

Abstract
We investigated the role of the glial water channel protein aquaporin-4 in brain edema in a mouse model of subarachnoid haemorrhage in which thirty microliters of blood was injected into the basal cisterns. Brain water content, intracranial pressure and neurological score were compared in wildtype and aquaporin-4 null mice. We also measured blood-brain barrier permeability, and the osmotic permeability of the glia limitans, one of the routes of edema elimination. Wildtype and aquaporin-4 null mice had comparable baseline brain water content, intracranial pressure and neurological score. At six hours after blood injection, aquaporin-4 null mice developed more brain swelling than wildtype mice. Brain water content increased by 1.5 ± 0.1 vs. 0.5 ± 0.2 % (Mean ± Standard Error, P < 0.0005) and intracranial pressure by 36 ± 5 vs. 21 ± 3 mmHg (P < 0.05) above pre-injection baseline, and neurological score was worse at 18.0 vs. 24.5 (median, P < 0.05), respectively. Although subarachnoid hemorrhage produced comparable increases in blood-brain barrier permeability in wildtype and aquaporin-4 null mice, aquaporin-4 null mice had a twofold reduction in glia limitans osmotic permeability. We conclude that aquaporin-4 null mice manifest increased brain edema following subarachnoid hemorrhage as a consequence of reduced elimination of excess brain water.
Keywords: Aquaporin-4, astrocyte, brain edema, mouse model, subarachnoid hemorrhage, water channel
Aneurysmal rupture releases blood at high pressure into the cerebrospinal fluid (CSF). Within hours, subarachnoid hemorrhage (SAH) produces brain edema (Claassen et al., 2002, Liu et al., 2007). Severe brain edema is associated with a large CSF blood load, with reduced consciousness at presentation being an independent predictor of poor outcome (Claassen et al., 2002).
Brain edema is detrimental by causing elevation in intracranial pressure (ICP), brain ischemia, herniation, and ultimately death. There are three major types of brain edema, which may coexist to different extents following brain injury (Tait et al., 2008): Vasogenic edema, as seen in brain tumors, results from fluid leak by hydrostatic pressure from the blood into the brain through a damaged blood-brain barrier (BBB). In cytotoxic edema, excess fluid passes from the blood into the brain parenchyma through an intact BBB because of an osmotic gradient across the BBB, as seen in hyponatremia, or astrocyte damage, as seen in ischemic stroke. Obstruction to the flow of CSF produces interstitial oedema, as seen in hydrocephalus. The protein aquaporin-4 (aqp4) plays a key role in vasogenic, cytotoxic and interstitial edema (Tait et al., 2008).
Aqp4 belongs to a family of water channels (Agre et al., 2002) and is the most abundant brain aquaporin. Aqp4 is expressed in the plasma membranes of pericapillary astrocyte foot processes, glia limitans and ependyma and facilitates water flow into and out of the brain (Manley et al., 2000, Papadopoulos et al., 2004, Bloch et al., 2005, Papadopoulos and Verkman, 2005). Aqp4-null (KO) mice have better outcome than wildtype (WT) mice after insults causing cytotoxic edema, such as water intoxication, cerebral ischemia and meningitis (Manley et al., 2000, Papadopoulos and Verkman, 2005). Aqp4 deletion reduces cytotoxic brain swelling by limiting water movement into the brain across an intact BBB. In mouse models of vasogenic edema such as cortical freeze, brain tumor and intracerebral fluid infusion (Papadopoulos et al., 2004), KO mice develop more brain swelling than WT mice because aqp4 facilitates the elimination of edema fluid. Aqp4 also mediates transparenchymal CSF absorption in hydrocephalus (Bloch et al., 2006).
Here we used a mouse model of SAH, which involves injection of autologous blood into the basal cisterns to mimic the high-pressure release of blood into the CSF that occurs in human SAH. We report greater brain swelling and worse clinical outcome in the aqp4 null mice in this model, which was attributed to impaired clearance of excess brain water that accumulated through a disrupted BBB.
