwitz LD. Estrogen receptors alpha and beta: prevalence of estrogen receptor beta mRNA in human vascular smooth muscle and transcriptional effects. Circulation. 2000;101(15):1792–1298. doi: 10.1161/01.cir.101.15.1792. [DOI] [PubMed] [Google Scholar]
- 63.Andersson C, Lydrup ML, Fernö M, Idvall I, Gustafsson J, Nilsson BO. Immunocytochemical demonstration of oestrogen receptor beta in blood vessels of the female rat. J. Endocrinol. 2001;169(2):241–247. doi: 10.1677/joe.0.1690241. [DOI] [PubMed] [Google Scholar]
- 64.Beato M, Klug J. Steroid hormone receptors: an update. Hum. Reprod. Update. 2000;6:225–236. doi: 10.1093/humupd/6.3.225. [DOI] [PubMed] [Google Scholar]
- 65.Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70(5-7):407–417. doi: 10.1016/j.steroids.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 66.Hirata S, Shoda T, Kato J, Hoshi K. The multiple untranslated first exons system of the human estrogen receptor beta (ER beta) gene. J. Steroid Biochem. Mol. Biol. 2001;78(1):33–40. doi: 10.1016/s0960-0760(01)00071-1. [DOI] [PubMed] [Google Scholar]
- 67.Kuiper GG, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138(3):863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 68.Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol. Endocrinol. 2002;16(5):938–946. doi: 10.1210/mend.16.5.0827. [DOI] [PubMed] [Google Scholar]
- 69.Alevizaki M, Saltiki K, Cimponeriu A, Kanakakis I, Xita N, Alevizaki CC, Georgiou I, Sarika HL. Severity of cardiovascular disease in postmenopausal women: associations with common estrogen receptor alpha polymorphic variants. Eur. J. Endocrinol. 2007;156(4):489–496. doi: 10.1530/EJE-06-0685. [DOI] [PubMed] [Google Scholar]
- 70.Emre A, Sahin S, Erzik C, Nurkalem Z, Oz D, Cirakoglu B, Yesilcimen K, Ersek B. Effect of hormone replacement therapy on plasma lipoproteins and apolipoproteins, endothelial function and myocardial perfusion in postmenopausal women with estrogen receptor-alpha IVS1-397 C/C genotype and established coronary artery disease. Cardiology. 2006;106(1):44–50. doi: 10.1159/000092598. [DOI] [PubMed] [Google Scholar]
- 71.Herrington DM, Howard TD, Hawkins GA, Reboussin DM, Xu J, Zheng SL, Brosnihan KB, Meyers DA, Bleecker ER. Estrogen-receptor polymorphisms and effects of estrogen replacement on high-density lipoprotein cholesterol in women with coronary disease. N. Engl. J. Med. 2002;346(13):967–974. doi: 10.1056/NEJMoa012952. [DOI] [PubMed] [Google Scholar]
- 72.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem. Biophys. Res. Commun. 2005;336(4):1023–1027. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- 73.Lewandowski S, Kalita K, Kaczmarek L. Estrogen receptor beta. Potential functional significance of a variety of mRNA isoforms. FEBS. Lett. 2002;524(1-3):1–5. doi: 10.1016/s0014-5793(02)03015-6. [DOI] [PubMed] [Google Scholar]
- 74.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321(6067):209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 75.Post WS, Goldschmidt-Clermont PJ, Wilhide CC, Heldman AW, Sussman MS, Ouyang P, Milliken EE, Issa JP. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc. Res. 1999;43(4):985–991. doi: 10.1016/s0008-6363(99)00153-4. [DOI] [PubMed] [Google Scholar]
- 76.Ying AK, Hassanain HH, Roos CM, Smiraglia DJ, Issa JJ, Michler RE, Caligiuri M, Plass C, Goldschmidt-Clermont PJ. Methylation of the estrogen receptor-alpha gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovasc. Res. 2000;46(1):172–179. doi: 10.1016/s0008-6363(00)00004-3. [DOI] [PubMed] [Google Scholar]
- 77.Wynne FL, Payne JA, Cain AE, Reckelhoff JF, Khalil RA. Age-related reduction in estrogen receptor-mediated mechanisms of vascular relaxation in female spontaneously hypertensive rats. Hypertension. 2004;43(2):405–412. doi: 10.1161/01.HYP.0000111833.82664.0c. [DOI] [PubMed] [Google Scholar]
- 78.Ogueta SB, Schwartz SD, Yamashita CK, Farber DB. Estrogen receptor in the human eye: influence of gender and age on gene expression. Invest. Ophthalmol. Vis. Sci. 1999;40:1906–1911. [PubMed] [Google Scholar]
- 79.Chakraborty TR, Ng L, Gore AC. Age-related changes in estrogen receptor beta in rat hypothalamus: a quantitative analysis. Endocrinology. 2003;144(9):4164–4171. doi: 10.1210/en.2003-0052. [DOI] [PubMed] [Google Scholar]
- 80.Kalesnykas G, Roschier U, Puoliväli J, Wang J, Miettinen R. The effect of aging on the subcellular distribution of estrogen receptor-alpha in the cholinergic neurons of transgenic and wild-type mice. Eur. J. Neurosci. 2005;21(5):1437–1442. doi: 10.1111/j.1460-9568.2005.03953.x. [DOI] [PubMed] [Google Scholar]
