

Abstract
Blood incompatibility reactions caused by surfaces often involve platelet activation and subsequent platelet-initiated activation of the coagulation and complement cascades. The goal of this study was to immobilize apyrase on a biomaterial surface in order to develop an enzymatically active surface that would have the capacity to inhibit platelet activation by degrading ADP. We were able to immobilize apyrase on a polystyrene surface with preservation of the enzymatic activity. We then analyzed the hemocompatibility of the apyrase surface and of control surfaces by incubation with platelet-rich plasma (PRP) or whole blood. Monitoring of markers of platelet, coagulation, and complement activation and staining of the surfaces revealed decreased levels of platelet and coagulation activation parameters on the apyrase surface. The formation of antithrombin-thrombin and antithrombin-factor XIa complexes and the extent of platelet consumption were significantly lower on the apyrase surface than on any of the control surfaces. No significant differences were seen in complement activation (C3a levels). Staining of the apyrase surface revealed low platelet adherence and no formation of granulocyte-platelet complexes. These results demonstrate that it is possible to create an anti-thrombotic surface targeting the ADP amplification of platelet activation by immobilizing apyrase.
1. Introduction
Introducing a biomaterial into the blood circulation or extracorporeal circulation is associated with blood incompatibility reactions. The foreign surface triggers activation of the cascade systems of the blood (the complement, contact, and coagulation systems), which leads to activation of platelets and leukocytes. These events initiate inflammation and occasionally lead to thrombotic complications [1, 2]. To minimize these incompatibility reactions and to prolong the durability of implanted devices, a number of materials and surface coatings have been investigated in an attempt to improve the outcome of treatments. However, there is still no totally blood-compatible artificial material with the ideal blood-compatibility properties of the endothelial lining.
Platelets are intimately involved in the incompatibility processes that occur on foreign surfaces. They are directly activated by the surface itself as a secondary response to activation of the coagulation and complement cascade systems [3, 4]. Platelets are closely connected to the coagulation cascade and can also interact with leukocytes and trigger activation of the complement system, thereby acting as an important hub that mediates the crosstalk between these components [4, 5]. Upon platelet activation, a multi-step process begins that involves adhesion, aggregation, contraction, and secretion. ADP is released from platelet-dense granules and acts as a paracrine activator of platelets in the vicinity; thus, it is essential for recruitment and aggregation of platelets. For this reason, ADP is an obvious target for platelet inhibition [6]. To regulate platelet activation under conditions of homeostasis, vascular ADP is continuously degraded by CD39 (NTPDase-1, EC 3.6.1.5), an enzyme present on endothelial cells. This process is an essential means of maintaining platelets in a resting state under physiological conditions [7].
CD39 belongs to the family of apyrases, enzymes that share the same catalytic mechanism for degrading triphospho- and diphosphonucleosides to their equivalent monophosphonucleosides. Several hematophagous arthropods express soluble apyrases in their mouthparts for this purpose. By excreting this enzyme, they can feed on their hosts’ blood without having blood-clotting problems. The ADP depletion is postulated to be an essential factor in the host-parasite interplay [8]. Both the CD39 and the arthropod variants of apyrase have been expressed in recombinant form as soluble enzymes and successfully used as platelet inhibitors in various settings [9–12].
We have previously immobilized regulators of complement activation on a surface, thereby decreasing surface-generated complement activation [13, 14]. The aim of the current project was to develop a surface with the capacity to regulate blood incompatibility reactions at the platelet level. Here, we have applied the principle of ADP catalysis on a biomaterial surface, immobilizing apyrase on the surface in order to regulate platelet activation generated by a biomaterial. The surface was evaluated by incubating whole blood and platelet-rich plasma (PRP) in experimental settings designed to simulate the vascular milieu.
2. Materials and Methods
2.1 Reagents
2.1.1 Biotinylation of apyrase and human serum albumin (HSA)
Apyrase from potato (≥ 200 U/mg; Sigma Aldrich, St. Louis, MO, USA) with a high ADPase/ATPase ratio (≥ 1) or HSA (Flexbumin; Baxter AG, Vienna, Austria) was dissolved in 0.1 M phosphate buffer, pH 7.6 (1.5 mg protein/mL buffer). Biotin amidohexanoic acid N-hydroxysuccinimide ester (Sigma-Aldrich) was dissolved in DMSO (50 mg/ml), and 12.5 μl was added to every mL of the protein solution. The reaction was allowed to proceed for 90 min with rotation at room temperature (RT). Each mixture was dialyzed at 4°C overnight in Float-a-lyzer (MWCO 3.5 kDa; Spectrum Labs, Breda, The Netherlands): HSA against PBS (10 mM phosphate buffer, pH 7.4, with 0.15 M NaCl), and apyrase against PBS, pH 6.5.
