

Abstract
AIM: To assess serum concentrations of prohepcidin in chronic hepatitis C individuals and evaluate their associations with disease activity and efficacy of pegylated interferon (PEG-IFN)/ribavirin (RBV) therapy.
METHODS: Prohepcidin was measured in sera of 53 chronic hepatitis C patients. Concentrations of prohepcidin and other iron metabolism markers were analyzed at 9 time points before, during and after the end of antiviral therapy.
RESULTS: In hepatitis C virus (HCV) genotype 1-infected individuals, a gradual decrease of prohepcidin during antiviral therapy was observed in responders (88.8 ± 14.7 ng/mL before therapy vs 60.6 ± 0.3 ng/mL in the 48th wk, P = 0.04). In contrast, no decrease was observed in non-responders. A similar association was observed in HCV genotype 3a individuals, with a statistically significant decline in serum prohepcidin only in the responder group (99.5 ± 5.2 ng/mL at baseline vs 72.7 ± 6.1 ng/mL in the 24th wk, P = 0.01). Moreover, HCV-RNA at week 12 of therapy was positively correlated with baseline (R = 0.63, P < 0.005) and week 12 (R = 0.60, P = 0.01) serum prohepcidin concentrations in HCV genotype 1 infection.
CONCLUSION: Successful PEG-IFN/RBV therapy results in a decline of serum prohepcidin concentration in chronic hepatitis C, which may suggest a direct effect of HCV on iron metabolism at the prohormonal level of hepcidin.
Keywords: Iron metabolism, Hepcidin, Hepatitis C virus, Interferon, Sustained viral response
INTRODUCTION
Liver iron overload is a well described, but not completely understood, feature of hepatitis C virus (HCV) infection. Several lines of evidence have suggested its negative influence on chronic hepatitis C outcome. Recent reports have emphasized the strong association between hepatic iron storage markers and oxidative DNA damage in persistent HCV infection. In the study by Fujita et al[1], phlebotomy resulted in a significant decrease of oxidatively generated DNA damage markers with concomitant reduction in serum transaminases as well as iron-related markers in chronic hepatitis C. Iron accumulation has also been linked with insulin resistance and liver steatosis in HCV-infected patients[2,3]. Furthermore, experimental data have shown that iron deposits may trigger hepatic stellate cell activation and thus induce liver fibrosis[4]. Among other potential links between iron metabolism and chronic hepatitis C, the possibility of enhancing HCV replication by serum iron has been postulated in experimental settings[5].
Fujita et al[1] found that total iron score correlated positively with transaminase activity, histological grading and staging in chronic hepatitis C subjects. Interestingly, in this study baseline iron metabolism alterations were more pronounced in non-sustained viral response (non-SVR) than in SVR to interferon (IFN)/ribavirin (RBV) treatment, therefore suggesting the association between iron deposition and resistance to anti-HCV therapy. This observation has been further confirmed in a recent study[6] showing inadequate hepcidin expression in chronic HCV, which could be restored by eradication of the virus.
The mechanisms of iron metabolism deregulation in chronic hepatitis C are not fully elucidated. Takeo et al[7] showed significantly higher liver mRNA expression of transferrin receptor 2 (TfR2) and ferroportin in chronic hepatitis C than in hepatitis B virus (HBV)-infected patients, which additionally correlated with the total hepatic iron score during the HCV infection. Authors have suggested that upregulation of hepatic iron transporters may contribute to the hepatic iron accumulation. Recently discovered iron regulatory hormone, hepcidin, synthesized predominantly in the liver, has changed the understanding of iron metabolism regulation[8]. Hepcidin has been found to suppress intestinal absorption of iron through the binding to ferroportin, which is robustly expressed by enterocytes and liver macrophages[9]. Synthesis of hepcidin is increased by iron overload and decreased by anemia, hypoxia-inducible factors and reactive oxygen species. Moreover, it may be induced by infection and inflammatory responses, which result in rapid plasma iron decrease[10-12]. Recent results have shown hepcidin deregulation in chronic hepatitis C and have suggested the pivotal role of this hormone in the pathogenesis of iron overload[1,6,13].