- 81.Schultz TW, Sinks GD, Cronin MT. Structure-activity relationships for gene activation oestrogenicity: evaluation of a diverse set of aromatic chemicals. Environ. Toxicol. 2002;17(1):14–23. doi: 10.1002/tox.10027. [DOI] [PubMed] [Google Scholar]
- 82.Pike AC. Lessons learnt from structural studies of the oestrogen receptor. Best. Pract. Res. Clin. Endocrinol. Metab. 2006;20(1):1–14. doi: 10.1016/j.beem.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 83.Cheskis BJ, Greger JG, Nagpal S, Freedman LP. Signaling by estrogens. J. Cell Physiol. 2007;213(3):610–617. doi: 10.1002/jcp.21253. [DOI] [PubMed] [Google Scholar]
- 84.Wiese TE, Polin LA, Palomino E, Brooks SC. Induction of the estrogen specific mitogenic response of MCF-7 cells by selected analogues of estradiol-17 beta: a 3D QSAR study. J. Med. Chem. 1997;40(22):3659–3669. doi: 10.1021/jm9703294. [DOI] [PubMed] [Google Scholar]
- 85.Aliau S, El Garrouj D, Yasri A, Katzenellenbogen BS, Borgna JL. 17a-(Haloacetamidoalkyl) estradiols alkylate the human estrogen receptor at cysteine residues 417 and 530. Biochemistry. 1997;36:5861–5867. doi: 10.1021/bi963111c. [DOI] [PubMed] [Google Scholar]
- 86.Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D, Norman AW. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2-vitamin D3 signaling. Proc. Natl. Acad. Sci. U. S. A. 2004;101(35):12876–12881. doi: 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Hoorn WP. Identification of a second binding site in the estrogen receptor. J. Med. Chem. 2002;45(3):584–589. doi: 10.1021/jm0109661. [DOI] [PubMed] [Google Scholar]
- 88.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652):753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 89.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 90.Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i) J. Biol. Chem. 2001;276(29):27071–27076. doi: 10.1074/jbc.M100312200. [DOI] [PubMed] [Google Scholar]
- 91.O'Dowd BF, Nguyen T, Marchese A, Cheng R, Lynch KR, Heng HH, Kolakowski LF, Jr., George SR. Discovery of three novel G-protein-coupled receptor genes. Genomics. 1998;47(2):310–313. doi: 10.1006/geno.1998.5095. [DOI] [PubMed] [Google Scholar]
- 92.Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007;148(7):3236–3245. doi: 10.1210/en.2006-1605. [DOI] [PubMed] [Google Scholar]
- 93.Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, Heigl A, Lachat M, Genoni M, Barton M. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension. 2007;49(6):1358–1363. doi: 10.1161/HYPERTENSIONAHA.107.089995. [DOI] [PubMed] [Google Scholar]
- 94.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun. 2006;346(3):904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 95.Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr. Rev. 2007;28(7):726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- 96.Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006;20(9):1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- 97.Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology. 2008;149(10):4846–4856. doi: 10.1210/en.2008-0269. [DOI] [PubMed] [Google Scholar]
- 98.Simoncini T, Mannella P, Fornari L, Caruso A, Varone G, Genazzani AR. Genomic and non-genomic effects of estrogens on endothelial cells. Steroids. 2004;69(8-9):537–542. doi: 10.1016/j.steroids.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 99.Jacob J, Sebastian KS, Devassy S, Priyadarsini L, Farook MF, Shameem A, Mathew D, Sreeja S, Thampan RV. Membrane estrogen receptors: genomic actions and post transcriptional regulation. Mol. Cell Endocrinol. 2006;246(1-2):34–41. doi: 10.1016/j.mce.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 100.Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S. Ligand-, cell-, and estrogen receptor subtype (alpha/beta)-dependent activation at GC-rich (Sp1) promoter elements. J. Biol. Chem. 2000;275(8):5379–5387. doi: 10.1074/jbc.275.8.5379. [DOI] [PubMed] [Google Scholar]
- 101.Gougelet A, Mueller SO, Korach KS, Renoir JM. Oestrogen receptors pathways to oestrogen responsive elements: the transactivation function-1 acts as the keystone of oestrogen receptor (ER)beta-mediated transcriptional repression of ER alpha. J. Steroid Biochem. Mol. Biol. 2007;104(3-5):110–122. doi: 10.1016/j.jsbmb.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 102.O'Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, Bienkowska J, Mendelsohn ME, Hansen U. Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol. Endocrinol. 2007;21(6):1281–1296. doi: 10.1210/me.2006-0497. [DOI] [PubMed] [Google Scholar]