Experimental Procedures
Mice
Experiments were approved by the University of California at San Francisco Committee on Animal Research and the British Home Office. We used WT and KO mice with a CD1 genetic background. KO mice have normal blood chemistries, urine output (except for slight defect in maximal urine concentrating ability), body weight, lifespan, temperature, brain anatomy, BBB permeability, ICP and bulk intracranial compliance (Ma et al., 1997, Papadopoulos et al., 2004, Saadoun et al., 2009). We used weight-matched (30 – 35 g) male mice, aged 6 – 8 w. Investigators were blinded to mouse genotype during the experiments. Mice were anesthetized using 2,2,2-tribromoethanol (Avertin, Sigma-Aldrich, Poole, UK) by injecting i.p. a 6.7 ml/kg bolus followed by 1.7 ml/kg every 20 min.
SAH model
We modified an established rodent model of SAH (Prunell et al., 2002, Prunell et al., 2005). Anaesthetized mice were immobilized in a stereotactic frame (myNeurolab, St Louis, MO). Rectal temperature was maintained at 37 – 38 °C using a heat lamp. A midline scalp incision was made and two 0.8 mm diameter burr holes were drilled using a Foredom drill (Bethel, CT), 3 mm left of the bregma on the coronal suture, and immediately posterior to the right vein (Figure 1A). Autologous venous blood (30 µl) was withdrawn from the tail, heparinized, and injected through a 27 G needle inserted through the anterior burr hole at 30° pointing posteriorly. The blood was injected over 7.5 s using a syringe driver (IVAC, San Diego, CA). ICP was monitored through the right-sided burr hole. After injection, the needle was left in situ for 10 min to prevent back flow. Once the needle and ICP monitor were removed, both burr holes were plugged with bone wax (Eastbourne DGH pharmaceuticals, Eastbourne, UK) and the scalp was sutured. In pilot experiments (not shown) we characterized the effect of different blood injection volumes on outcome. We chose a 30 µL injection volume, which was best for demonstrating the difference in brain edema between KO and WT mice. When 40 or 50 µL of blood was injected, most KO mice died thus precluding accurate comparisons of brain edema in WT vs. KO mice.
Figure 1. Mouse SAH model.

A. (Top) Mouse calvarium. Burr hole 1 is for injection and burr hole 2 for ICP monitoring. (Bottom) The injecting needle is advanced 30° to the perpendicular into the basal cisterns. B. Exposed dura overlying cisterna magna and CSF at T6 before and after injecting blood into the basal cisterns. C. ICP traces. Arrowheads show onset of blood injection. Inset 1: prominent respiratory waves. Inset 2: cardiac ICP pulsations. D. Coronal brain section at 2.8 mm from the frontal poles stained with hematoxylin-eosin showing normal-sized 3rd ventricle 24 h after sham injection or SAH. Bar = 0.5 mm. E. Brain sections 24 h after needle insertion (Sham) or SAH. Mice had i.v. injection of 10 kDa rhodamine-dextran (red) and 2 MDa FITC-dextran (green). Bar = 50 µm.
ICP measurements
A 0.8 mm diameter burrhole was made 2 mm laterally on the left coronal suture (Fig. 1A) and an ICP monitoring micro-probe (SPC-320; 600 µm diameter; Millar, Houston, TX) was inserted intraparenchymally 2 mm deep from the brain surface. ICP recordings were taken using a computer interface (MP 35; Biopac, Santa Barbara, CA) throughout the injection of blood. Mice also had ICP monitoring for 10 min at 6 or 24 h after surgery.
Brain water content
Mice were killed by anaesthetic overdose and the brain was rapidly removed. Brains were weighed (w1), placed in an oven (95 °C, 48 h) and reweighed (w2). Percent brain water content was computed as 100 × [1 – (w1 – w2) / w1]. Comparisons between KO and WT mice were made at 6 and 24 h after SAH.
Neurological assessment
We modified a standard neurological scale (5 – 27 points), previously used for assessing mice after SAH (Parra et al., 2002), summarized in Fig. 3A.
Figure 3. Neurological score after SAH.

A. Neurological scale. B. Neurological score at 6 and 24 h after needle insertion or SAH in WT and KO mice. ● = WT (+/+), ○ = KO (−/−), X = dead mice. — = median. *P < 0.05 for WT vs. KO.