In the current study, levels of prohepcidin, a hepcidin precursor protein predominantly synthesized in the liver, were measured. In the hepatocytes, prohepcidin undergoes two cleavages and is rapidly secreted from cells as the mature, 25 amino acid peptide, hepcidin. It has been shown that, to some extent, prohepcidin is also secreted by hepatocytes[14]. Recently, we have suggested the association between serum prohepcidin levels and liver function impairment in liver cirrhosis patients[15]. The aim of the study was to assess the serum concentration of this hepcidin prohormone in chronic hepatitis C and evaluate its possible association with the disease activity as well as the efficacy of pegylated IFN (PEG-IFN)/RBV therapy.
MATERIALS AND METHODS
Patients
Prohepcidin levels were measured in sera of 53 chronic hepatitis C patients (15 females and 38 males; median age: 46.5 years, min: 20 years, max: 67 years) before and during PEG-IFN-α/RBV therapy. Twenty-nine of them were infected with HCV genotype 1 and 24 with HCV genotype 3a. All patients had chronic hepatitis C proven through the presence of anti-HCV antibodies and HCV-RNA in sera for at least 6 mo. Quantitative HCV-RNA and HCV genotyping were performed by two-step real-time quantitative RT-PCR using TaqMan. Disease activity was evaluated by liver biopsy (Hepafix System, Braun, Melsungen, Germany) performed before the start of anti-HCV therapy. Paraffin-embedded biopsy specimens were stained and evaluated using Scheuer’s scoring system[16]. None of the included patients had liver cirrhosis diagnosed, nor HBV, hepatitis delta virus or human immunodeficiency virus co-infections. Clinical characteristics of the studied population are presented in Table 1.
Table 1.
Baseline characteristics of studied population (mean ± SE)
| Control group (n = 15) |
Genotype 1 |
Genotype 3a |
|||
| R (n = 13) | NR (n = 16) | R (n = 21) | NR (n = 3) | ||
| Age [median (min-max), yr] | 40 (26-54) | 47 (21-67) | 48 (23–58) | 47 (25-64)a | 24 (20-26) |
| Sex (M/F) | 7/8 | 11/2 | 13/3 | 12/9 | 2/1 |
| HCV-RNA log10 (cps/mL) | - | 6.01 ± 5.74 | 5.86 ± 5.73 | 3.95 ± 3.65 | 2.95 ± 2.30 |
| ALT (U/L) | - | 110.4 ± 29.3 | 75.1 ± 8.7 | 111.5 ± 23.6 | 133.0 ± 25.2 |
| Bilirubin (mg/dL) | - | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 | 1.1 ± 0.1 |
| Liver inflammatory activity [median (min-max), pts] | - | 3 (1-3) | 2 (1-4) | 2 (1-3) | 2.5 (2-3) |
| Liver fibrosis [median (min-max), pts] | - | 1 (1-3) | 1 (1-3) | 1 (1-2) | 1.5 (1-2) |
| Fe (μg/dL) | - | 138.9 ± 18.3 | 162.2 ± 19.9 | 127.3 ± 10.9 | 165.0 ± 29.0 |
| TIBC (μg/dL) | - | 352.3 ± 20.7 | 316.5 ± 21.1 | 345.5 ± 13.5 | 361.3 ± 25.2 |
| Transferrin saturation (%) | - | 42.5 ± 6.9 | 39.1 ± 5.9 | 38.0 ± 4.1 | 43.7 ± 10.1 |
| Ferritin (ng/mL) | - | 255.4 ± 50.2 | 204.5 ± 35.1 | 193.4 ± 58.1 | 134.8 ± 61.8 |
| Hemoglobin (g/dL) | - | 15.0 ± 0.32 | 14.5 ± 0.4 | 14.6 ± 0.3 | 15.4 ± 0.9 |
| Prohepcidin (ng/mL) | 84.1 ± 7.8 | 88.8 ± 14.7 | 88.3 ± 11.4 | 99.5 ± 5.2 | 94.1 ± 7.9 |
P < 0.05 in comparison to non-responders, all of the other differences are non-significant. R: Responders; NR: Non-responders; HCV: Hepatitis C virus; ALT: Alanine aminotransferase; TIBC: Total iron binding capacity.
Patients received a combination therapy with a weekly dose of PEG-IFN-α2a administered subcutaneously and RBV administered orally at daily doses of 1000 or 1200 mg/d based on body weight (< 75 or ≥ 75 kg, respectively) for genotype 1 and 800 mg/d for genotype 3a. The total duration of treatment was 48 wk for genotype 1 and 24 wk for genotype 3a. Patients were divided into two groups: responders, defined as undetectable HCV-RNA at week 24 after the end of therapy, and non-responders, defined as HCV-RNA positive at week 24 after the end of therapy.