- 103.Katayama S, Ashizawa K, Fukuhara T, Hiroyasu M, Tsuzuki Y, Tatemoto H, Nakada T, Nagai K. Differential expression patterns of Wnt and beta-catenin/TCF target genes in the uterus of immature female rats exposed to 17alpha-ethynyl estradiol. Toxicol. Sci. 2006;91(2):419–430. doi: 10.1093/toxsci/kfj167. [DOI] [PubMed] [Google Scholar]
- 104.Shim WS, Conaway M, Masamura S, Yue W, Wang JP, Kmar R, Santen RJ. Estradiol hypersensitivity and mitogen-activated protein kinase expression in long-term estrogen deprived human breast cancer cells in vivo. Endocrinology. 2000;141(1):396–405. doi: 10.1210/endo.141.1.7270. [DOI] [PubMed] [Google Scholar]
- 105.Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER. Plasma membrane estrogen receptors exist and functions as dimers. Mol. Endocrinol. 2004;18(12):2854–2865. doi: 10.1210/me.2004-0115. [DOI] [PubMed] [Google Scholar]
- 106.Moriarty K, Kim KH, Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology. 2006;147(12):5557–5563. doi: 10.1210/en.2006-0729. [DOI] [PubMed] [Google Scholar]
- 107.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146(2):624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 108.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402(6764):884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 109.Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol. Metab. 2005;16(8):362–367. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 110.Geraldes P, Sirois MG, Bernatchez PN, Tanguay JF. Estrogen regulation of endothelial and smooth muscle cell migration and proliferation: role of p38 and p42/44 mitogen-activated protein kinase. Arterioscler. Thromb. Vasc. Biol. 2002;22(10):1585–1590. doi: 10.1161/01.atv.0000035393.11854.6a. [DOI] [PubMed] [Google Scholar]
- 111.Filardo EJ, Quinn JA, Frackelton AR, Jr., Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 112.Miller VM, Mulvagh SL. Sex steroids and endothelial function: translating basic science to clinical practice. Trends Pharmacol. Sci. 2007;28(6):263–270. doi: 10.1016/j.tips.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 113.Kublickiene K, Svedas E, Landgren BM, Crisby M, Nahar N, Nisell H, Poston L. Small artery endothelial dysfunction in postmenopausal women: in vitro function, morphology, and modification by estrogen and selective estrogen receptor modulators. J. Clin. Endocrinol. Metab. 2005;90(11):6113–6122. doi: 10.1210/jc.2005-0419. [DOI] [PubMed] [Google Scholar]
- 114.Kauser K, Rubanyi GM. Gender difference in endothelial dysfunction in the aorta of spontaneously hypertensive rats. Hypertension. 1995;25(4 Pt 1):517–523. doi: 10.1161/01.hyp.25.4.517. [DOI] [PubMed] [Google Scholar]
- 115.Widder J, Pelzer T, von Poser-Klein C, Hu K, Jazbutyte V, Fritzemeier KH, Hegele-Hartung C, Neyses L, Bauersachs J. Improvement of endothelial dysfunction by selective estrogen receptor-alpha stimulation in ovariectomized SHR. Hypertension. 2003;42(5):991–996. doi: 10.1161/01.HYP.0000098661.37637.89. [DOI] [PubMed] [Google Scholar]
- 116.Nilsson BO, Ekblad E, Heine T, Gustafsson JA. Increased magnitude of relaxation to oestrogen in aorta from oestrogen receptor beta knock-out mice. J. Endocrinol. 2000;166(2):R5–R9. doi: 10.1677/joe.0.166r005. [DOI] [PubMed] [Google Scholar]
- 117.Darblade B, Pendaries C, Krust A, Dupont S, Fouque MJ, Rami J, Chambon P, Bayard F, Arnal JF. Estradiol alters nitric oxide production in the mouse aorta through the alpha-, but not beta-, estrogen receptor. Circ. Res. 2002;90(4):413–419. doi: 10.1161/hh0402.105096. [DOI] [PubMed] [Google Scholar]
- 118.Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J. Biol. Chem. 2005;280(20):19704–19710. doi: 10.1074/jbc.M501244200. [DOI] [PubMed] [Google Scholar]
- 119.Barchiesi F, Jackson EK, Gillespie DG, Zacharia LC, Fingerle J, Dubey RK. Methoxyestradiols mediate estradiol-induced antimitogenesis in human aortic SMCs. Hypertension. 2002;39(4):874–879. doi: 10.1161/01.hyp.0000013863.25970.ba. [DOI] [PubMed] [Google Scholar]
- 120.Koledova VV, Khalil RA. Sex hormone replacement therapy and modulation of vascular function in cardiovascular disease. Expert Rev. Cardiovasc. Ther. 2007;5(4):777–789. doi: 10.1586/14779072.5.4.777. [DOI] [PubMed] [Google Scholar]
- 121.Nelson SH, Steinsland OS, Wang Y, Yallampalli C, Dong YL, Sanchez JM. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ. Res. 2000;87(5):406–411. doi: 10.1161/01.res.87.5.406. [DOI] [PubMed] [Google Scholar]