2.1.2 Preparation of protein surfaces
Microtiter plates (Nunc Maxisorb Immunoplates, Nunc, Copenhagen, Denmark) or polystyrene microscope slides (Tedpella Inc, Redding, CA) were coated with 2% HSA in PBS by overnight incubation at 4°C. The HSA layer was subsequently biotinylated with biotin amidohexanoic acid N-hydroxysuccinimide ester (25 ng/cm2) dissolved in PBS and allowed to react overnight at 4°C. Avidin (0.2 mg/mL; Sigma Aldrich) was dissolved in PBS and linked to the biotinylated HSA by a 60-min incubation at RT. For platelet-rich plasma (PRP) and whole blood experiments, 10 IU/cm2 of biotinylated apyrase or HSA (75 μg/cm2) was incubated on the avidin-layer for 60 min at RT. To minimize exposure of the underlying avidin on the apyrase surface, the surface was saturated with biotinylated HSA (approximately 75 μg/cm2) for 30 min at RT.
2.1.3 Enzymatic activity of surface-bound apyrase
The enzymatic activity of the immobilized apyrase was examined by measuring the catalysis of ATP using the luciferase-based ATP Kit SL (BioThema, Handen, Sweden). Microtiter plates with apyrase immobilized at four different concentrations (10, 5, 2.5, and 1.25 IU/well) or HSA was agitated in the presence of 1 μM or 10μM ATP (Sigma Aldrich) in PBS for 30, 60, and 90 min at 37°C. The remaining ATP was measured according to the manufacturer’s protocol.
2.2 Preparation of whole blood and PRP
Whole blood was obtained from healthy volunteers who had received no medication for at least 10 days. Blood was drawn into an open system into 50-mL isopropylene tubes (Sarstedt, Nümbrecht, Germany) supplemented with 0.5 IU heparin/mL (whole blood experiments) or without anticoagulant (PRP experiments). The 50-mL tubes and all blood-collecting materials were heparinized with the Corline heparin surface (CHS) according to the manufacturer’s recommendation (Corline Systems AB, Uppsala, Sweden). The blood was either used immediately or further processed to generate PRP. To produce PRP, whole blood was centrifuged at 180 × g for 10 min at 22°C, and the PRP was transferred to a heparinized 15-mL (Sarstedt) tube. Alternatively (in the platelet adhesion experiments), whole blood was drawn into 4.5-mL Monovette® sampling tubes (Sarstedt) containing citrate. PRP was prepared by centrifuging whole blood at 140 × g for 20 min at room temperature.
2.3 Functional studies in whole blood and PRP
2.3.1 Whole blood incubation
The slide chamber model [15] was used for the whole blood experiments. The slide chamber consisted of a polymethylmetaacrylate microscopic slide with two rings, forming two circular wells. The slide chamber wells were heparinized (with CHS). Whole blood (1.3 mL) was added to each well, and the test surface was used as a lid, creating two enclosed chambers. The test surfaces consisted of microscopic slides with immobilized apyrase or HSA, polystyrene, or glass. The polystyrene and glass surfaces were prewashed in 5% ammonium persulfate (Sigma Aldrich) and extensively rinsed with milliQ water prior to blood exposure. The device was rotated vertically at 25 rpm in a 37°C water bath for 60 min. After incubation, the chamber was dismantled, and the whole blood from each chamber was transferred to tubes with EDTA (10 mM final concentration) to stop further activation. Cell counts were monitored using a Coulter® AC.T diff™ Analyzer (Coulter Corporation, Miami, FL, USA). In order to obtain plasma, the blood was centrifuged at 3000 × g for 20 min at RT.
2.3.2 Platelet adhesion
Platelet adhesion to immobilized apyrase or HSA was investigated using citrate-anticoagulated PRP, with or without preactivation with ADP. PBS or ADP (10 μM final concentration, Sigma Aldrich) was added to PRP, and 100 μl PRP was transferred and incubated on microscope slides, conjugated with apyrase or HSA, for 60 min at RT. After incubation, the slides were carefully rinsed with PBS. Celltracker™ Green CMFDA (Invitrogen, San Diego, CA, USA) was incubated with the surfaces for 20 min, and the platelets were then fixed to the surface by incubation in 4% paraformaldehyde (Sigma-Aldrich) in PBS for 5 min. The slides were evaluated in an AxioObserver D1 (Zeiss, Oberkochen, Germany) inverted fluorescence microscope.
2.3.3 PRP incubation
PRP was added to microtiter plates (100 μl/well) containing immobilized apyrase, HSA, avidin, or exposed polystyrene, and incubated for 60 min at 37°C with mild agitation. The experiment was stopped by adding 100 μl EDTA (final concentration, 10 mM). The remaining platelets in PRP were counted in a Swelab AC920 EO autocounter (Boule Medical AB, Stockholm, Sweden). Plasma was obtained from PRP by centrifuging at 3000 × g for 20 min at 8°C.