Prohepcidin serum concentrations were measured at baseline, in the 4th, 12th, 24th and 48th (genotype 1) wk of antiviral therapy and in the 24th wk after termination of the treatment (week 72). During the same visits, liver function tests and the serum iron metabolism markers, serum iron, total iron binding capacity (TIBC), transferrin saturation, and ferritin, were measured. Moreover, serum iron saturation was calculated using the following formula: (serum Fe/TIBC) × 100%. Serum prohepcidin values were compared with those collected from 15 healthy volunteers (7 females and 8 males, median age: 40 years).
Hepcidin prohormone measurement
Venous blood was collected on ice using vacutainer tubes and centrifuged at 2500 × g at 4°C within 30 min of collection. Serum samples were assayed for prohepcidin with the quantitative sandwich enzyme immunoassay kit (DRG Instrument GmbH, Marburg, Germany) according to manufacturer instructions. The employed antibody detects both the pro-region and pro-hepcidin (aa 25-84). The sensitivity of assay is 3.95 ng/mL, intra-assay variation coefficient of variation (CV) 4.69% and inter-assay variation CV 4.82%.
Statistical analysis
Values were expressed as mean ± SE of mean. The significance of differences was calculated by non-parametric Mann-Whitney U, Kruskall-Wallis and Friedman ANOVA tests. For correlation analysis, the Spearman non-parametric correlation was used. A P < 0.05 value was considered as statistically significant. Statistical analyses were performed with Statistica 7.0 for Windows software (Statsoft Inc., Tulsa, USA).
Informed consent was obtained from each patient. The procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 1983. The study was approved by the Bioethical Committee of the Medical University of Bialystok.
RESULTS
Serum concentration of prohepcidin measured before PEG-IFN/RBV therapy in chronic hepatitis C individuals slightly, but not significantly, exceeded values observed in healthy controls (93.2 ± 5.3 ng/mL vs 84.1 ± 7.8 ng/mL, respectively, P = 0.46). Interestingly, we observed a higher concentration of serum prohepcidin in HCV genotype 3a compared to genotype 1 (98.8 ± 4.7 ng/mL vs 88.5 ± 8.9 ng/mL, respectively, P = 0.02). In the latter group the values were also significantly higher than in controls (P = 0.04) (Figure 1).
Figure 1.
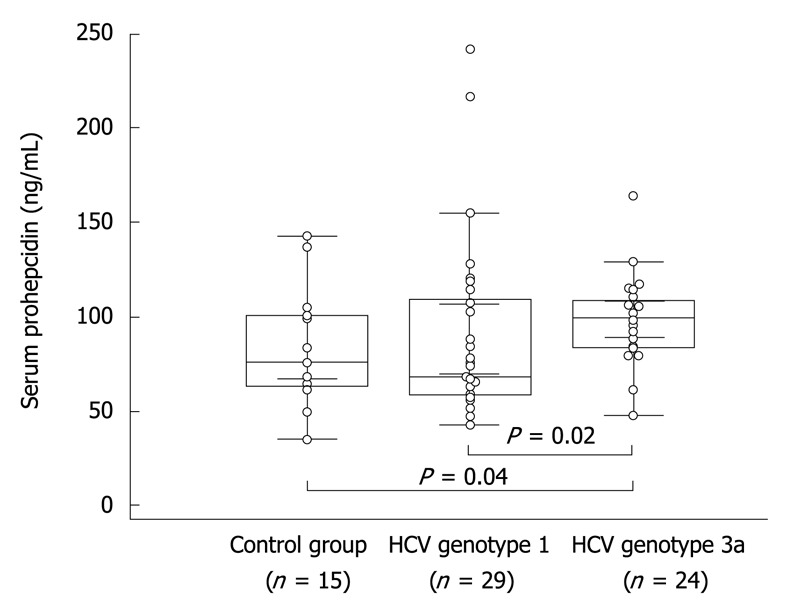
Baseline concentration of prohepcidin in healthy individuals as well as in hepatitis C virus (HCV) genotype 1 and genotype 3 patients. Dots indicate individual values in studied groups, boxes depict mean and standard error of mean, bars show standard deviations.