- 122.Rahimian R, Chan L, Goel A, Poburko D, van Breemen C. Estrogen modulation of endothelium-derived relaxing factors by human endothelial cells. Biochem. Biophys. Res. Commun. 2004;322(2):373–379. doi: 10.1016/j.bbrc.2004.07.137. [DOI] [PubMed] [Google Scholar]
- 123.Knot HJ, Lounsbury KM, Brayden JE, Nelson MT. Gender differences in coronary artery diameter reflect changes in both endothelial Ca2+ and ecNOS activity. Am. J. Physiol. 1999;276(3 Pt 2):H961–H969. doi: 10.1152/ajpheart.1999.276.3.H961. [DOI] [PubMed] [Google Scholar]
- 124.Kähönen M, Tolvanen JP, Sallinen K, Wu X, Pörsti I. Influence of gender on control of arterial tone in experimental hypertension. Am. J. Physiol. 1998;275(1 Pt 2):H15–H22. doi: 10.1152/ajpheart.1998.275.1.H15. [DOI] [PubMed] [Google Scholar]
- 125.Egami R, Tanaka Y, Nozaki M, Koera K, Okuma A, Nakano H. Chronic treatment with 17beta-estradiol increases susceptibility of smooth muscle cells to nitric oxide. Eur. J. Pharmacol. 2005;520(1-3):142–149. doi: 10.1016/j.ejphar.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 126.Ba ZF, Lu A, Shimizu T, Szalay L, Schwacha MG, Rue LW, 3rd., Bland KI, Chaudry IH. 17beta-Estradiol modulates vasoconstriction induced by endothelin-1 following trauma-hemorrhage. Am. J. Physiol. Heart Circ. Physiol. 2007;292(1):H245–H250. doi: 10.1152/ajpheart.00809.2006. [DOI] [PubMed] [Google Scholar]
- 127.Hernández I, Delgado JL, Díaz J, Quesada T, Teruel MJ, Llanos MC, Carbonell LF. 17beta-estradiol prevents oxidative stress and decreases blood pressure in ovariectomized rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;279(5):R1599–R1605. doi: 10.1152/ajpregu.2000.279.5.R1599. [DOI] [PubMed] [Google Scholar]
- 128.Case J, Davison CA. Estrogen alters relative contributions of nitric oxide and cyclooxygenase products to endothelium-dependent vasodilation. J. Pharmacol. Exp. Ther. 1999;291(2):524–530. [PubMed] [Google Scholar]
- 129.Calkin AC, Sudhir K, Honisett S, Williams MR, Dawood T, Komesaroff PA. Rapid potentiation of endothelium-dependent vasodilation by estradiol in postmenopausal women is mediated via cyclooxygenase 2. J. Clin. Endocrinol. Metab. 2002;87(11):5072–5075. doi: 10.1210/jc.2002-020057. [DOI] [PubMed] [Google Scholar]
- 130.O'Sullivan MG, Goodrich JA, Adams MR. Increased prostacyclin synthesis by atherosclerotic arteries from estrogen-treated monkeys. Life Sci. 2001;69(4):395–401. doi: 10.1016/s0024-3205(01)01131-6. [DOI] [PubMed] [Google Scholar]
- 131.Egan KM, Lawson JA, Fries S, Koller. B.; Rader DJ, Smyth EM, Fitzgerald GA. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306(5703):1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- 132.Barber DA, Miller VM. Gender differences in endothelium-dependent relaxations do not involve NO in porcine coronary arteries. Am. J. Physiol. 1997;273(5 Pt 2):H2325–H2332. doi: 10.1152/ajpheart.1997.273.5.H2325. [DOI] [PubMed] [Google Scholar]
- 133.Jiang CW, Sarrel PM, Lindsay DC, Poole-Wilson PA, Collins P. Endothelium-independent relaxation of rabbit coronary artery by 17 beta-oestradiol in vitro. Br. J. Pharmacol. 1991;104(4):1033–1037. doi: 10.1111/j.1476-5381.1991.tb12545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hertrampf T, Schmidt S, Laudenbach-Leschowsky U, Seibel J, Diel P. Tissue-specific modulation of cyclooxygenase-2 (Cox-2) expression in the uterus and the v. cava by estrogens and phytoestrogens. Mol. Cell Endocrinol. 2005;243(1-2):51–57. doi: 10.1016/j.mce.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 135.Tamura M, Deb S, Sebastian S, Okamura K, Bulun SE. Estrogen up-regulates cyclooxygenase-2 via estrogen receptor in human uterine microvascular endothelial cells. Fertil. Steril. 2004;81(5):1351–1356. doi: 10.1016/j.fertnstert.2003.09.076. [DOI] [PubMed] [Google Scholar]
- 136.Sherman TS, Chambliss KL, Gibson LL, Pace MC, Mendelsohn ME, Pfister SL, Shaul PW. Estrogen acutely activates prostacyclin synthesis in ovine fetal pulmonary artery endothelium. Am. J. Respir. Cell Mol. Biol. 2002;26(5):610–616. doi: 10.1165/ajrcmb.26.5.4528. [DOI] [PubMed] [Google Scholar]
- 137.García-Martínez MC, Hermenegildo C, Tarín JJ, Cano A. Phytoestrogens increase the capacity of serum to stimulate prostacyclin release in human endothelial cells. Acta Obstet. Gynecol. Scand. 2003;82(8):705–710. doi: 10.1080/j.1600-0412.2003.00178.x. [DOI] [PubMed] [Google Scholar]
- 138.Oviedo PJ, Hermenegildo C, Tarín JJ, Cano A. Raloxifene promotes prostacyclin release in human endothelial cells through a mechanism that involves cyclooxygenase-1 and -2. Fertil. Steril. 2005;83(6):1822–1829. doi: 10.1016/j.fertnstert.2004.11.075. [DOI] [PubMed] [Google Scholar]