BBB permeability
Qualitative and quantitative studies were done to assess BBB permeability, which is associated with vasogenic edema formation. For qualitative studies 2 MDa FITC-dextran and 10 kDa Rhodamine-dextran (0.2 mL of 25 mg/ml in 0.9 % saline each, Sigma-Aldrich, Poole, UK) were injected into the internal jugular vein. After 30 min the mice were killed, the brains were extracted, fixed in formalin, cryoprotected in 30 % sucrose and embedded in OCT (Raymond Lamb, Eastbourne, UK). Frozen sections (7 µm thick) of cerebral cortex distant from the needle and ICP monitoring sites was visualized using a BX-51 Olympus microscope.
The Evans blue dye (EBD) assay was used as a quantitative measure of BBB opening. Mice were anaesthetized and EBD (4% in saline, 160 mg/kg; Sigma-Aldrich, Poole, UK) was injected into the right jugular vein. After 30 min the left cardiac ventricle was perfused with 30 ml 0.9 % saline. Brains were removed and placed in 2 ml N,N-dimethylformamide (Sigma-Aldrich, Poole, UK) at 55 °C overnight. The concentration of extracted EBD in formamide was calculated by comparing optical absorbance at 610 nm against EBD-formamide standards.
Glia limitans osmotic permeability
One of the routes of edema elimination is across the aqp4-rich glia limitans into the CSF (Reulen et al., 1977, Marmarou et al., 1994). To estimate glia limitans osmotic permeability, a right-sided craniectomy was performed. Craniectomy margins were the coronal suture anteriorly, neck muscles posteriorly, sagittal suture medially, and 4 mm from the midline laterally. After carefully removing the dura, a cylinder (5 mm diameter, 3 mm high) of 4 % agarose in water (hypotonic) or normal saline (isotonic) was placed over the brain surface for 30 min, in a humid atmosphere. Mice were killed and the % water content of the right minus left cerebral hemisphere was determined.
Histology
Brains were fixed in formalin, dehydrated in alcohols, and embedded in paraffin. Sections (7 µm thick) were deparaffinized, unmasked in citrate and exposed to a polyclonal rabbit anti-rat aqp4 antibody (Millipore-Chemicon, Watford, UK) followed by biotinylated goat anti-rabbit antibody (Vector Labs, Peterborough, UK) and streptavidin-HRP. Aqp4 immunoreactivity was visualized brown with DAB/H2O2 and was graded by consensus by two observers who were unaware of the experimental condition associated with each specimen. Grading was 0 = no aqp4; + = aqp4 in glia limitans and around vessels; ++ = high levels of aqp4 in glia limitans, perivascularly and adjacent brain parenchyma. To visualize the ventricles, coronal brain sections at 2.8 mm from the frontal poles were stained with hematoxylin-eosin.
Statistical analysis
Parametric data, presented as Mean ± SEM, were compared using the Student t test. Non-parametric data, shown as Median, were compared using the Mann Witney U test. WinSTAT (v2001.1, A-Prompt, Lehigh Valley, PA) was used. Significance was taken at P < 0.05.
Results
SAH model
Fig. 1A shows the experimental setup including the injection (burr hole 1) and ICP monitoring (burr hole 2) burr holes, needle tract, and injection site (basal cisterns). Before injecting the blood, the cisternal and spinal CSF was colorless. Within 5 min of the blood injection, blood appeared in the cisterna magna and around the spinal cord (Fig. 1B). No blood was seen in the CSF when saline was injected (not shown). Three mice were killed 10 min after blood injection and coronal brain sections were cut through the needle tract – no intraparenchymal hematoma was detected (not shown). Fig. 1C shows representative ICP traces from two mice. Peri-injection ICP was monitored in 22 WT vs. 22 KO mice and is presented as mean ± SEM. Pre-injection ICP was 7.6 ± 2.7 vs. 6.8 ± 3.6 mmHg (difference not significant). Blood injection caused comparable ICP changes in WT and KO mice. Peak injection pressures were 94 ± 6 vs. 96 ± 5 mmHg, respectively (difference not significant). Following the initial rapid rise, the ICP stabilized within 2 min at 26 ± 3 vs. 30 ± 3 mmHg, respectively (difference not significant). During blood injection, respiratory rate became slower and breaths were deeper, producing large spikes on the ICP trace (Fig. 1C, Mouse 1 inset). Breathing normalized as the ICP dropped towards the plateau. ICP had characteristic cardiac fluctuations (Fig. 1C, Mouse 2 inset).