Baseline serum prohepcidin concentration showed a positive correlation with serum ferritin in chronic hepatitis C (R = 0.50, P = 0.02) (Table 2). In HCV genotype 1 the positive correlation between prohepcidin and ferritin was even stronger (R = 0.63, P = 0.001). Furthermore, an association with alanine aminotransferase (ALT) activity (R = 0.38, P = 0.04) was observed. We did not notice correlations between serum prohepcidin and either age, sex, liver histology, baseline HCV viral load, serum iron or TIBC.
Table 2.
Correlations between baseline serum prohepcidin (ng/mL), ALT activity, HCV-RNA, and iron metabolism parameters in studied population
|
Genotype 1 |
Genotype 3a |
|||
| R | P | R | P | |
| HCV-RNA (cp/mL) | -0.09 | 0.620 | 0.08 | 0.750 |
| ALT (U/L) | 0.38 | 0.0401 | -0.06 | 0.810 |
| Fe (μg/dL) | -0.06 | 0.510 | -0.48 | 0.0401 |
| Hgb (g/dL) | -0.03 | 0.850 | 0.18 | 0.420 |
| TIBC (μg/dL) | 0.07 | 0.730 | -0.11 | 0.640 |
| Ferritin (ng/mL) | 0.63 | 0.0011 | 0.25 | 0.330 |
Statistical significance calculated by use of spearman rank test.
Serum prohepcidin concentrations showed an association with PEG-IFN/RBV therapy effectiveness. In the first 7 d after the introduction of PEG-IFN/RBV, a gradual and reversible decrease in serum prohepcidin was observed regardless of HCV genotype (Tables 3 and 4). In the following phase, a further decrease of prohepcidin during antiviral therapy was noted in the HCV genotype 1 responder group, from 88.8 ± 14.7 ng/mL before therapy to 60.6 ± 2.3 ng/mL in the 48th wk (Friedman ANOVA test, χ2 = 9.6, P = 0.04). Such an association was not revealed in non-responders, with serum prohepcidin showing stable values (88.3 ± 11.4 ng/mL at baseline vs 82.3 ± 4.4 ng/mL in 48th wk) (Table 3 and Figure 2). A similar relationship was observed in genotype 3a individuals, with a statistically significant decrease in serum prohepcidin only in responders (99.5 ± 5.2 ng/mL at baseline vs 72.7 ± 6.1 ng/mL in the 24th wk, P = 0.01) (Table 4 and Figure 2).
Table 3.
Serum prohepcidin concentrations in genotype 1 HCV-infected patients during antiviral therapy with respect to SVR (mean ± SE)
|
Prohepcidin (ng/mL) |
P | ||
| Responders (n = 13) | Non-responders (n = 16) | ||
| Baseline | 88.8 ± 14.7 | 88.3 ± 11.4 | 0.980 |
| 6 h | 75.2 ± 10.2 | 79.5 ± 7.2 | 0.620 |
| 24 h | 72.6 ± 6.8 | 68.4 ± 4.6 | 1.000 |
| 48 h | 67.9 ± 7.6 | 64.8 ± 4.3 | 0.930 |
| 7th d | 87.9 ± 8.6 | 85.9 ± 9.6 | 0.690 |
| 4th wk | 77.7 ± 9.8 | 76.0 ± 7.8 | 0.920 |
| 12th wk | 71.6 ± 6.7 | 76.1 ± 5.2 | 0.350 |
| 24th wk | 63.9 ± 4.8 | 82.4 ± 4.3 | 0.004 |
| 48th wk | 60.6 ± 2.3 | 82.3 ± 4.4 | < 0.001 |
| ANOVA1 | χ2 = 9.6, P = 0.040 | χ2 = 2.8, P = 0.590 | |
Calculated from data collected at baseline and weeks 4, 12, 24, 48. SVR: Sustained viral response.
Table 4.