- 139.Nawate S, Fukao M, Sakuma I, Soma T, Nagai K, Takikawa O, Miwa S, Kitabatake A. Reciprocal changes in endothelium-derived hyperpolarizing factor- and nitric oxide-system in the mesenteric artery of adult female rats following ovariectomy. Br. J. Pharmacol. 2005;144(2):178–189. doi: 10.1038/sj.bjp.0706091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Woodman OL, Boujaoude M. Chronic treatment of male rats with daidzein and 17 beta-oestradiol induces the contribution of EDHF to endothelium-dependent relaxation. Br. J. Pharmacol. 2004;141(2):322–328. doi: 10.1038/sj.bjp.0705603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.David FL, Carvalho MH, Cobra AL, Nigro D, Fortes ZB, Rebouças NA, Tostes RC. Ovarian hormones modulate endothelin-1 vascular reactivity and mRNA expression in DOCA-salt hypertensive rats. Hypertension. 2001;38(3 Pt 2):692–696. doi: 10.1161/01.hyp.38.3.692. [DOI] [PubMed] [Google Scholar]
- 142.Bilsel AS, Moini H, Tetik E, Aksungar F, Kaynak B, Ozer A. 17Beta-estradiol modulates endothelin-1 expression and release in human endothelial cells. Cardiovasc. Res. 2000;46(3):579–584. doi: 10.1016/s0008-6363(00)00046-8. [DOI] [PubMed] [Google Scholar]
- 143.Dubey RK, Jackson EK, Keller PJ, Imthurn B, Rosselli M. Estradiol metabolites inhibit endothelin synthesis by an estrogen receptor-independent mechanism. Hypertension. 2001;37(2 Part 2):640–644. doi: 10.1161/01.hyp.37.2.640. [DOI] [PubMed] [Google Scholar]
- 144.Méthot J, Hamelin BA, Arsenault M, Bogaty P, Plante S, Poirier P. The ACE-DD genotype is associated with endothelial dysfunction in postmenopausal women. Menopause. 2006;13(6):959–966. doi: 10.1097/01.gme.0000243576.09065.93. [DOI] [PubMed] [Google Scholar]
- 145.Lewis DA, Avsar M, Labreche P, Bracamonte M, Jayachandran M, Miller VM. Treatment with raloxifene and 17beta-estradiol differentially modulates nitric oxide and prostanoids in venous endothelium and platelets of ovariectomized pigs. J. Cardiovasc. Pharmacol. 2006;48(5):231–238. doi: 10.1097/01.fjc.0000247800.34991.a1. [DOI] [PubMed] [Google Scholar]
- 146.Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem. Pharmacol. 2005;70(11):1537–1547. doi: 10.1016/j.bcp.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Crews JK, Khalil RA. Gender-specific inhibition of Ca2+ entry mechanisms of arterial vasoconstriction by sex hormones. Clin. Exp. Pharmacol. Physiol. 1999;26(9):707–715. doi: 10.1046/j.1440-1681.1999.03110.x. [DOI] [PubMed] [Google Scholar]
- 148.Crews JK, Khalil RA. Antagonistic effects of 17β-estradiol, progesterone, and testosterone on Ca2+ entry mechanisms of coronary vasoconstriction. Arterioscler. Thromb. Vasc. Biol. 1999;19(4):1034–1040. doi: 10.1161/01.atv.19.4.1034. [DOI] [PubMed] [Google Scholar]
- 149.Murphy JG, Khalil RA. Decreased [Ca2+]i during inhibition of coronary smooth muscle contraction by 17β-estradiol, progesterone, and testosterone. J. Pharmacol. Exp. Ther. 1999;291(1):44–52. [PubMed] [Google Scholar]
- 150.Zhang F, Ram JL, Standley PR, Sowers JR. 17β-Estradiol attenuates voltage-dependent Ca2+ currents in A7r5 vascular smooth muscle cell line. Am. J. Physiol. 1994;266(4 Pt 1):C975–C980. doi: 10.1152/ajpcell.1994.266.4.C975. [DOI] [PubMed] [Google Scholar]
- 151.Murphy JG, Khalil RA. Gender-specific reduction in contractility and [[Ca2+]i in vascular smooth muscle cells of female rat. Am. J. Physiol. Cell Physiol. 2000;278(4):C834–C844. doi: 10.1152/ajpcell.2000.278.4.C834. [DOI] [PubMed] [Google Scholar]
- 152.Kanashiro CA, Khalil RA. Gender-related distinctions in protein kinase C activity in rat vascular smooth muscle. Am. J. Physiol. Cell Physiol. 2001;280(1):C34–C45. doi: 10.1152/ajpcell.2001.280.1.C34. [DOI] [PubMed] [Google Scholar]
- 153.Hiroki J, Shimokawa H, Mukai Y, Ichiki T, Takeshita A. Divergent effects of estrogen and nicotine on Rho-kinase expression in human coronary vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2005;326(1):154–159. doi: 10.1016/j.bbrc.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 154.Shimokawa H, Takeshita A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2005;25(9):1767–1775. doi: 10.1161/01.ATV.0000176193.83629.c8. [DOI] [PubMed] [Google Scholar]
- 155.Chrissobolis S, Budzyn K, Marley PD, Sobey CG. Evidence that estrogen suppresses rho-kinase function in the cerebral circulation in vivo. Stroke. 2004;35(9):2200–2205. doi: 10.1161/01.STR.0000136951.85586.c8. [DOI] [PubMed] [Google Scholar]