Because hydrocephalus may occur after SAH, we examined the ventricles in 3 WT mice 24 h after needle insertion (Sham) vs. 3 WT mice 24 h after SAH. The lateral ventricles were 0.07 ± 0.02 vs. 0.06 ± 0.02 mm2 and the 3rd ventricle was 0.15 ± 0.03 vs. 0.14 ± 0.02 mm2 in coronal brain sections at 1.6 mm from the frontal poles (differences not significant) (Fig. 1D). Leakage of fluorescent dextrans across the BBB was used to qualitatively detect microvascular damage (Fig. 1E). In WT and KO mice at 24 h after SAH, the 2 MDa dextran remained within the capillaries whereas the 10 kDa dextran entered the brain parenchyma throughout both cerebral hemispheres. These findings indicate comparable BBB disruption in WT and KO mice at 24 h after SAH. There was no leakage of 2 MDa (green) or 10 kDa (red) dextran from the vasculature into the brain parenchyma at 24 h after needle insertion without injection in WT or KO mice. WT and AQP4−/− mice also had comparable serum sodium concentrations at baseline, 6 h and 24 h after SAH (data not shown).
Brain edema
Brain edema was quantified by measuring brain water content (Fig. 2A). There was no difference in whole brain water content between WT and KO mice 6 h or 24 h after needle insertion. Injection of blood into the subarachnoid space caused an increase in brain water content at 6 h by 0.8 % in WT and 1.8 % in KO mice above baseline. Six hours after injecting 30 µL normal saline into the subarachnoid space, there was a significant (P < 0.05) increase in brain water content of 0.7 ± 0.1 % (mean ± SEM) compared with needle insertion in 3 KO mice, with no brain edema in 3 WT mice. The saline was injected with the same ICP profile as the blood injection. Within 24 h of injecting blood into the subarachnoid space 3/10 KO mice died. By 24 h, mean brain water content in 10 WT mice was almost normal, whereas 7 KO mice still had markedly increased mean brain water content of 1.5 %, above baseline.
Figure 2. Brain edema after SAH.

A. Whole brain water content measured at 6 and 24 h after needle insertion or SAH. B. (Top) Typical ICP recordings from 2 WT and 2 KO mice at 6 h after SAH and (Bottom) summary of ICP data measured at 6 and 24 h after needle insertion or SAH. ● = WT (+/+), ○ = KO (−/−), X = dead mice. ■ = Mean of WT, □ = Mean of KO, Error lines = ±SEM. *P < 0.05, **P < 0.01, #P < 0.0005 for WT vs. KO.
ICP traces from 2 KO and 2 WT mice recorded at 6 h after SAH are shown in Fig. 2B (top) and summary of the ICP data is in Fig. 2B (bottom). Baseline ICP at 6 and 24 h after needle insertion without infusion was comparable in WT and KO mice. At 6 h after SAH, mean ICP was significantly higher than baseline by 19 mmHg in 10 WT vs. 32 mmHg in 10 KO mice. At 24 h after SAH, mean ICP was also significantly higher than baseline by 16 mmHg in 10 WT vs. 29 mmHg in 7 KO mice. By 24 h after SAH, 3/10 KO mice died and were thus excluded from the ICP measurements.
Neurological assessment
Fig. 3A summarizes the neurological scale used. Normal neurological scores were found in 3 WT and 3 KO mice at 6 and 24 h after needle insertion without injection. SAH reduced the neurological scores of 12 WT and 12 KO mice at 6 h (Fig. 3B). Compared with the WT mice, the KO mice had significantly lower neurological scores at 6 h. At 24 h after SAH, we compared 10 WT vs. 7 KO mice (because 3/10 KO mice had died) and found that the KO mice had significantly lower neurological scores. No focal neurological signs, such as hemiparesis, were observed in any mouse.
