

Abstract
The structure of recombinant P. aeruginosa bacterioferritin B (Pa BfrB) has been solved from crystals grown from protein devoid of core mineral iron (as-isolated) and from protein mineralized with ~ 600 iron atoms (mineralized). Structures were also obtained from crystals grown from mineralized BfrB after soaking them in FeSO4 solution (Fe soak) and in separate experiments after soaking them in FeSO4 solution followed by soaking in crystallization solution (double soak). Although the structures consist of a typical bacterioferritin fold comprised of a nearly spherical 24-mer assembly that binds 12 heme molecules, comparison of microenvironments observed in the distinct structures provided interesting insights: The ferroxidase center in the as-isolated, mineralized and double soak structures is empty. The ferroxidase ligands (except His130) are poised to bind iron with minimal conformational changes. The His130 side chain, on the other hand, must rotate toward the ferroxidase center to coordinate iron. In comparison, the structure obtained from crystals soaked in an FeSO4 solution display a fully occupied ferroxidase center and iron bound to the internal, Fe(in), and external, Fe(out), surfaces of Pa BfrB. The conformation of His130 in this structure is rotated toward the ferroxidase center and coordinates an iron ion. The structures also revealed a pore on the surface of Pa BfrB that likely serves as an entry port for Fe2+ to the ferroxidase center. On its opposite end the pore is capped by the side chain of His130 when it adopts its “gate closed” conformation that enables coordination to a ferroxidase iron. A change to its “gate-open”, non-coordinative conformation, creates a path for the translocation of iron from the ferroxidase center to the interior cavity. These structural observations, together with findings obtained from iron incorporation measurements in solution suggest that the ferroxidase pore is the dominant entry route for the uptake of iron by Pa BfrB. These findings, which are clearly distinct from those made with E. coli Bfr (Crow, A. C., Lawson, T. L., Lewin, A., Moore, G. R., and Le Brun, N. E. (2009) J. Am. Chem. Soc. 131, 6808–6813) indicate that not all bacterioferritins operate in the same manner.
The chemical properties of the biologically relevant oxidation states of iron, i. e. the insolubility of the Fe3+ ion and the propensity of the soluble Fe2+ ion to react with O2 to produce reactive oxygen species, likely influenced the development of iron storage proteins as integral components of the iron homeostasis machinery in living cells. These iron storage proteins, termed ferritin and ferritin-like proteins, are Nature's solution to overcome the insolubility of the Fe3+ ion and the toxicity of the Fe2+ ion in aerobic environments, because they function by oxidizing Fe2+ utilizing O2 as electron acceptors and storing the resultant Fe3+ ions in the form of an iron mineral isolated from the aqueous cell environment. Studies with bacterioferritin from E. coli (1) and Azotobacter vinelandii (2) showed that O2 is reduced to H2O, whereas in the case of eukaryotic ferritins two mechanisms are observed; at iron loads less than 50 Fe2+/ferritin O2 is reduced to H2O2 and at higher loads O2 is reduced to H2O (3). Eukaryotic ferritins are composed of 24 subunits that assemble into a spherical protein with a hollow internal cavity, approximately 80 Å in diameter, where the iron mineral is stored (4). The arrangement of the subunits produces a structure that possesses 432 point symmetry and results in the formation of pores along the 4-fold and 3-fold symmetric molecular axes. Bacterioferritins (Bfr) also assemble from 24 subunits into a similar architecture, except that these molecules bind 12 heme molecules. In contrast the Dps (DNa protection during starvation) proteins assemble from 12 subunits to form a central cavity of smaller proportions compared to those formed by ferritins and bacterioferritins (5).
The process of iron uptake starts when Fe2+ is captured by the protein to form a diferrous center, known as the ferroxidase center, where it is oxidized to Fe3+. In mammalian ferritins the dinuclear iron site, which is present in the H-chain subunits but not in the L-subunits, is central to the mineralization process (6). The ferroxidase center of Bfr is also central to Fe2+ ion oxidation (1, 7, 8) and its structurally symmetric architecture consisting of two bridging Glu ligands (Glu51 and Glu127) and His and Glu residues as capping ligands (Glu18/His54 and Glu94/His130) (4) (Figure 1-A), resemble the architecture of di-iron type II proteins such as soluble methane monooxygenase and ribonucleotide reductase (9), where the di-iron center is stable and acts as a cofactor. In comparison, in the ferroxidase center of mammalian ferritins only one iron is coordinated by a capping His ligand (His65) and the two iron ions are bridged only by Glu62 (1, 4) (Figure 1-B).