- 156.Simoncini T, Scorticati C, Mannella P, Fadiel A, Giretti MS, Fu XD, Baldacci C, Garibaldi S, Caruso A, Fornari L, Naftolin F, Genazzani AR. Estrogen receptor alpha interacts with Galpha13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol. Endocrinol. 2006;20(8):1756–1771. doi: 10.1210/me.2005-0259. [DOI] [PubMed] [Google Scholar]
- 157.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 2008;75(2):346–359. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lewandowski KC, Komorowski J, Mikhalidis DP, Bienkiewicz M, Tan BK, O'Callaghan CJ, Lewinski A, Prelevic G, Randeva HS. Effects of hormone replacement therapy type and route of administration on plasma matrix metalloproteinases and their tissue inhibitors in postmenopausal women. J. Clin. Endocrinol. Metab. 2006;91(8):3123–3130. doi: 10.1210/jc.2005-2789. [DOI] [PubMed] [Google Scholar]
- 159.McVeigh GE, Hamilton PK, Morgan DR. Evaluation of mechanical arterial properties: clinical, experimental and therapeutic aspects. Clin. Sci. (Lond) 2002;102(1):51–67. [PubMed] [Google Scholar]
- 160.Jani B, Rajkumar C. Ageing and vascular ageing. Postgrad. Med. J. 2006;82(968):357–362. doi: 10.1136/pgmj.2005.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.O'Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a clinical perspective. J. Am. Coll. Cardiol. 2007;50(1):1–13. doi: 10.1016/j.jacc.2006.12.050. [DOI] [PubMed] [Google Scholar]
- 162.Karim R, Hodis HN, Stanczyk FZ, Lobo RA, Mack WJ. Relationship between serum levels of sex hormones and progression of subclinical atherosclerosis in postmenopausal women. J. Clin. Endocrinol. Metab. 2008;93(1):131–138. doi: 10.1210/jc.2007-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lew R, Komesaroff P, Williams M, Dawood T, Sudhir K. Endogenous estrogens influence endothelial function in young men. Circ. Res. 2003;93(11):1127–1133. doi: 10.1161/01.RES.0000103633.57225.BC. [DOI] [PubMed] [Google Scholar]
- 164.Gonzales RJ, Ansar S, Duckles SP, Krause DN. Androgenic/estrogenic balance in the male rat cerebral circulation: metabolic enzymes and sex steroid receptors. J. Cereb. Blood Flow Metab. 2007;27(11):1841–1852. doi: 10.1038/sj.jcbfm.9600483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pritchard KI, Abramson BL. Cardiovascular health and aromatase inhibitors. Drugs. 2006;66(13):1727–1740. doi: 10.2165/00003495-200666130-00005. [DOI] [PubMed] [Google Scholar]
- 166.Goletiani NV, Keith DR, Gorsky SJ. Progesterone: review of safety for clinical studies. Exp. Clin. Psychopharmacol. 2007;15(5):427–444. doi: 10.1037/1064-1297.15.5.427. [DOI] [PubMed] [Google Scholar]
- 167.Cox MW, Fu W, Chai H, Paladugu R, Lin PH, Lumsden AB, Yao Q, Chen C. Effects of progesterone and estrogen on endothelial dysfunction in porcine coronary arteries. J. Surg. Res. 2005;124(1):104–111. doi: 10.1016/j.jss.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 168.Wassmann K, Wassmann S, Nickenig G. Progesterone antagonizes the vasoprotective effect of estrogen on antioxidant enzyme expression and function. Circ. Res. 2005;97(10):1046–1054. doi: 10.1161/01.RES.0000188212.57180.55. [DOI] [PubMed] [Google Scholar]
- 169.Nickenig G, Strehlow K, Wassmann S, Bäumer AT, Albory K, Sauer H, Böhm M. Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation. 2000;102(15):1828–1833. doi: 10.1161/01.cir.102.15.1828. [DOI] [PubMed] [Google Scholar]
- 170.Dean SA, Tan J, O'Brien ER, Leenen FH. 17beta-estradiol downregulates tissue angiotensin-converting enzyme and ANG II type 1 receptor in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288(3):R759–R766. doi: 10.1152/ajpregu.00595.2004. [DOI] [PubMed] [Google Scholar]
- 171.Sunday L, Tran MM, Krause DN, Duckles SP. Estrogen and progestagens differentially modulate vascular proinflammatory factors. Am. J. Physiol. Endocrinol. Metab. 2006;291(2):E261–E267. doi: 10.1152/ajpendo.00550.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Register TC, Adams MR, Golden DL, Clarkson TB. Conjugated equine estrogens alone, but not in combination with medroxyprogesterone acetate, inhibit aortic connective tissue remodeling after plasma lipid lowering in female monkeys. Arterioscler. Thromb. Vasc. Biol. 1998;18(7):1164–1171. doi: 10.1161/01.atv.18.7.1164. [DOI] [PubMed] [Google Scholar]
- 173.Adams MR, Register TC, Golden DL, Wagner JD, Williams JK. Medroxyprogesterone acetate antagonizes inhibitory effects of conjugated equine estrogens on coronary artery atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1997;17(1):217–221. doi: 10.1161/01.atv.17.1.217. [DOI] [PubMed] [Google Scholar]