Edema formation
BBB opening, the hallmark of vasogenic edema formation, was quantified using EBD. There is no baseline difference in EBD extravasation between WT and KO mice (Papadopoulos et al., 2004). At 24 h after SAH, WT and KO mice had 2 – 3×more EBD extravasation into the brain parenchyma than control mice that had intracerebral needle insertion without injection (Fig. 4A). There was no difference in EBD extravasation between WT and KO mice at 24 h after SAH. Comparable increase in EBD extravasation in WT vs. KO mice was also found at 6 h after SAH (not shown).
Figure 4. Blood-brain barrier and glia limitans.

A. (Left) Mouse brains in formamide. WT (+/+) and KO (−/−) mice had needle insertion (Sham) or SAH. (Right) Summary of Evans blue data. N = 4 (Sham, 2 +/+ and 2 −/−), 3 (SAH, +/+), 3 (SAH, −/− ). Mean ± SEM. *P < 0.05 compared with sham. B. Setup for measuring glia limitans osmotic permeability. C. Difference in % water content between right and left hemispheres (ΔH2O) in WT (●, +/+) and KO (○, −/−) mice exposed to 4 % agarose gel in water (hypotonic) or 0.9 % saline (isotonic). ■ = Mean of WT, □ = Mean of KO, Error lines = ±SEM. *P < 0.02.
Edema elimination
The technique for estimating glia limitans osmotic permeability is illustrated in Fig. 4B and the data are summarized in Fig. 4C. We computed ΔH2O, the % water content of the right minus left hemisphere. Without an osmotic gradient across the glia limitans, by using 4 % agarose in isotonic saline, there was no difference in ΔH2O between 4 WT vs. 4 KO mice. An osmotic gradient across the glia limitans, produced using 4 % agarose in distilled water (hypotonic), caused a 2 – 3 fold increase in ΔH2O above baseline in WT, with no corresponding change in KO mice. Since the brain surface is covered with blood at 6 h and 24 h after SAH, it impossible to measure water elimination across the glia limitans by placing an agarose disc on the brain surface at these time points.
AQP4 expression
Three patterns of aqp4 immunoreactivity were seen (Fig. 5A): No aqp4 (0), aqp4 around blood vessels and in glia limitans (+), prominent aqp4 perivascularly, parenchymally and in glia limitans (++). There was increased aqp4 immunoreactivity throughout the brain in 3 WT mice 24 h after SAH, but not in 3 WT mice 24 h after needle insertion (Fig. 5B). Aqp4 upregulation after SAH was more marked in the basal cerebral cortex, including the glia limitans, around microvessels and, in a punctate pattern, throughout the cerebral cortex. Aqp4 upregulation at 24 h after SAH was less prominent in the superior cerebral cortex. Aqp4 immunoreactivity was also increased in 3 WT mice at 6 h after SAH in a similar distribution to the increased expression that was seen at 24 h (Fig. 5B). No aqp4 was detected in 3 KO mice (Fig. 5B) or when the primary antibody was omitted in WT mice (not shown).
Figure 5. Aqp4 immunoreactivity after SAH.