Figure 1.

Schematic representation of: (A) The symmetrical ferroxidase center typical of bacterioferritin. Bridging glutamates are Glu51 and Glu127 and the capping residue pairs are Glu18/His54 and Glu94/His130. (B) The ferroxidase center of human H-chain ferritin adapted from the crystal structure of the Tb3+ derivative (43).
The process of mineralization has been a major focus of studies aimed at investigating ferritin-like molecules over approximately the last two decades (4, 5, 10). The wealth of information derived from kinetic and more recently structural studies have been used to support at least two models with three distinct kinetic phases (4, 7, 11): In the first model, phase 1 is the binding of Fe2+ to a vacant ferroxidase center, phase 2 is oxidation of the Fe2+, creating an unstable diferric species, and phase 3 is movement of the unstable Fe3+ into the internal cavity where it is incorporated into the core; subsequent binding of Fe2+ to the ferroxidase center starts a new cycle. Hence, in this model, the ferroxidase center serves a dual role of substrate binding site for the oxidation of iron and the pore for its internalization. In the second model phases 1 and 2 are the same as in model one; phase 3, however, differs in that the diferric center is stable and functions strictly as a cofactor. Fe2+ ions are assumed to enter the Bfr cavity via channels in the structure of the protein where they are oxidized at nucleating centers so that electrons released from their oxidation are relayed to the ferroxidase center for the reduction of O2 and H2O2. Kinetic studies conducted with eukaryotic ferritins have shown that at low flux (< 50 Fe2+ ions per/ferritin molecule) the Fe2+ is oxidized by the ferroxidase center with stoichiometry Fe2+:O2 of 2:1. At higher flux, however, the mechanism changes, as indicated by a change in stoichiometry to Fe2+:O2 = 4:1 (3). In comparison most of the information regarding the process of mineralization by Bfr has been obtained from studies of Bfr from Escherichia coli (Ec Bfr) (1, 4, 7, 11). Information derived from kinetic studies strongly suggest that the ferroxidase center in Ec Bfr is stable in its oxidized form and does not revert spontaneously to the apo-form (devoid of ferroxidase iron) (1, 7). In other words, the second model, where the ferroxidase center is stable and functions only as a cofactor appears to be operative in Ec Bfr. Additional support for these ideas has been obtained from recent structural work conducted with Ec Bfr (12). It was observed that the apo form of the ferroxidase center is preformed since the ferroxidase ligands bind Fe2+ with minimal reorganization. Soaking crystals of the apo-ferroxidase form in a solution of Fe2+ enables assembly of a ferroxidase center, where the di-Fe2+ center oxidizes to a stable di-Fe3+ species that appears to be μ-oxo or μ-hydroxo bridged. Moreover, the structure does not reveal a possible route for translocating iron from the ferroxidase iron to the internal cavity. These structural observations, together with observations made from kinetic studies conducted with Ec Bfr, led to the suggestion that the ferroxidase center in Bfrs operates as a true catalytic center, rather than a pore for iron entry into the Bfr cavity (12), as is the case for eukaryotic ferritins.
In addition to the structure of Ec Bfr (12, 13), the structures of Bfrs from Azotobacter vinelandii (Av) (14), Mycobacterium smegmatis (Ms) (15), Desulfovibrio desulfuricans (Dd) (16) and Rhodobacter capsulatus (Rc) (17) have been reported. The ferroxidase center in the structure of Ms Bfr contains zinc instead of iron and is empty in the structure of Rc Bfr. In the structure of Dd Bfr the ferroxidase center is occupied by iron in the “as isolated” Bfr but if the protein is reduced and then allowed to oxidize in air, one of the ferroxidase iron ions is lost. This observation led to the suggestion that the ferroxidase site in Dd Bfr may function as a pore for the internalization of iron (16) although a path for the translocation of iron from the ferroxidase center to the internal cavity was not apparent in the structures. In the structure of Av Bfr the ferroxidase center is occupied by iron but the Fe2 position (nomenclature as in figure 1-A) has low occupancy, an observation that was also interpreted to suggest that Fe3+ is translocated from the ferroxidase center to the internal cavity (14). It is also interesting that the side chain of His130 in the Av Bfr structure moved away from Fe2, while the side chain of Glu47 moved closer, suggesting that the concerted motion of these side chains may constitute a gating mechanism for the translocation of iron from the ferroxidase center to the interior cavity (14).