2.4. ELISAs
2.4.1 C3a activation
In all ELISAs, PBS containing 0.05% Tween 20, 1% bovine serum albumin (BSA), and 10 mM EDTA was used as the working buffer; PBS with 0.05% Tween as the washing buffer; and 1,2-phenylenediamine dihydrochloride in 0.1 M citrate, pH 5, as the color substrate. Complement activation was measured using a C3a sandwich ELISA. Plasma from PRP and whole blood experiments, diluted in working buffer, was incubated in microtiter plates coated with monoclonal antibody 4SD17.3. Captured C3a was detected with biotinylated polyclonal rabbit anti-C3a, followed by horseradish peroxidase (HRP)-conjugated streptavidin (GE Healthcare, Uppsala, Sweden). Zymosan-activated serum, calibrated against purified C3a, served as standard. Results are given as ng/mL.
2.4.2 Thrombin-antithrombin (TAT) complexes
Plasma levels of TAT were measured with a sandwich ELISA. TAT was captured in wells coated with anti-human thrombin, and HRP-coupled anti-human antithrombin antibody was used for detection (both antibodies from Enzyme Research Laboratories, South Bend, IN, USA). A standard prepared by diluting pooled human serum in normal citrate-phosphate-dextrose plasma was used. Values were expressed as mg/L.
2.4.3 FXIIa-C1INH, FXIa-C1INH, FXIIa-AT, and FXIa-AT complexes
These complexes were measured by sandwich ELISA essentially according to the method of Sanchez et al., except that affinity-purified antibodies were used for capture and detection [16]. Microtiter plates were coated with either goat anti-human FXII (Enzyme Research Laboratories) or sheep anti-human FXI (The Binding Site, Birmingham, UK), and the bound complexes detected with either goat anti-human C1INH (Enzyme Research Laboratories) or rabbit anti-human AT antibodies (Dako). The secondary antibodies were biotinylated and detected using streptavidin-HRP (Amersham, Little Chalford, UK). Standards for each assay were prepared and evaluated as described by Sanchez et al. [16]. In short, purified FXIIa or FXIa were mixed with a molar excess of AT or C1INH. In order to ascertain that all protease activity was inhibited, residual FXIIa or FXIa activity was detected using the relevant chromogenic substrates. These standard solutions were diluted in normal plasma and included in each analysis, and the values were expressed as nM. The reactivity of each antibody preparation used in these assays was verified by western blot analysis. The coefficient of variation for all assays was approximately 10%.
2.4.4 Kallikrein-AT and kallikrein-C1INH complexes
Kallikrein complexes with C1INH and AT were measured in a manner analogous to the FXIIa-C1INH/AT and FXIa/C1INH/AT complexes, but with capturing antibody directed against prekallikrein (The Binding Site). Purified kallikrein mixed with a molar excess of AT or C1INH was used as a standard. Data are given as AU/mL.
2.5 Immunostaining of surface-bound cells
The test surface, acting as a lid on the whole blood chamber, was stained for nucleated blood cells in general and platelets in particular. The slide was washed three times for 5 min in PBS, fixed in 4% paraformaldehyde (Apoteket, Sweden) for 15 min, and blocked 30 min in 2% BSA (Sigma Aldrich). Nuclei and CD42a were stained by incubating propidium iodide (Sigma Aldrich) and anti-CD42a-FITC (BD Pharminogen, BD Biosciences, San José, CA, USA) on the slides for 60 min at RT. The slides were washed with PBS-Tween and examined in a confocal microscope (Zeiss, 510 Meta Confocal, Carl Zeiss, Jena, Germany).
2.6 Statistical analysis
Results are presented as means ± SEM. For statistical analysis, values from control surfaces were compared to those for the apyrase surface. Statistical significance was calculated using one-way ANOVA with Dunnett’s post hoc-test in GraphPad Prism, version 5.01 for Windows (GraphPad Software, San Diego CA, USA). The data were assumed to be normally distributed, and those related to FXIIa-AT complex formation were transformed to a logarithmic scale to meet this criterion.
3. Results
3.1 Enzymatic activity of surface-bound apyrase
Apyrase with a high ADPase/ATPase ratio was immobilized onto surfaces using biotin/avidin chemistry. To ascertain that the enzymatic activity still remained after biotinylation and surface attachment, the ATPase activity in apyrase wells was measured at four different concentrations of apyrase and two concentrations of ATP (1μM [Fig 1A] and 10 μM [Fig. 1B]). HSA wells were used as controls. After 60 min the amount of ATP in the wells containing the lower concentration had decreased from 0.1 nmol to 0.001–0.044 nmol, and after 90 min of ATP incubation, the amount of ATP in the wells containing the higher concentration had decreased from 1 nmol to <0.1 nmol in all apyrase wells. No ATP decay was observed in the HSA wells. We saw a concentration-dependent reaction, with the largest decrease in ATP occurring in the wells treated with 10U of apyrase and the least in those with 1.25U of apyrase. Based on the velocity of the ATP breakdown by the biotinylated immobilized apyrase in relation to soluble native enzyme, the actual activity of the immobilized form corresponded to approximately 1% of that of the native enzyme. By measuring the enzymatic activity of biotinylated apyrase in solution and comparing it to the soluble native form, a drop in activity was detected after biotinylation. This lost activity was estimated to be approximately two thirds (data not shown). The relative enzymatic activity of immobilized apyrase biotinylated at three different molar ratios (5:1, 15:1, or 45:1 biotin over apyrase) showed no differences in activity (data not shown).