Serum prohepcidin concentrations in genotype 3a HCV-infected patients during antiviral therapy with respect to SVR (mean ± SE)
|
Prohepcidin (ng/mL) |
P | ||
| Responders (n = 21) | Non-responders (n = 3) | ||
| Baseline | 99.5 ± 5.2 | 94.1 ± 7.9 | 0.620 |
| 6 h | 87.6 ± 5.1 | 83.1 ± 4.7 | 0.760 |
| 24 h | 84.3 ± 4.0 | 78.6 ± 6.5 | 0.730 |
| 48 h | 76.8 ± 7.0 | 97.3 ± 22.1 | 0.630 |
| 7th d | 99.6 ± 6.1 | 96.2 ± 6.3 | 0.940 |
| 4th wk | 97.5 ± 6.3 | 84.9 ± 18.8 | 0.640 |
| 12th wk | 83.7 ± 4.7 | 90.1 ± 13.3 | 0.750 |
| 24th wk | 72.7 ± 6.1 | 103.5 ± 12.4 | 0.080 |
| ANOVA1 | χ2 = 14.8, P = 0.010 | - | |
Calculated from data collected at baseline and weeks 4, 12, 24. ANOVA was not calculated for non-responders because of insufficient number of patients.
Figure 2.
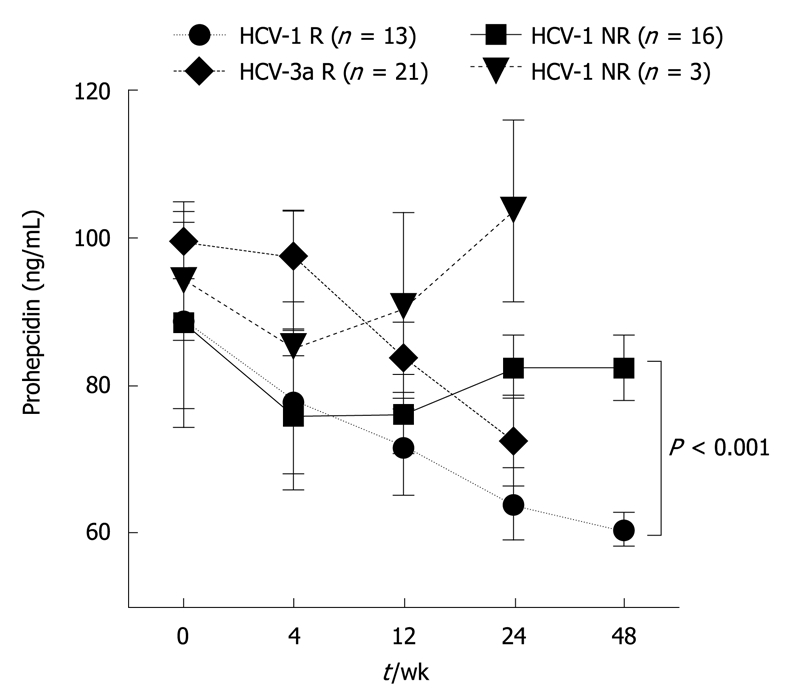
Serum prohepcidin concentrations in chronic hepatitis C during antiviral therapy with respect to the treatment response. R: Responders [HCV-RNA undetectable 6 mo after the end of pegylated interferon (PEG-IFN) + ribavirin (RBV) therapy]; NR: Non-responders (HCV-RNA positive 6 mo after the end of PEG-IFN + RBV therapy).
The above mentioned observations were further confirmed by the significant association between serum prohepcidin and HCV viral load during the treatment. HCV-RNA at the 12th wk was positively correlated with baseline (R = 0.63, P < 0.005) and 12th wk (R = 0.60, P = 0.01) serum prohepcidin concentrations in genotype 1 HCV-infected individuals. A similar analysis was not possible for genotype 3a since HCV-RNA was detectable only in 6 of 24 patients at the 12th wk.
To assess the possibility of the interference of RBV-induced anemia with prohepcidin synthesis we performed a correlation analysis between hemoglobin concentration and prohepcidin. We found no association between these parameters at baseline (Table 1) or at different time points during the PEG-IFN/RBV therapy (data not shown).
DISCUSSION
Recent years have brought to light new facts concerning the pathogenesis of iron metabolism alterations in chronic liver diseases. The discovery of hepcidin has linked inflammatory disorders and iron turnover disturbances[8]. The name of hepcidin originated from its antimicrobial properties and the predominant production of this protein in the liver. Following infectious, inflammatory stimuli or iron-overload its production increases, resulting in the decrease of plasma iron by negative regulation of iron uptake by duodenal enterocytes and sequestration by macrophages[12,17]. In contrast, down-regulation of hepcidin is an important factor facilitating iron deposition in parenchymal organs, especially in the liver. The regulation of hepcidin production is multilateral and still not fully understood. Main pathways involved include iron-storage, hypoxic and inflammatory-related[18]. Hemochromatosis protein (HFE) seems to maintain the basal expression of hepcidin, probably via TfR2 and hemojuvelin, acting as a body iron store sensor[19]. In contrast, inflammatory-related hepcidin induction appears to be independent of HFE, TfR2 and mainly relies on interleukin-6 (IL-6) via STAT-3 activation[20,21].