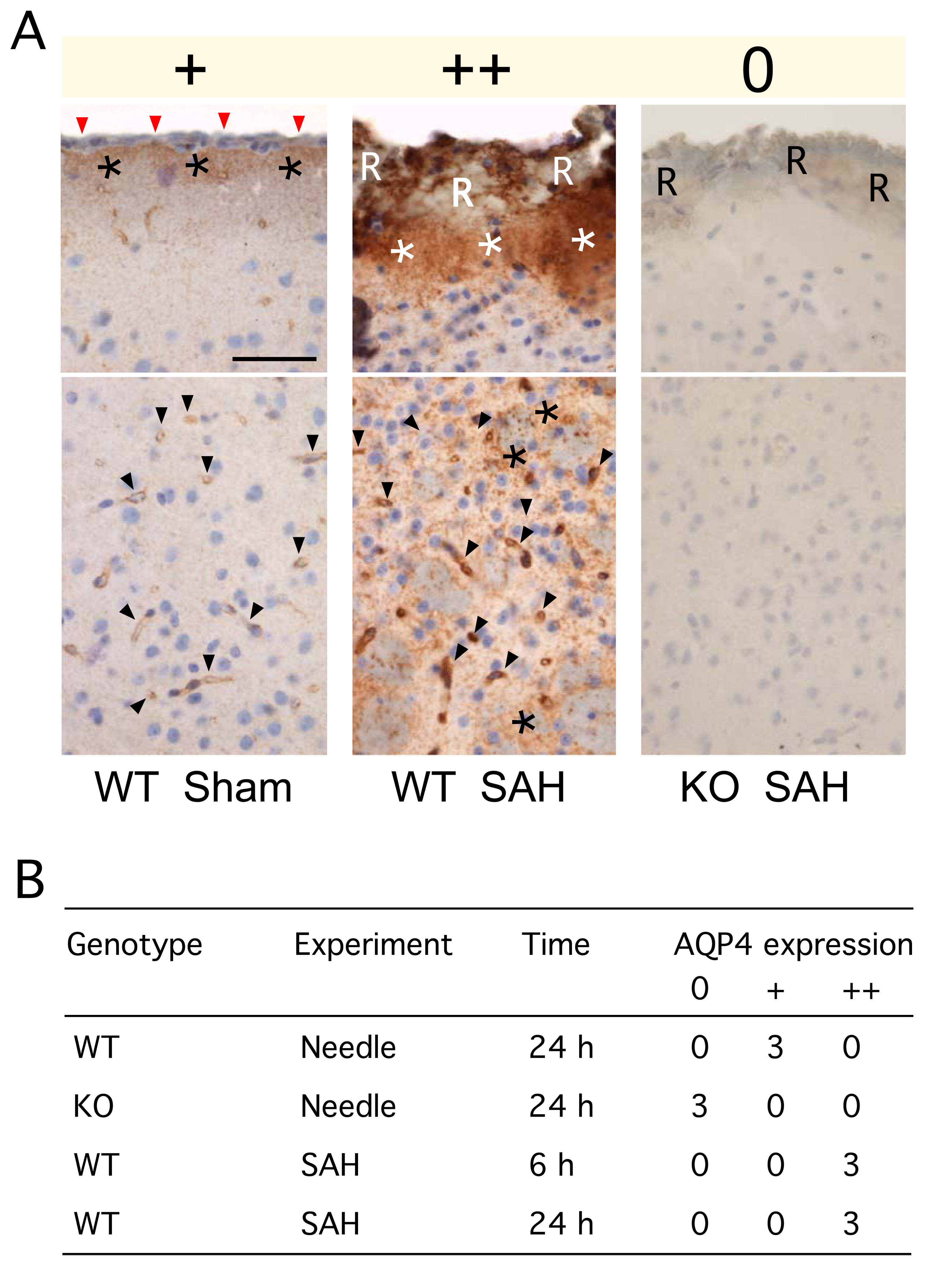
A. (top) Glia limitants and (bottom) basal cerebral cortex. In the sham WT mouse (+, left), red arrows indicate the pia, asterisks the glia limitants, and arrowheads show microvessels. In the WT mouse 24 h after SAH (++, middle), ‘R’ indicates red blood cells overlying the pia, white asterisks the subpial region, arrowheads show microvessels, and black asterisks show aqp4 immunoreactivity in the brain parenchyma. In the KO mouse 24 h after SAH (0, right), ‘R’ indicates red blood cells covering the pia. Bar = 50 µm. B. Summary of immunohistochemical staining, graded as 0, +, ++ as described in A.
Discussion
Our principal finding is that aqp4 KO mice develop more brain edema after experimental SAH than WT mice. KO mice had increased brain water content, higher ICP and lower neurological score than WT mice at 6 and 24 h after SAH. The data also suggest that aqp4 reduces brain edema after SAH by facilitating the elimination of brain edema fluid.