Fig. 1.

Enzymatic activity of apyrase, measured as ATPase activity. Apyrase (AP) at four concentrations and human serum albumin (HSA) were immobilized in mitrotiter wells. ATP was added to the wells and incubated for 60 (ATP =1 μM, Fig. 1A) or 90 min (10 μM, Fig. 1B), respectively. The remaining ATP was measured using a luciferase-based ATP kit.
3.2 Effect of surface-bound apyrase on platelet binding to a material surface
To examine the binding of platelets to the surfaces, citrate-anticoagulated PRP was incubated on microscope slides with immobilized apyrase and HSA, with or without preactivation with ADP. In citrate-anticoagulated blood without preactivation of the platelets, only a small number of separate platelets adhered to the surface, and no difference was seen between the HSA (Fig. 2A) and apyrase (Fig. 2B) results. Preactivation of the platelets with 10μM ADP caused an increased binding of platelets, including formation of platelet aggregates on the HSA surface (Fig. 2C), but not on the apyrase surface (Fig. 2D). The adhesion of preactivated platelets to the apyrase surface was similar to that on the apyrase surface without ADP preactivation, with only a small number of bound platelets. By scratching the apyrase surface and thereby exposing the underlying polystyrene layer, we could visualize the difference in thrombogenicity between the surfaces (Fig. 2E). Platelets were deposited on the damaged area with the exposed polystyrene, whereas the apyrase-covered surface revealed no platelet deposition.
Fig. 2.

Platelet binding to polystyrene with either immobilized apyrase or HSA. The platelets were stained with Celltracker™ Green. PRP incubated on HSA- (A and C) or apyrase- (B and D) surfaces in the absence (A and B) or presence (C and D) of ADP (10 μM). Panel E shows an apyrase surface damaged with a scratch, resulting in exposure of the underlying polystyrene surface. The bar equals 100 μM.
3.3 Effect of surface-bound apyrase in PRP on the activation of platelets and the coagulation and complement systems
PRP without anticoagulant was obtained from five blood donors and tested in separate experiments. PRP samples were incubated with apyrase- and control surfaces (HSA, avidin, and polystyrene) for 60 min, and markers of platelet, coagulation, and complement activation were measured. Platelet consumption on the apyrase surface was 37±7% (Fig. 3A), significantly lower (p<0.01) than that seen on any of the control surfaces (HSA, 75±4%; avidin, 65±7%; and PS, 90±2%). Thrombin-antithrombin (TAT) complexes were found to be a sensitive parameter reflecting coagulation activation (Fig. 3B.). Apyrase surfaces showed significantly lower TAT levels (p<0.05) than did any of the control surfaces (apyrase, 4.1±0.7 μg/mL; HSA, 16.0±1.6 μg/mL; avidin, 11.0±1.0 μg/mL; and PS, 20.7±2.3 μg/mL). Contact system activation initiated by platelets or artificial surfaces has previously been shown to generate AT complexes and C1INH complexes, respectively [17]. The apyrase surfaces also had significantly lower levels of FXIa-AT than did the control surfaces (p<0.01) (Fig. 3C: apyrase, 2.81 ±0.12 nM; HSA, 3.88 ±0.06 nM; avidin, 3.83 ±0.06 nM; and PS, 3.84±0.03 nM). Complex formation between antithrombin and FXIIa (Fig. 3D) was significantly lower in the apyrase wells than in those with HSA (<0.05) but not with avidin or PS (apyrase, 9.0 ±1.5 nM; HSA, 156 ±33 nM; avidin, 19 ±1.6 nM; and PS, 91±27 nM). In contrast, no significant differences were seen between surfaces with regard to the C1INH or AT complexes formed with kallikrein (data not shown). Complement activation, as reflected by the generation of C3a (Fig. 3E) was significantly lower for the apyrase surface than for the PS (p< 0.05), but not for the other control surfaces (apyrase, 822 ±92 nM; HSA, 956 ±114 nM; avidin, 1012 ±114 nM; and PS, 1433±182 nM).
Fig. 3.