The contrasting observations made with the structures of Ec Bfr and those from Av and Dd suggest that some Bfrs harbor a stable ferroxidase center that functions exclusively as a cofactor for the oxidation of Fe2+ that gained access into the cavity via channels in the structure, whereas other Bfrs form an “unstable” ferroxidase center that functions in the oxidation of Fe2+ and as a pore for the translocation of Fe3+ into the internal cavity. Although the function of the ferroxidase center in Ec Bfr seems almost certain, the function of the ferroxidase center in Bfrs harboring an unstable ferroxidase center is still uncertain. Herein we report that the high-resolution structure of Pseudomonas aeruginosa bacterioferritin B (Pa BfrB) reconstituted with ~ 600 iron atoms in its core (mineralized Pa BfrB) harbors an empty ferroxidase center, which indicates that the ferroxidase iron needed to mineralize ~ 600 core iron ions is labile. Soaking crystals of mineralized Pa BfrB results in a Pa BfrB structure (Fe soak Pa BfrB) harboring a ferroxidase center, iron bound to the external surface of the protein and iron bound to the internal surface of the protein, which indicates that Pa BrB can capture and mineralize iron in the crystal. Comparison of the structures reveals a ferroxidase pore for the entry of iron to the ferroxidase center, and upon reorganization of the His130 side chain (a ligand to Fe2) a clear route for the translocation of ferroxidase iron to the internal cavity. These structural observations taken together strongly support the idea the ferroxidase center in Pa BfrB functions in a dual role of cofactor and pore for the oxidation and internalization of iron.
Experimental Procedures
Protein Preparation and Purification
Recombinant Pa BfrB was prepared and purified as previously described (18); the pure protein, as isolated, binds 12 heme groups and contains ~ 5–10 atoms of non-heme iron in its core. Analysis of heme and non-heme iron was carried out as reported previously (18). The integrity of the 24-mer assembly was probed by estimating the molecular mass of the protein with the aid of a high-resolution size exclusion column (Superdex 200 16/60 Prep; GE Healthcare) utilizing previously reported conditions (18). Pure protein was dialyzed against 100 mM potassium phosphate buffer containing 5 mM TCEP and stored at −80 °C. When appropriate, the core of Pa Bfr B was reconstituted (mineralized) with ~ 600 iron atoms per protein molecule. To this end, a solution of ferrous ammonium sulfate was prepared inside an anaerobic chamber (Coy Laboratories, Grass lake, MI), placed in a container capped with a rubber septum, and removed from the chamber. Concentrated HCl (50 μL/100 mL) was added to the anaerobic ferrous ammonium sulfate solution through the septum with the aid of a Hamilton microsyringe and a needle. The resultant solution was added to a stirred solution of 0.002 mM Pa BfrB and 5.0 mM TCEP in 100 mM potassium phosphate buffer, pH 7.6 in aliquots delivering 50 Fe2+ ion/Pa BfrB, until 600 Fe2+/Pa BfrB had been added. Ten minutes were allowed after the addition of one aliquot before the next was added. The mineralization process was monitored spectrophotometrically following the absorbance near 320 nm with the aid of a USB 2000 spectrophotometer (Ocean Optics, Dunedin, FL).