Normal saline injection into the subarachnoid space did not alter the brain water content in WT mice at 6 h, but increased brain water content in KO mice by 0.7 %. It is likely that the high-pressure injection forced some normal saline into the brain extracellular space thus causing some interstitial edema. The WT mice were able to eliminate this excess fluid within 6 h but the KO mice were not, thus explaining the 0.7 % rise in brain water content in KO but not in WT mice. The high-pressure entry of fluid into the CSF only produces part of the brain edema seen in SAH at 6 h. We detected extravasation of intravascular macromolecules, including 10 kDa dextran and EBD, into the brain parenchyma after SAH, which indicates increased BBB permeability. We, therefore, suggest that the remaining brain edema forms by fluid extravasation from the blood into the brain through the damaged BBB. It is well established that vasogenic cerebral edema forms through an aqp4-independent route (Tait et al., 2008). Since WT and KO mice had similar blood injection pressure profile and subsequent BBB opening, we propose that the formation rate of brain edema is the same in WT and KO mice.
The elimination of vasogenic brain edema occurs through three routes where aqp4 is highly expressed: the glia limitans, ependyma, and BBB (Tait et al., 2008). KO mice have reduced BBB osmotic permeability evidenced by slowed brain swelling after acute water intoxication in KO vs. WT mice (Manley et al., 2000, Papadopoulos and Verkman, 2005). Reduced water permeability of the ependyma in KO mice is evidenced by slower water clearance from the ventricles into the brain in obstructive hydrocephalus in KO mice (Bloch et al., 2006). Reduced osmotic permeability of the glia limitans in KO mice was shown here using an agarose gel technique. Taken together, these findings suggest reduced edema elimination in KO mice via all elimination routes – glia limitans (present study), ependyma (Bloch et al., 2006) and capillaries (Manley et al., 2000, Papadopoulos and Verkman, 2005).
Our conclusions are based on a mouse SAH model that involves using a syringe driver to reproducibly inject blood into the basal cisterns at high pressure with no intraparenchymal hematoma formation. The brain around the needle prevented ‘back-bleeding’. This model, based on a previous rat model of SAH (Prunell et al., 2002, Prunell et al., 2005), allowed us to standardize two key parameters, injection pressure and injection volume. Other rodent SAH models include endovascular perforation of the basilar (Barry et al., 1979) or internal carotid arteries (Veelken et al., 1995), or cisterna magna injection of autologous blood (Solomon et al., 1985). Endovascular perforation does not allow control of injected blood volume or ICP and cisterna magna injection does not deliver blood into the correct anatomical site (basal cisterns) and has the key problem of back-bleeding when high-pressure injections are attempted. The severity of human SAH is often graded between 1 and 5 using the WFNS clinical scale. Our chosen mouse model produced little neurological deficit in WT mice at 24 h and may thus be relevant to a good WFNS grade (1 or 2) in humans. Increasing the volume of blood injected, increases the severity of SAH and may make the mouse model relevant to poor WFNS grades (3 – 5).
Aqp4 was upregulated in WT mice at 6 h and 24 h after SAH at key sites of edema elimination including the glia limitans and pericapillary astrocyte processes (Fig. 5). Increased aqp4 immunoreactivity was also found in human brain after SAH (Badaut et al., 2003, Saadoun et al., 2003). We suggest that aqp4 upregulation is an adaptive response that increases the rate of edema elimination after SAH.
We conclude that aqp4 deletion increases SAH-induced brain edema, most likely by reducing edema fluid elimination from the brain. Therefore, drugs that activate or upregulate AQP4 may reduce SAH-associated brain edema.
Acknowledgments
Funded by the Royal College of Surgeons of England (MJT), London Deanery (MJT), Harrison Fund (SS), Neurosciences Research Foundation (MCP, BAB), National Institutes of Health (ASV) and the Guthy-Jackson Foundation (ASV, MCP, SS).
Abbreviations
- aqp4
Aquaporin-4 protein
- aqp4
Aquaporin-4 gene
- BBB
Blood-brain barrier
- CSF
Cerebrospinal fluid
- DAB
Diaminobenzidine
- EBD
Evans Blue Dye
- FITC
Fluoroisothiocyanate
- HRP
Horseradish peroxidase
- ICP
Intracranial pressure
- i.p.