Complement, coagulation, and platelet activation in PRP on apyrase and control surfaces. PRP was incubated for 60 min at 37°C in untreated polystyrene (PS) microtiter wells or PS wells with HSA, apyrase, or avidin. The figures show (A) platelet consumption, presented as a percentage of the starting value; (B) complexes between antithrombin and thrombin (TAT); (C), antithrombin and factor XIa (FXIa-AT); (D) antithrombin and factor XIIa (FXIIa-AT); and (E) generation of C3a. Data are presented as means ± SEM; n = 5; *p<0.05, **p<0.01, ***p<0.001. The apyrase surface shows significantly lower levels of platelet consumption, TAT, and FXIa-AT, when compared to all of the control surfaces (p<0.05). The apyrase surface demonstrates significantly lower (p<0.05) levels of FXIIa-AT and C3a than the HSA and PS surfaces respectively.
3.4 Effect of surface-bound apyrase in whole blood on platelet consumption and platelet binding
Whole blood supplemented with 0.5 IU heparin/mL was drawn from five blood donors and tested in separate experiments. The blood was incubated with the test surfaces in the slide chamber model for 60 min. Platelet consumption was measured, and the surface was stained for nucleated blood cells and platelets. Blood incubated on the glass and PS surfaces showed clotting and an almost complete platelet consumption (glass, 95% ± 3%; PS, 95% ± 3%; Fig. 4). The apyrase surface showed a significantly lower platelet consumption (p<0.001) when compared to these surfaces. No significant difference was detected between the two protein-containing surfaces, which showed only a limited platelet consumption (HSA: 30% ± 10%, apyrase: 13% ± 4%). Staining of cell nuclei and platelets revealed heterogeneity in the cell-binding pattern among the various surfaces (Fig. 5). The glass surface showed a large abundance of adhered leukocytes, most of which exhibited complex formations with platelets. The PS surface displayed the largest abundance of bound leukocytes. This surface also showed a large quantity of platelets smeared onto the surface. The HSA surface displayed some complex formation between leukocytes and platelets, and overall the number of cells was lower than on the glass and PS surfaces. The apyrase surface showed leukocyte adhesion similar to that of HSA, but no complex formation with platelets. Some platelets were present at the surface as small aggregates.
Fig. 4.
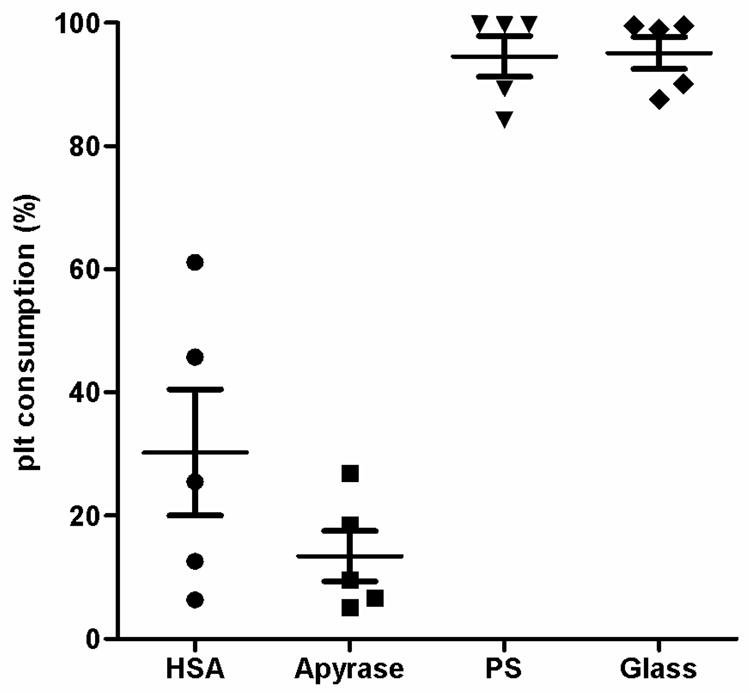
Platelet consumption, presented as a percentage of the starting value. Whole blood anticoagulated with 0.5IU heparin per mL was incubated on each surface in the slide chamber model for 60 min at 37°C. Data are presented as means ± SEM; n = 5. The apyrase surface shows significantly less platelet consumption than do the polystyrene and glass surfaces, which demonstrate an almost complete platelet consumption. No significantly difference was detected between the apyrase and HSA surfaces.
Fig. 5.

Confocal microscope images showing cell adhesion to slides with HSA (A), apyrase (B), PS (C), and glass (D) after incubation in the slide chamber model for 60 min at 37°C. Nucleated blood cells were stained with propidium iodine (red) and platelets with FITC-labeled monoclonal antibody against CD42a (green). Representative images from five separate experiments are shown.
4. Discussion
Here we have introduced a new approach to improving the blood compatibility of a biomaterial. By immobilizing apyrase, an ADP-degrading enzyme, on a biomaterial surface, we have succeeded in down-regulating platelet activation, presumably by abolishing the ADP-dependent feedback activation loop. By connecting apyrase to a surface, we were able to significantly inhibit platelet activation and platelet-dependent activation of the coagulation system. The system was successfully tested in both PRP and whole blood.