It is only in the last years that some insights into the role of hepcidin in chronic hepatitis C have been presented. Nagashima et al[13] found significantly lower concentrations of serum prohepcidin in chronic hepatic C compared to chronic HBV infection and healthy individuals, suggesting that failure of prohepcidin regulation may be induced by HCV infection. More recently, Fujita et al[22] evaluated hepatic hepcidin mRNA expression in chronic hepatitis B and C individuals. They showed significant positive correlations between hepatic hepcidin expression, serum iron, and ferritin, as well as liver total iron score. However, liver hepcidin mRNA expression was comparable in chronic hepatitis B and C in this study. Only after adjustment for serum ferritin were hepcidin indices significantly lower in HCV infection. The same group[23] found a positive correlation between hepatic oxidatively generated DNA damage and serum ferritin, total iron score and liver hepcidin mRNA in chronic hepatitis C. Interestingly, they also showed more prominent baseline hepatic oxidative stress markers in non-SVR to IFN/ribavirin therapy.
We investigated the association between PEG-IFN/ribavirin therapy and iron metabolism parameters, especially hepcidin prohormone, prohepcidin, concentrations in chronic hepatitis C. We found comparable levels of serum prohepcidin in chronic hepatitis C and healthy individuals. Interestingly enough, baseline prohepcidin was significantly higher in HCV genotype 3a than in HCV-1. This new finding could be possibly explained by differences in iron metabolism in patients with genotype 3. Sebastiani et al[3] showed that hepatic iron deposits were significantly more frequent in HCV-3-infected individuals and strongly associated with viral-induced hepatic steatosis.
Baseline serum prohepcidin in this current study showed a strong positive correlation with serum ferritin. Moreover, an association between baseline serum prohepcidin and ALT activity was found. These observations are in accordance with other reports[22] and probably reflect above mentioned pathways of hepcidin regulation. The new finding of this study is the association between serum prohepcidin and PEG-IFN/ribavirin efficacy in chronic hepatitis C. We showed a statistically significant, gradual decrease of serum prohepcidin during successful antiviral treatment in HCV-1 and 3a individuals. The reduction in prohepcidin concentration for genotype 1 was 31% in the 48th wk of treatment (Table 3) and 27% in the 24th wk for HCV-3a (Table 4) in the SVR groups. In contrast, in non-SVR the prohepcidin serum concentrations did not change significantly compared to baseline values. This finding seems to contradict findings of a recent clinical study performed by Fujita et al[6], however it has to be underlined that the current study measured a hepcidin precursor, not an active hormone. In the aforementioned work[6], serum hepcidin was measured by mass spectrometry in 73 untreated chronic hepatitis C patients and in 27 patients during PEG-IFN + RBV therapy. The baseline serum hepcidin was higher in chronic hepatitis C compared to healthy individuals but only in those with hyperferritinemia. On the other hand, HCV-infected individuals with normal ferritinemia had comparable serum prohepcidin with the control group. Interestingly, the most recent analysis by Girelli et al[24] showed that in a rigorously selected population with HFE genotyping performed, serum hepcidin was significantly reduced in chronic hepatitis C in comparison to healthy controls and strongly associated with body iron deposits, regardless of IL-6 concentrations.
The current study applied a different approach. The assay used detected a hepcidin prohormone, not a mature peptide. It has been shown that serum assessment of prohepcidin does not reflect the levels of biologically active mature hepcidin peptide[14]. On the contrary, it may give some insight into the mechanisms of hepcidin maturation. Hepcidin is almost exclusively synthesized in the liver as a 84 amino acid prepropeptide, which is further processed in hepatocytes to the mature form[25]. Recent evidence showed that the proteolytic cleavage of prohepcidin to hepcidin is regulated by the hepatic prohormone convertase furin[14]. The mechanism involved in the transport of hepcidin to the extracellular zone is not fully understood. Valore at al[14] showed that a larger hepcidin precursor protein undergoes two cleavages (the signal sequence, then the pro-region) and is rapidly secreted from the cell. Moreover, in primary human hepatocytes, prohepcidin was transiently detected in culture supernatant immediately after labeling but it appeared to be present at low levels compared to mature hepcidin. Of interest, the inhibition of furin activity prevented the conversion of prohepcidin to hepcidin but also appeared to stabilize the prohepcidin peptide both in the cell and the media. In situations of liver function impairment the prohepcidin synthesis as well as activity or expression of converting enzymes might be altered and affect circulating prohepcidin concentrations. This finding could also suggest that HCV interference with hepcidin synthesis may occur at the step of prohormone synthesis or maturation in the liver.