Intraperitoneal
- KO
Aquaporin-4 null (knockout)
- SAH
Subarachnoid hemorrhage
- SEM
Standard error of the mean
- WT
Wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, Engel A, Nielsen S. Aquaporin water channels--from atomic structure to clinical medicine. J Physiol. 2002;542:3–16. doi: 10.1113/jphysiol.2002.020818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Brunet JF, Grollimund L, Hamou MF, Magistretti PJ, Villemure JG, Regli L. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir Suppl. 2003;86:495–498. doi: 10.1007/978-3-7091-0651-8_101. [DOI] [PubMed] [Google Scholar]
- Barry KJ, Gogjian MA, Stein BM. Small animal model for investigation of subarachnoid hemorrhage and cerebral vasospasm. Stroke. 1979;10:538–541. doi: 10.1161/01.str.10.5.538. [DOI] [PubMed] [Google Scholar]
- Bloch O, Auguste KI, Manley GT, Verkman AS. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J Cereb Blood Flow Metab. 2006;26:1527–1537. doi: 10.1038/sj.jcbfm.9600306. [DOI] [PubMed] [Google Scholar]
- Bloch O, Papadopoulos MC, Manley GT, Verkman AS. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J Neurochem. 2005;95:254–262. doi: 10.1111/j.1471-4159.2005.03362.x. [DOI] [PubMed] [Google Scholar]
- Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33:1225–1232. doi: 10.1161/01.str.0000015624.29071.1f. [DOI] [PubMed] [Google Scholar]
- Liu Y, Soppi V, Mustonen T, Kononen M, Koivisto T, Koskela A, Rinne J, Vanninen RL. Subarachnoid hemorrhage in the subacute stage: elevated apparent diffusion coefficient in normal-appearing brain tissue after treatment. Radiology. 2007;242:518–525. doi: 10.1148/radiol.2422051698. [DOI] [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Marmarou A, Hochwald G, Nakamura T, Tanaka K, Weaver J, Dunbar J. Brain edema resolution by CSF pathways and brain vasculature in cats. Am J Physiol. 1994;267:H514–H520. doi: 10.1152/ajpheart.1994.267.2.H514. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. Faseb J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem. 2005;280:13906–13912. doi: 10.1074/jbc.M413627200. [DOI] [PubMed] [Google Scholar]
- Parra A, McGirt MJ, Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Mouse model of subarachnoid hemorrhage associated cerebral vasospasm: methodological analysis. Neurol Res. 2002;24:510–516. doi: 10.1179/016164102101200276. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Mathiesen T, Svendgaard NA. A new experimental model in rats for study of the pathophysiology of subarachnoid hemorrhage. Neuroreport. 2002;13:2553–2556. doi: 10.1097/00001756-200212200-00034. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Svendgaard NA, Alkass K, Mathiesen T. Inflammation in the brain after experimental subarachnoid hemorrhage. Neurosurgery. 2005;56:1082–1092. discussion 1082–1092. [PubMed] [Google Scholar]
- Reulen HJ, Graham R, Spatz M, Klatzo I. Role of pressure gradients and bulk flow in dynamics of vasogenic brain edema. J Neurosurg. 1977;46:24–35. doi: 10.3171/jns.1977.46.1.0024. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Krishna S. Water transport becomes uncoupled from K+ siphoning in brain contusion, bacterial meningitis, and brain tumours: immunohistochemical case review. J Clin Pathol. 2003;56:972–975. doi: 10.1136/jcp.56.12.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Tait MJ, Reza A, Davies DC, Bell BA, Verkman AS, Papadopoulos MC. AQP4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience. 2009 doi: 10.1016/j.neuroscience.2009.03.069. [DOI] [PubMed] [Google Scholar]
- Solomon RA, Antunes JL, Chen RY, Bland L, Chien S. Decrease in cerebral blood flow in rats after experimental subarachnoid hemorrhage: a new animal model. Stroke. 1985;16:58–64. doi: 10.1161/01.str.16.1.58. [DOI] [PubMed] [Google Scholar]
- Tait MJ, Saadoun S, Bell BA, Papadopoulos MC. Water movements in the brain: role of aquaporins. Trends Neurosci. 2008;31:37–43. doi: 10.1016/j.tins.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Veelken JA, Laing RJ, Jakubowski J. The Sheffield model of subarachnoid hemorrhage in rats. Stroke. 1995;26:1279–1283. doi: 10.1161/01.str.26.7.1279. discussion 1284. [DOI] [PubMed] [Google Scholar]