Crystallization and Data Collection
Mineralized or non-mineralized (as-isolated) Pa BfrB were concentrated to 10 mg/mL and 9 mg/mL, respectively in 100 mM potassium phosphate pH 7.6, 5 mM TCEP for crystallization. Crystals were grown under aerobic conditions in Compact Jr. (Emerald biosystems) sitting drop vapor diffusion plates using 1μL of crystallization solution and 1 μL of protein equilibrated against 100 μL of crystallization solution at 20 °C. Red prisms were obtained within 24 hours from the following conditions: Mineralized BfrB (Wizard 2 #2, Emerald biosystems) 35% (v/v) 2-methyl-2,4-pentanediol, 100 mM MES pH 6.0, 200 mM Li2SO4; as isolated Pa BfrB (Cryo 1 #11, Emerald biosystems) 35% (v/v) 2-methyl-2,4-pentanediol, 100 mM Tris pH 8.5, 200 mM (NH4)2SO4. To prepare samples with iron bound, crystals of mineralized BfrB were soaked aerobically for 15 min in crystallization solution (35% v/v 2-methyl-2,4-pentanediol, 100 mM MES pH 6.0, 200 mM Li2SO4) containing 50 mM FeSO4. Doubly soaked crystals were prepared by aerobically soaking mineralized BfrB crystals in the same crystallization solution containing 50 mM FeSO4, followed by aerobically soaking for 15 min in the same crystallization solution without FeSO4. Single crystals were transferred to a fresh drop of crystallization solution which also served as a cryoprotectant and frozen in liquid nitrogen for data collection. Data were collected at 100 K at the Advanced Photon Source (APS) beamline 17-BM (IMCACAT) using an ADSC Quantum 210 CCD detector at λ = 1.0000 Å. Additional data were collected from the same crystals for mineralized and Fe soak crystals at λ = 1.6314 Å in order to measure an anomalous signal from potentially bound Fe ions. Although the X-ray fluorescence K edge emission peak of Fe has a maximum approximately at 1.73 Å, at 1.6314 Å a prominent anomalous signal can be observed for Fe ions.
Structure Solution and Refinement
Intensities were integrated and scaled using the HKL2000 package (19). Structure solution was carried out by molecular replacement with Molrep (20) using the full 24 subunit assembly of Ec BfrB as the search model (PDB: 1BFR). Rotation and translation searches for the 24-subunit assembly yielded a clear solution with a correlation coefficient of 0.735. The refined model of Pa BfrB was used as the search model for subsequent data sets. For the as isolated Pa BfrB, a single subunit from the final mineralized Pa BfrB structure was used as the search model. Six molecules were positioned with Molrep yielding a correlation coefficient of 0.703. Structure refinement and manual model building were performed with Refmac (21) and Coot (22) respectively. Medium NCS restraints were applied to the double soaked model. Figures were prepared using the Ribbons (23) and CCP4mg (24) software packages. Structure validation was performed with Molprobity (25).
Iron Uptake in Solution
Pa BfrB to be used in these experiments was prepared by extensively dialyzing the pure, as-isolated protein against 100 mM MES, 100 mM KCl, pH 6.5. Fe2+ oxidation by Pa-BfrB was measured with the aid of a conventional diode-array UV-vis spectrophotometer (Ocean Optics, Dunedin, FL), following addition of 50 Fe2+ ions/Pa BfrB molecule to a solution of Pa BfrB (0.8 μM) at 30 °C in 100 mM MES buffer, 100 mM KCl, pH 6.5 in a 1.0 cm path length cuvette equipped with a stirring bar. The process was also studied with the aid of a Applied Photophysics SX.18MV-R Stopped-Flow Spectrophotometer (Applied Photophysics Ltd., Leatherhead, UK), which was used to study the iron uptake process by mixing solutions of ferrous ammonium sulfate and Pa BfrB in Fe2+:protein ratios of 30, 50, 100, 200 and 300. These experiments were also carried out at 30 °C in 100 mM MES buffer, 100 mM KCl, pH 6.5; protein concentration was 1.0 μM (after mixing) and the path length was 1.0 cm. Progress curves, monitored at 320 nm, were obtained from averaging 6 replicates for each Fe2+ concentration. Rate constants were calculated by fitting the first 5 s of progress curves obtained upon addition of 30, 50 and 100 Fe2+ ions, which allowed distinction between the fast and slow phases, to a single exponential function.
Results and Discussion
Initial crystallization trials of recombinant mineralized Pa BfrB rendered red prisms from various crystallization conditions. The primitive orthorhombic crystal form described here, containing 24 molecules of Pa BfrB in the asymmetric unit, diffracted to the highest resolution (Table 1). In addition, an orthorhombic I crystal form containing six molecules in the asymmetric cell was obtained later from the as-isolated Pa BfrB. The crystals obtained from mineralized Pa BfrB were also used to acquire diffraction data from crystals soaked for 15 minutes in an iron solution (Fe soak Pa BrfB) and in a separate set of experiments to acquire data after soaking them for 15 minutes in an iron solution, followed by a 15 minute soak in crystallization solution without iron present (double-soak Pa BfrB). Although the coarse properties of the structures determined from all these experiments are very similar, there are important differences brought about by the binding of iron to the ferroxidase center, as well as to the external and internal surfaces of the protein. These results and their potential implications to the function of Pa BfrB are presented below.