- 174.Allolio B, Arlt W. DHEA treatment: myth or reality? Trends. Endocrinol. Metab. 2002;13(7):288–294. doi: 10.1016/s1043-2760(02)00617-3. [DOI] [PubMed] [Google Scholar]
- 175.Reckelhoff JF. Sex steroids, cardiovascular disease, and hypertension: unanswered questions and some speculations. Hypertension. 2005;45(2):170–174. doi: 10.1161/01.HYP.0000151825.36598.36. [DOI] [PubMed] [Google Scholar]
- 176.Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocr. Rev. 2003;24(3):313–340. doi: 10.1210/er.2003-0005. [DOI] [PubMed] [Google Scholar]
- 177.Reckelhoff JF, Yanes LL, Iliescu R, Fortepiani LA, Granger JP. Testosterone supplementation in aging men and women: possible impact on cardiovascular-renal disease. Am. J. Physiol. Renal Physiol. 2005;289(5):F941–F948. doi: 10.1152/ajprenal.00034.2005. [DOI] [PubMed] [Google Scholar]
- 178.Hinojosa-Laborde C, Lange DL, Haywood JR. Role of female sex hormones in the development and reversal of dahl hypertension. Hypertension. 2000;35(1 Pt 2):484–489. doi: 10.1161/01.hyp.35.1.484. [DOI] [PubMed] [Google Scholar]
- 179.English KM, Steeds RP, Jones TH, Diver MJ, Channer KS. Low-dose transdermal testosterone therapy improves angina threshold in men with chronic stable angina: A randomized, double-blind, placebo-controlled study. Circulation. 2000;102(16):1906–1911. doi: 10.1161/01.cir.102.16.1906. [DOI] [PubMed] [Google Scholar]
- 180.Alexandersen P, Haarbo J, Byrjalsen I, Lawaetz H, Christiansen C. Natural androgens inhibit male atherosclerosis: a study in castrated, cholesterol-fed rabbits. Circ. Res. 1999;84(7):813–819. doi: 10.1161/01.res.84.7.813. [DOI] [PubMed] [Google Scholar]
- 181.Rosano GM, Leonardo F, Pagnotta P, Pelliccia F, Panina G, Cerquetani E, della Monica PL, Bonfigli B, Volpe M, Chierchia SL. Acute anti-ischemic effect of testosterone in men with coronary artery disease. Circulation. 1999;99(13):1666–1670. doi: 10.1161/01.cir.99.13.1666. [DOI] [PubMed] [Google Scholar]
- 182.Webb CM, Adamson DL, de Zeigler D, Collins P. Effect of acute testosterone on myocardial ischemia in men with coronary artery disease. Am. J. Cardiol. 1999;83(3):437–439. doi: 10.1016/s0002-9149(98)00880-7. A9. [DOI] [PubMed] [Google Scholar]
- 183.Jiroutek MR, Chen MH, Johnston CC, Longcope C. Changes in reproductive hormones and sex hormone-binding globulin in a group of postmenopausal women measured over 10 years. Menopause. 1998;5(2):90–94. [PubMed] [Google Scholar]
- 184.Laughlin GA, Barrett-Connor E, Kritz-Silverstein D, von Mühlen D. Hysterectomy, oophorectomy, and endogenous sex hormone levels in older women: the Rancho Bernardo Study. J. Clin. Endocrinol. Metab. 2000;85(2):645–651. doi: 10.1210/jcem.85.2.6405. [DOI] [PubMed] [Google Scholar]
- 185.Quinkler M, Bumke-Vogt C, Meyer B, Bähr V, Oelkers W, Diederich S. The human kidney is a progesterone-metabolizing and androgen-producing organ. J. Clin. Endocrinol. Metab. 2003;88(6):2803–2809. doi: 10.1210/jc.2002-021970. [DOI] [PubMed] [Google Scholar]
- 186.Diamanti-Kandarakis E. Polycystic ovarian syndrome: pathophysiology, molecular aspects and clinical implications. Expert. Rev. Mol. Med. 2008;10(2):e3. doi: 10.1017/S1462399408000598. [DOI] [PubMed] [Google Scholar]
- 187.Pallottini V, Bulzomi P, Galluzzo P, Martini C, Marino M. Estrogen regulation of adipose tissue functions: involvement of estrogen receptor isoforms. Infect. Disord. Drug Targets. 2008;8(1):52–60. doi: 10.2174/187152608784139631. [DOI] [PubMed] [Google Scholar]
- 188.Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic Review. Cancer Epidemiol. Biomarkers Prev. 2002;11(12):1531–1543. [PubMed] [Google Scholar]
- 189.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr. Rev. 2000;21(6):697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 190.Peraldi P, Spiegelman B. TNF-alpha and insulin resistance: summary and future prospects. Mol. Cell Biochem. 1998;182(1-2):169–175. [PubMed] [Google Scholar]
- 191.Bondy CA. Heart disease in Turner syndrome. Minerva Endocrinol. 2007;32(4):245–261. [PubMed] [Google Scholar]
- 192.Gravholt CH. Epidemiological, endocrine and metabolic features in Turner syndrome. Eur. J. Endocrinol. 2004;151(6):657–687. doi: 10.1530/eje.0.1510657. [DOI] [PubMed] [Google Scholar]
- 193.Grodstein F, Stampfer MJ, Falkeborn M, Naessen T, Persson I. Postmenopausal hormone therapy and risk of cardiovascular disease and hip fracture in a cohort of Swedish women. Epidemiology. 1999;10(5):476–480. [PubMed] [Google Scholar]