In conclusion, we found an association between successful PEG-IFN/ribavirin therapy and serum prohepcidin concentration decrease in chronic hepatitis C patients. This may suggest a direct link between HCV and hepcidin maturation in the liver.
COMMENTS
Background
Iron accumulation in the liver has a negative influence on chronic hepatitis C outcome. It has been linked with oxidative DNA damage, insulin resistance and liver steatosis, but may also trigger hepatic stellate cell activation and thus induce liver fibrosis. Recently, a key iron regulatory hormone, hepcidin, was discovered. This hormone has been found to suppress intestinal absorption of iron through its binding to ferroportin. Hepcidin is synthesized in the liver from its precursor protein, prohepcidin.
Research frontiers
The mechanisms of iron metabolism deregulation in chronic hepatitis C are not fully elucidated. Hepcidin, a versatile regulator of iron homeostasis is likely engaged in this process. Nevertheless, the factors influencing maturation of hepcidin in the setting of chronic liver injury are unclear. Recently, the authors have suggested the association between serum prohepcidin levels and liver function impairment in liver cirrhosis patients. The aim of the current study was to assess the serum concentration of hepcidin prohormone in chronic hepatitis C and evaluate its possible association with disease activity as well as with the efficacy of pegylated interferon (PEG-IFN)/ribavirin therapy.
Innovations and breakthroughs
In the present study, comparable levels of serum prohepcidin in chronic hepatitis C and healthy individuals were noted. Interestingly, baseline prohepcidin was significantly higher in hepatitis C virus (HCV) genotype 3a than in HCV genotype 1. The new finding of the study is the association between serum prohepcidin and PEG-IFN/ribavirin efficacy in chronic hepatitis C. A statistically significant, gradual decrease of serum prohepcidin during successful antiviral treatment in HCV-1 and 3a individuals was shown, which was not the case in patients who did not respond to anti-HCV therapy.
Applications
The results of this study may suggest that in situations of liver function impairment prohepcidin synthesis, as well as activity or expression of converting enzymes, might be altered and affect circulating prohepcidin concentrations. This finding could also suggest that HCV interference with hepcidin synthesis may occur at the step of prohormone synthesis or maturation in the liver.
Peer review
This study was conducted to elucidate the effect of HCV and the influence of PEG-IFN/ribavirin combination therapy on the iron metabolism in patients with chronic hepatitis C. The manuscript was well prepared. The data analysis was appropriate. The authors obtained a reasonable result and the discussion was good.
Footnotes
Supported by A Scientific Grant from the Medical University in Bialystok, No. 356978-L and a Polpharma Foundation For Development of Polish Pharmacy and Medicine
Peer reviewer: Wan-Long Chuang, MD, PhD, MS, Professor, Director, Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University, No. 100 Shih-Chuan 1st Road, Kaohsiung 807, Taiwan, China
S- Editor Wang JL L- Editor Logan S E- Editor Ma WH
References
- 1.Fujita N, Sugimoto R, Urawa N, Tanaka H, Konishi M, Kobayashi Y, Iwasa M, Watanabe S, Kaito M. Influence of phlebotomy on iron-related gene expression levels in the livers of patients with chronic hepatitis C. J Gastroenterol. 2007;42:326–327. doi: 10.1007/s00535-007-2004-5. [DOI] [PubMed] [Google Scholar]
- 2.Sumida Y, Kanemasa K, Fukumoto K, Yoshida N, Sakai K. Hepatic iron accumulation may be associated with insulin resistance in patients with chronic hepatitis C. Hepatol Res. 2007;37:932–940. doi: 10.1111/j.1872-034X.2007.00152.x. [DOI] [PubMed] [Google Scholar]
- 3.Sebastiani G, Vario A, Ferrari A, Pistis R, Noventa F, Alberti A. Hepatic iron, liver steatosis and viral genotypes in patients with chronic hepatitis C. J Viral Hepat. 2006;13:199–205. doi: 10.1111/j.1365-2893.2005.00662.x. [DOI] [PubMed] [Google Scholar]