A commercially available apyrase with high ADPase/ATPase activity was used for these experiments, but an ideal protein for this purpose would be an apyrase with exclusively ADPase activity. In our system, apyrase was conjugated to a protein-coated polystyrene surface using biotin-avidin chemistry. Initially, HSA was absorbed to a polystyrene surface and then the HSA layer was biotinylated, with avidin being used as a linker protein to bridge biotinylated apyrase to the surface.
In experiments evaluating the biological function of the novel surface, the apyrase surface was compared with a clean polystyrene surface, a clean glass surface (whole blood experiments), a surface with adsorbed avidin (PRP experiments), and an HSA surface. The polystyrene and glass surfaces were used as positive controls, both activating the blood cascade systems. Both surfaces triggered the contact system, with subsequent coagulation and platelet activation. HSA, which is abundantly present in plasma and on biomaterials after plasma exposure, served as an inert control. The HSA was biotinylated and connected to the surface via avidin in the same manner as apyrase. After the attachment of biotinylated apyrase, there was still a significant exposure of available biotin binding sites on avidin.
Therefore, biotinylated HSA was used to block the remaining binding sites. To eliminate the possibility that some of the avidin layer was still exposed, we introduced a background control with avidin alone to the PRP experiments to verify that this surface had not contributed to the platelet regulation or initiated platelet activation.
After conjugation of the apyrase to the surface, the enzymatic activity was evaluated. Apyrase was biotinylated via primary amines using a 45-molar excess of biotin over apyrase. A change in the protein structure elicited by introducing biotin or by attaching it to a surface may inactivate the protein. Therefore, we assessed the activity of the enzyme and found that it was fully active at a substrate concentration of 1 to 10 μM, although the activity of the immobilized enzyme was estimated to be approximately 1% of the corresponding native soluble form. A comparison of biotinylated enzyme with the native form demonstrated an activity loss of approximately two thirds after biotinylation. There are difficulties to estimate the enzyme activity in a microtiter well since the interaction between the enzyme and the substrate depends solely on diffusion of the substrate while the enzyme is immobilized. Therefore a more precise assessment of the enzyme activity was not carried out. The importance of this experiment was to verify enzymatic activity after surface attachment.
Platelet adherence was investigated by incubating citrate-anticoagulated PRP on surfaces treated with apyrase and HSA in the same manner. The results showed that there was no platelet adherence to either of the two surfaces if the platelets were unstimulated, demonstrating the inert property of both surfaces. When the platelets were preactivated with ADP before incubation on the surfaces, platelet activation and adherence took place only on the HSA surface. The apyrase surface was able to down-regulate this activation, and there was no visible platelet adherence. By scratching the surface and exposing the underlying polystyrene surface, we produced conditions resembling a vascular injury that exposed the subendothelial layer. This exposure of the polystyrene led to activation of the platelets, resulting in the formation of platelet aggregates on the polystyrene surface. Importantly, next to the site of damage, platelet activation was down-regulated, just as it was on an undamaged endothelial surface.
In order to mimic near-physiological conditions, the PRP experiments were performed without the addition of anticoagulants. This was possible because the blood was collected using equipment coated with CHS. After incubation on the test surfaces, less platelet consumption and lower levels of TAT complexes were found on the apyrase-conjugated surface than on the control surfaces. We have recently shown that by measuring complexes between factor XI, factor XII, and kallikrein and the protease inhibitors C1INH and antithrombin, it is possible to determine whether contact activation is triggered by platelet activation or artificial surface activation, respectively [17]. Using this approach, we were able to see a significant difference between the surfaces with regard to coagulation initiated by platelet activation. The levels of antithrombin complexes, but not C1INH complexes, were lower on the apyrase surface than on the control surfaces.
Whole blood experiments were performed with 0.5 IU heparin per mL of blood. Unfortunately, these experiments could not be performed without anticoagulants, since platelets are much more responsive in whole blood than in PRP because of the presence of erythrocytes and other blood cells [18]. The addition of heparin caused a significant attenuation of the system, with low platelet consumption on both the apyrase and HSA surfaces. Despite this attenuation in the response, a difference in cell adherence was detected between the protein-covered surfaces. Neutrophilc granulocytes and monocytes were present to the same extent on both surfaces, but cells formed complexes with platelets on the HSA surface, indicating that the leukocytes were much more activated on this surface. This increased formation of leukocyte-platelet complexes reflects the up-regulation of CD11b on leukocytes and an interaction with fibrin on the platelets [19].
Regulation of platelet activation by irreversibly blocking the platelet ADP receptor with non steroidal anti-inflammatory drugs (NSAIDs) is well established. Marcus et al. have expressed a soluble form of recombinant human CD39 by truncating the N- and C-terminal transmembrane domains; this soluble form of CD39 has shown promising results in terms of cerebrovascular and cardiovascular protection [20]. One advantage of using this approach is that the platelets remain unmodified, and the drug affects only the prothrombotic ADP. Smith and colleagues have expressed a recombinant human apyrase, homologous to the apyrase of the bed bug, and successfully designed it to increase the ADPase activity [12, 21]. Despite the limited activity of the enzyme used in our study, we were in our in vitro system, able to achieve a significant attenuation of platelet activation. Given a more optimized immobilization technique and a more potent apyrase, an even better surface could be generated. The reports cited above support this encouraging possibility.