- 194.Mikkola TS, Clarkson TB. Estrogen replacement therapy, atherosclerosis, and vascular function. Cardiovasc. Res. 2002;53(3):605–619. doi: 10.1016/s0008-6363(01)00466-7. [DOI] [PubMed] [Google Scholar]
- 195.Mori T, Durand J, Chen Y, Thompson JA, Bakir S, Oparil S. Effects of short-term estrogen treatment on the neointimal response to balloon injury of rat carotid artery. Am. J. Cardiol. 2000;85(10):1276–1279. doi: 10.1016/s0002-9149(00)00748-7. [DOI] [PubMed] [Google Scholar]
- 196.Finking G, Krauss N, Römer S, Eckert S, Lenz. C.; Kamenz J, Menke A, Brehme U, Hombach V, Hanke H. 17beta-estradiol, gender independently, reduces atheroma development but not neointimal proliferation after balloon injury in the rabbit aorta. Atherosclerosis. 2001;154(1):39–49. doi: 10.1016/s0021-9150(00)00446-9. [DOI] [PubMed] [Google Scholar]
- 197.Dubey RK, Jackson EK, Gillespie DG, Zacharia LC, Imthurn B, Keller PJ. Clinically used estrogens differentially inhibit human aortic smooth muscle cell growth and mitogen-activated protein kinase activity. Arterioscler. Thromb. Vasc. Biol. 2000;20(4):964–972. doi: 10.1161/01.atv.20.4.964. [DOI] [PubMed] [Google Scholar]
- 198.Geary RL, Adams MR, Benjamin ME, Williams JK. Conjugated equine estrogens inhibit progression of atherosclerosis but have no effect on intimal hyperplasia or arterial remodeling induced by balloon catheter injury in monkeys. J. Am. Coll. Cardiol. 1998;31(5):1158–1164. doi: 10.1016/s0735-1097(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 199.Hodis HN, Mack WJ. Atherosclerosis imaging methods: assessing cardiovascular disease and evaluating the role of estrogen in the prevention of atherosclerosis. Am. J. Cardiol. 2002;89(12A):19E–27E. doi: 10.1016/s0002-9149(02)02407-4. discussion 27E. [DOI] [PubMed] [Google Scholar]
- 200.Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann. Intern. Med. 2000;133(12):933–941. doi: 10.7326/0003-4819-133-12-200012190-00008. [DOI] [PubMed] [Google Scholar]
- 201.Koh KK, Jin DK, Yang SH, Lee SK, Hwang HY, Kang MH, Kim W, Kim DS, Choi IS, Shin EK. Vascular effects of synthetic or natural progestagen combined with conjugated equine estrogen in healthy postmenopausal women. Circulation. 2001;103(15):1961–1966. doi: 10.1161/01.cir.103.15.1961. [DOI] [PubMed] [Google Scholar]
- 202.Wakatsuki A, Okatani Y, Ikenoue N, Fukaya T. Effect of medroxyprogesterone acetate on endothelium-dependent vasodilation in postmenopausal women receiving estrogen. Circulation. 2001;104(15):1773–1778. doi: 10.1161/hc4001.097035. [DOI] [PubMed] [Google Scholar]
- 203.Clarkson TB, Anthony MS, Wagner JD. A comparison of tibolone and conjugated equine estrogens effects on coronary artery atherosclerosis and bone density of postmenopausal monkeys. J. Clin. Endocrinol. Metab. 2001;86(11):5396–5404. doi: 10.1210/jcem.86.11.8021. [DOI] [PubMed] [Google Scholar]
- 204.Williams JK, Anthony MS, Honoré EK, Herrington D.M., Morgan, T.M., Register TC, Clarkson TB. Regression of atherosclerosis in female monkeys. Arterioscler. Thromb. Vasc. Biol. 1995;15(7):827–836. doi: 10.1161/01.atv.15.7.827. [DOI] [PubMed] [Google Scholar]
- 205.Levine RL, Chen SJ, Durand J, Chen YF, Oparil S. Medroxyprogesterone attenuates estrogen-mediated inhibition of neointima formation after balloon injury of the rat carotid artery. Circulation. 1996;94(9):2221–2227. doi: 10.1161/01.cir.94.9.2221. [DOI] [PubMed] [Google Scholar]
- 206.Decensi A, Omodei U, Robertson C, Bonanni B, Guerrieri-Gonzaga A, Ramazzotto F, Johansson H, Mora S, Sandri MT, Cazzaniga M, Franchi M, Pecorelli S. Effect of transdermal estradiol and oral conjugated estrogen on C-reactive protein in retinoid-placebo trial in healthy women. Circulation. 2002;106(10):1224–1228. doi: 10.1161/01.cir.0000028463.74880.ea. [DOI] [PubMed] [Google Scholar]
- 207.Davison S, Davis SR. New markers for cardiovascular disease risk in women: impact of endogenous estrogen status and exogenous postmenopausal hormone therapy. J. Clin. Endocrinol. Metab. 2003;88:2470–2478. doi: 10.1210/jc.2002-021929. [DOI] [PubMed] [Google Scholar]
- 208.Brinton EA, Hodis HN, Merriam GR, Harman SM, Naftolin F. Can menopausal hormone therapy prevent coronary heart disease? Trends Endocrinol. Metab. 2008;19(6):206–212. doi: 10.1016/j.tem.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 209.Harman SM, Brinton EA, Cedars M, Lobo R, Manson JE, Merriam GR, Miller VM, Naftolin F, Santoro N. KEEPS: The Kronos Early Estrogen Prevention Study. Climacteric. 2005;8(1):3–12. doi: 10.1080/13697130500042417. [DOI] [PubMed] [Google Scholar]