- 4.Martinelli AL, Ramalho LN, Zucoloto S. Hepatic stellate cells in hepatitis C patients: relationship with liver iron deposits and severity of liver disease. J Gastroenterol Hepatol. 2004;19:91–98. doi: 10.1111/j.1440-1746.2004.03255.x. [DOI] [PubMed] [Google Scholar]
- 5.Kakizaki S, Takagi H, Horiguchi N, Toyoda M, Takayama H, Nagamine T, Mori M, Kato N. Iron enhances hepatitis C virus replication in cultured human hepatocytes. Liver. 2000;20:125–128. doi: 10.1034/j.1600-0676.2000.020002125.x. [DOI] [PubMed] [Google Scholar]
- 6.Fujita N, Sugimoto R, Motonishi S, Tomosugi N, Tanaka H, Takeo M, Iwasa M, Kobayashi Y, Hayashi H, Kaito M, et al. Patients with chronic hepatitis C achieving a sustained virological response to peginterferon and ribavirin therapy recover from impaired hepcidin secretion. J Hepatol. 2008;49:702–710. doi: 10.1016/j.jhep.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Takeo M, Kobayashi Y, Fujita N, Urawa N, Iwasa M, Horiike S, Tanaka H, Kaito M, Adachi Y. Upregulation of transferrin receptor 2 and ferroportin 1 mRNA in the liver of patients with chronic hepatitis C. J Gastroenterol Hepatol. 2005;20:562–569. doi: 10.1111/j.1440-1746.2005.03770.x. [DOI] [PubMed] [Google Scholar]
- 8.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 9.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 10.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi SO, Cho YS, Kim HL, Park JW. ROS mediate the hypoxic repression of the hepcidin gene by inhibiting C/EBPalpha and STAT-3. Biochem Biophys Res Commun. 2007;356:312–317. doi: 10.1016/j.bbrc.2007.02.137. [DOI] [PubMed] [Google Scholar]
- 12.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–2463. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 13.Nagashima M, Kudo M, Chung H, Ishikawa E, Hagiwara S, Nakatani T, Dote K. Regulatory failure of serum prohepcidin levels in patients with hepatitis C. Hepatol Res. 2006;36:288–293. doi: 10.1016/j.hepres.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Valore EV, Ganz T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol Dis. 2008;40:132–138. doi: 10.1016/j.bcmd.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaroszewicz J, Rogalska M, Flisiak R. Serum prohepcidin reflects the degree of liver function impairment in liver cirrhosis. Biomarkers. 2008;13:478–485. doi: 10.1080/13547500802033391. [DOI] [PubMed] [Google Scholar]
- 16.Scheuer PJ. Classification of chronic viral hepatitis: a need for reassessment. J Hepatol. 1991;13:372–374. doi: 10.1016/0168-8278(91)90084-o. [DOI] [PubMed] [Google Scholar]
- 17.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oates PS, Ahmed U. Molecular regulation of hepatic expression of iron regulatory hormone hepcidin. J Gastroenterol Hepatol. 2007;22:1378–1387. doi: 10.1111/j.1440-1746.2007.04950.x. [DOI] [PubMed] [Google Scholar]
- 19.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 20.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–3209. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109:353–358. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 22.Fujita N, Sugimoto R, Takeo M, Urawa N, Mifuji R, Tanaka H, Kobayashi Y, Iwasa M, Watanabe S, Adachi Y, et al. Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol Med. 2007;13:97–104. doi: 10.2119/2006-00057.Fujita. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujita N, Horiike S, Sugimoto R, Tanaka H, Iwasa M, Kobayashi Y, Hasegawa K, Ma N, Kawanishi S, Adachi Y, et al. Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis C patients. Free Radic Biol Med. 2007;42:353–362. doi: 10.1016/j.freeradbiomed.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Girelli D, Pasino M, Goodnough JB, Nemeth E, Guido M, Castagna A, Busti F, Campostrini N, Martinelli N, Vantini I, et al. Reduced serum hepcidin levels in patients with chronic hepatitis C. J Hepatol. 2009;51:845–852. doi: 10.1016/j.jhep.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loréal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]