Present surface immobilization works for in the in vitro system with limited amount of blood, but taking it into in vivo situations where the surface of a medical device would be exposed to large blood volumes, an optimized immobilization will be preferred. The enzymes robustness for conjugation and surface attachment would argue that it could be conjugated to already existing surface coatings to achieve active platelet regulation. It is presumably unrealistic to reach total blood compatibility with a foreign surface, but by mimicking self structures as with this enzyme, compatibility would be elevated. In the clinical situation, combining this surface modification with low dose soluble CD39, prothrombotic ADP will be affected whereas the platelet remains unmodified.
Apyrase has been immobilized before onto biomaterial surfaces in order to improve biocompatibility [22, 23]. In these studies apyrase has been adsorbed [22] or conjugated using glutaraldehyde [23]. These unspecific procedures have been evaluated in vivo and been shown to improve the biocompatibility of the material with regard to thrombogenicity. However, it is difficult to know whether the apyrase is improving the biocompatibility of the surface by its enzymatic activity or whether the mere alteration of the physico-chemical properties of the surface may explain the improvement. By contrast, we have demonstrated that a more specific conjugation of apyrase to the surface maintains its enzymatic activity which eliminates the possibility of this effect.
Metal stents used in coronary arteries etc. would be a suitable surface for immobilizing apyrase. Implantation of stents is associated with restenosis in the narrow lumen of the device [24]. The metal surface and damaged endothelial cells force thrombus formation and cell activation which fuel restenosis. An efficiently regulation of platelet activation of this limited area would improve the outcome of the stent.
Based on the scarcity of blood-biocompatible biomaterials, this new principle for modifying surfaces could represent a useful new approach to improving biocompatibility. We have previously shown that by introducing a small peptide derived from Streptococcus pyogenes, one can regulate complement by inhibiting the classical pathway [13]. This peptide uses the patient’s own regulators by capturing C4BP, a regulator of the classical pathway of complement, from the fluid phase and pulling it down to the surface. By combining different subsets of regulators, a broad specificity of biocompatibility of medical devices could be achieved.
5. Conclusions
We have shown that by immobilizing apyrase on a biomaterial surface, platelet activation and platelet-dependent activation of the coagulation cascade can be inhibited. This inhibition was achieved via the breakdown of ADP, an important molecule for platelet activation and aggregation. This concept was successfully evaluated in both PRP and whole blood. Our study has demonstrated that it is feasible to lower the platelet activation produced by an artificial surface by conjugating an apyrase to that surface. By combining this apyrase with regulators of coagulation and complement activation, blood-activating biomaterials can be converted into blood-compatible surfaces. Various types of metal stents would be a suitable matrices for immobilization of apyrase.
Acknowledgments
We are grateful to Dr. Graciela Elgue and Dr. Javier Sanchez for laboratory assistance, Dr. Jaan Hong for the whole blood experiment setup and Dr. Deborah McClellan for editorial assistance. This work was supported by grants from the Swedish Research Council (VR) 2009-4675 and 2009-4462, grant # EB003968 from the National Institute of Health (USA) and the Linneaus University in Kalmar, Sweden.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nilsson B, Korsgren O, Lambris JD, Ekdahl KN. Can cells and biomaterials in therapeutic medicine be shielded from innate immune recognition? Trends Immunol. 2010;31:32–38. doi: 10.1016/j.it.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25:5681–703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 3.Dormann D, Clemetson KJ, Kehrel BE. The GPIb thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood. 2000;96:2469–78. [PubMed] [Google Scholar]
- 4.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–92. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Hamad OA, Ekdahl KN, Nilsson PH, Andersson J, Magotti P, Lambris JD, et al. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thromb Haemost. 2008;6:1413–21. doi: 10.1111/j.1538-7836.2008.03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cattaneo M, Gachet C. ADP receptors and clinical bleeding disorders. Arterioscler Thromb Vasc Biol. 1999;19:2281–5. doi: 10.1161/01.atv.19.10.2281. [DOI] [PubMed] [Google Scholar]