Table 1.
Crystallographic data for Pa BfrB.
| BfrB (as isolated) | BfrB (mineralized) | BfrB (Fe Soak) | BfrB (Double Soak) | |
|---|---|---|---|---|
| Data Collection | ||||
| Unit-cell parameters (Å) | a = 122.76 | a = 125.81 | a = 125.72 | a = 125.71 |
| b = 126.27 | b = 202.76 | b = 203.28 | b = 203.21 | |
| c = 168.55 | c = 207.55 | c = 207.73 | c = 207.87 | |
| Space group | I222 | P212121 | P212121 | P212121 |
| Resolution (Å) | 50.0–2.07 (2.14–2.07) | 50.0–2.10 (2.18–2.10) | 50.0–2.25 (2.33–2.25) | 50.0–2.80 (2.90–2.80) |
| Wavelength (Å) | 1.6531 | 1.0000 | 1.0000 | 1.6531 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Observed reflections | 550,190 | 2,179,278 | 1,858,510 | 955,818 |
| Unique reflections | 79,588 | 309,226 | 251,658 | 131,241 |
| <I/σ(I)>1 | 20.3 (2.4) | 16.2 (2.0) | 13.1 (1.9) | 13.0 (2.3) |
| Completeness (%)1 | 99.5 (97.8) | 99.9 (99.4) | 99.7 (98.5) | 100 (100) |
| Redundancy1 | 6.9 (5.3) | 7.0 (6.3) | 7.4 (5.6) | 7.3 (6.5) |
| Rmerge (%)1,2 | 9.0 (50.4) | 10.9 (66.9) | 13.5 (69.2) | 17.0 (75.3) |
| Refinement | ||||
| Resolution (Å) | 50.0–2.07 | 50.0–2.10 | 50.0–2.25 | 50.0–2.80 |
| Molecules / asu | 6 | 24 | 24 | 24 |
| Reflections (working/test) | 75,351 / 3,973 | 291,584 / 15,512 | 237,971 / 12,585 | 124,548 / 6,600 |
| Rfactor / Rfree (%)3 | 19.8 / 24.5 | 19.3 / 24.4 | 17.7 / 22.6 | 19.5 / 23.7 |
| No. of atoms (protein / heme / Fe2+ / K+ / water) | 7703 / 172 / 0 / 2 / 363 | 30,491 / 516 / 0 / 6 / 1,512 | 30,805 / 516 / 96 / 6 / 1,500 | 30,414 / 516 / 24 / 6 / 0 |
| Model Quality | ||||
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.021 | 0.021 | 0.019 | 0.016 |
| Bond angles (°) | 1.871 | 1.824 | 1.683 | 1.553 |
| Average B factor (Å2) | ||||
| All Atoms | 38.9 | 27.3 | 25.0 | 39.5 |
| Protein | 38.6 | 26.9 | 24.5 | 39.4 |
| Heme | 39.4 | 27.8 | 28.8 | 46.6 |
| K+ / Fe2+ | 26.8 | 23.9 / NA | 29.4 / 30.3 | 31.4 / 49.4 |
| Water | 44.4 | 35.8 | 31.7 | |
| Coordinate error based on Rfree (Å) | 0.171 | 0.174 | 0.193 | 0.337 |
| Ramachandran Plot | ||||
| Favored (%) | 99.6 | 99.8 | 99.6 | 99.3 |
Values in parenthesis are for the highest resolution shell.
Rmerge = ΣhklΣi |Ii(hkl) − <I(hkl)>| / ΣhklΣi Ii(hkl), where Ii(hkl) is the intensity measured for the ith reflection and <I(hkl)> is the average intensity of all reflections with indices hkl.
Rfactor = Σhkl ||Fobs (hkl) | − |Fcalc (hkl) || / Σhkl |Fobs (hkl)|; Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
In common with the structure of ferritin and other bacterioferritins, the structure of Pa BfrB is comprised of 24 subunits that bind 12 heme molecules with an external diameter of ~ 118 Å and an internal cavity diameter of ~ 73 Å. Each monomeric subunit is built of four α-helices nearly parallel to one another and a short C'-terminal helix almost perpendicular to the four helix bundle. Two monomeric subunits assemble into a dimer that harbors one heme molecule axially coordinated by a methionine (Met52) from each subunit. For the as-isolated crystal form, two of the heme molecules were located on 2-fold crystallographic axes bisecting the CHA, FE and CHC atoms. Since a heme molecule is not 2-fold symmetric, it must necessarily adopt two orientations in a 50:50 ratio, in this case related by the crystallographic 2-fold axis. To model this statistical disorder, a single heme molecule was fit to the difference density map and all atoms set to occupancies of 0.5. The crystallographic 2-fold operation then generates the symmetry related mate at this site. A similar method was used to model the heme in Rd Bfr (17). Non-crystallographic three-fold and four-fold axes traverse six four-fold and four three-fold channels (Figure 2); potassium ions were found in the four-fold channels of Pa-BfrB in all structures reported herein (see below).
Figure 2.

Structure of Pa-BfrB viewed along four-fold axis (left) and along a non-crystallographic 3-fold axis. The green spheres represent potassium in each of the four-fold pores; the heme molecules are shown in gold.
The ferroxidase center in Pa-BfrB is unstable
Residues coordinating the di-iron center are highly conserved amongst bacterioferritins (Supplementary Material Figure S1). In the amino acid sequence of Pa BfrB these are predicted to be Glu18 and His54 as terminal ligands to Fe1, Glu94 and His130 as terminal ligands to Fe2, and Glu51 and Glu127 as bridging ligands. The electron density maps for these residues in the structure obtained from as isolated Pa BfrB were well defined. Nevertheless, no electron density consistent with metal ions in this site is present, indicating that the ferroxidase center of as isolated Pa BfrB is empty. It is also interesting to note that although most of the residues appear to be poised to bind iron, His130 is rotated away from the ferroxidase center and would need to undergo a conformational change in order to bind the Fe2 ion (Figure 3). As indicated in Experimental Procedures, anomalous difference Fourier analysis was conducted using data collected at 1.6314 Å. These experiments produced prominent anomalous signal originating from the Fe atoms at the heme sites but negligible electron density at the ferroxidase center, thus confirming that the ferroxidase center in as isolated Pa BfrB is empty.
Figure 3.
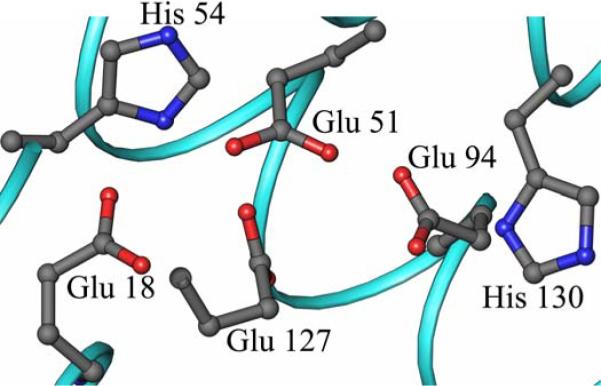
The ferroxidase center of mineralized Pa BfrB is devoid of iron. The side chains furnishing the ligands are poised to bind iron with minimal rearrangement, with the exception of His 130 which is rotated away from the ferroxidase center.
In an attempt to obtain a structure with an iron-filled ferroxidase center a sample of as isolated Pa BfrB was reconstituted (mineralized) with 600 iron ions. The family of spectra in Figure 4 shows a gradual increase in the absorption near 300 nm, associated with the growth of iron mineral in the core of ferritin-like molecules (26), with each aliquot of Fe2+. After addition of the last Fe2+ aliquot the resultant solution was incubated overnight at 4 °C and then sieved through a Sephadex G-25 column. Subsequent analysis revealed ~ 600 ± 40 iron ions per BfrB molecule, indicating that the core had been successfully reconstituted. The crystal structure of mineralized Pa BfrB, obtained from 2.1 Å diffraction data (Table 1), is nearly identical that obtained from as isolated Pa BfrB, including an empty ferroxidase center. In fact, the ligands thought to coordinate the di-iron center in the mineralized Pa BfrB structure have identical conformations to those depicted in Figure 3 for the as-isolated protein, including the rotated conformation of the His130 side chain. The absence of ferroxidase iron, despite the fact that the protein had been mineralized with ~600 iron ions previous to crystallization indicates that the di-iron center in Pa-BfrB is unstable.