- 7.Marcus AJ, Broekman MJ, Drosopoulos JH, Islam N, Alyonycheva TN, Safier LB, et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest. 1997;99:1351–60. doi: 10.1172/JCI119294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Champagne DE. Antihemostatic molecules from saliva of blood-feeding arthropods. Pathophysiol Haemost Thromb. 2005;34:221–7. doi: 10.1159/000092428. [DOI] [PubMed] [Google Scholar]
- 9.Pinsky DJ, Broekman MJ, Peschon JJ, Stocking KL, Fujita T, Ramasamy R, et al. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest. 2002;109:1031–40. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belayev L, Khoutorova L, Deisher TA, Belayev A, Busto R, Zhang Y, et al. Neuroprotective effect of SolCD39, a novel platelet aggregation inhibitor, on transient middle cerebral artery occlusion in rats. Stroke. 2003;34:758–63. doi: 10.1161/01.STR.0000056169.45365.15. [DOI] [PubMed] [Google Scholar]
- 11.Buergler JM, Maliszewski CR, Broekman MJ, Kaluza GL, Schulz DG, Marcus AJ, et al. Effects of solCD39, a novel inhibitor of platelet aggregation, on platelet deposition and aggregation after PTCA in a porcine model. J Thromb Thrombolysis. 2005;19:115–22. doi: 10.1007/s11239-005-1381-y. [DOI] [PubMed] [Google Scholar]
- 12.Dai J, Liu J, Deng Y, Smith TM, Lu M. Structure and protein design of a human platelet function inhibitor. Cell. 2004;116:649–59. doi: 10.1016/s0092-8674(04)00172-2. [DOI] [PubMed] [Google Scholar]
- 13.Engberg AE, Sandholm K, Bexborn F, Persson J, Nilsson B, Lindahl G, et al. Inhibition of complement activation on a model biomaterial surface by streptococcal M protein-derived peptides. Biomaterials. 2009;30:2653–9. doi: 10.1016/j.biomaterials.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersson J, Bexborn F, Klinth J, Nilsson B, Ekdahl KN. Surface-attached PEO in the form of activated Pluronic with immobilized factor H reduces both coagulation and complement activation in a whole-blood model. J Biomed Mater Res A. 2006;76:25–34. doi: 10.1002/jbm.a.30377. [DOI] [PubMed] [Google Scholar]
- 15.Hong J, Nilsson Ekdahl K, Reynolds H, Larsson R, Nilsson B. A new in vitro model to study interaction between whole blood and biomaterials. Studies of platelet and coagulation activation and the effect of aspirin. Biomaterials. 1999;20:603–11. doi: 10.1016/s0142-9612(98)00210-5. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez J, Lundquist PB, Elgue G, Larsson R, Olsson P. Measuring the degree of plasma contact activation induced by artificial materials. Thromb Res. 2002;105:407–12. doi: 10.1016/s0049-3848(02)00051-8. [DOI] [PubMed] [Google Scholar]
- 17.Back J, Lang MH, Elgue G, Kalbitz M, Sanchez J, Ekdahl KN, et al. Distinctive regulation of contact activation by antithrombin and C1-inhibitor on activated platelets and material surfaces. Biomaterials. 2009;30:6573–80. doi: 10.1016/j.biomaterials.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 18.Hong J, Larsson A, Ekdahl KN, Elgue G, Larsson R, Nilsson B. Contact between a polymer and whole blood: sequence of events leading to thrombin generation. J Lab Clin Med. 2001;138:139–45. doi: 10.1067/mlc.2001.116486. [DOI] [PubMed] [Google Scholar]
- 19.Goel MS, Diamond SL. Neutrophil enhancement of fibrin deposition under flow through platelet-dependent and -independent mechanisms. Arterioscler Thromb Vasc Biol. 2001;21:2093–8. doi: 10.1161/hq1201.100255. [DOI] [PubMed] [Google Scholar]
- 20.Marcus AJ, Broekman MJ, Drosopoulos JH, Olson KE, Islam N, Pinsky DJ, et al. Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin Thromb Hemost. 2005;31:234–46. doi: 10.1055/s-2005-869528. [DOI] [PubMed] [Google Scholar]
- 21.Smith TM, Hicks-Berger CA, Kim S, Kirley TL. Cloning, expression, and characterization of a soluble calcium-activated nucleotidase, a human enzyme belonging to a new family of extracellular nucleotidases. Arch Biochem Biophys. 2002;406:105–15. doi: 10.1016/s0003-9861(02)00420-4. [DOI] [PubMed] [Google Scholar]
- 22.Bakker WW, van der Lei B, Nieuwenhuis P, Robinson P, Bartels HL. Reduced thrombogenicity of artificial materials by coating with ADPase. Biomaterials. 1991;12:603–6. doi: 10.1016/0142-9612(91)90059-j. [DOI] [PubMed] [Google Scholar]
- 23.Marconi W, Bartoli F, Mantovani E, Pittalis F, Settembri L, Cordova C, et al. Development of new antithrombogenic surfaces by employing platelet antiaggregating agents: preparation and characterization. Trans Am Soc Artif Intern Organs. 1979;25:280–6. doi: 10.1097/00002480-197902500-00051. [DOI] [PubMed] [Google Scholar]
- 24.Kastrati A, Schomig A, Dietz R, Neumann FJ, Richardt G. Time course of restenosis during the first year after emergency coronary stenting. Circulation. 1993;87:1498–505. doi: 10.1161/01.cir.87.5.1498. [DOI] [PubMed] [Google Scholar]