Figure 4.
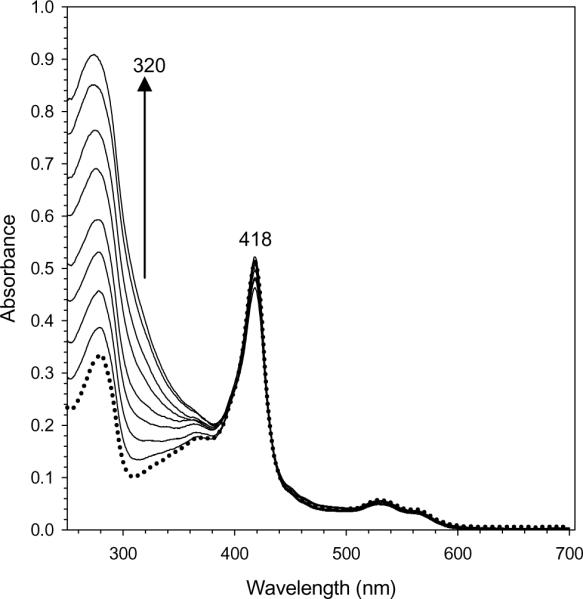
Reconstitution of core iron mineral in as isolated Pa BfrB with aliquots delivering 50 Fe2+ ions/Pa BfrB to protein dissolved in 100 mM potassium phosphate buffer, pH 7.6 at 25 °C. The spectra, which correspond to the addition of 0, 50, 100, 150, 200, 300, 400, 500 and 600 Fe2+ ions, were obtained 10 min after the corresponding aliquot was added. It is noteworthy that colloidal ferric hydrous oxide was not formed, as indicated by the conservation of baseline in the family of spectra. Time-dependent changes in intensity at 320 nm were used to monitor the kinetic experiments summarized in Figure 11.
The complete absence of metal ions in the ferroxidase center of Pa BfrB is in contrast with observations made with Bfr structures from other organisms. For instance, the structure of as isolated Ec Bfr was shown to bind Mn ions at the ferroxidase center (13) or a mixture of Fe and Zn ions if manganese is removed from the buffers used in protein purification and crystallization (27). The structure of Av Bfr isolated from a mutant strain of A vinelandii was shown to contain a partially occupied ferroxidase center consisting of a nearly fully occupied Fe1 site and a partially occupied Fe2 site (14, 28). The structure of Ms Bfr isolated from a Mycobacterium smegmatis strain showed Zn at the ferroxidase center, in spite of the fact that the only source of Zn during protein expression and purification was the growth medium (15). In comparison, the structure of Bfr isolated from the anaerobic bacterium Desulfovibrio desulfuricans was shown to harbor a ferroxidase center fully occupied with iron ions (16).
Pa BfrB in the crystal state binds and internalizes iron
In an attempt to incorporate iron in the ferroxidase center, crystals grown from mineralized Pa BfrB were soaked for 15 min in Fe2+ solution before they were flash frozen for data collection. The structure obtained from these crystals not only harbors a fully assembled ferroxidase center, but also reveals iron bound to a site on the exterior protein surface as well as to a site in the interior surface. Hence, Pa BfrB in the crystalline state is functional and incorporates iron from solution to the internal cavity, which suggests that the structure represents a snapshot of the iron uptake process. A view of a monomer subunit, which is representative of the 24-mer structure, illustrates the presence of three types of iron (Figure 5): (i) ferroxidase iron, located within the helix bundle, (ii) the iron ions bound on the external protein surface (Feout) and (iii) the iron ions bound to the internal surface (Fein).
Figure 5.

Views of a single subunit of Fe soaked Pa BfrB showing the iron atoms (gold spheres) located on the outer surface of the protein Feout, the inner surface Fein, and the ferroxidase iron atoms located in the center of the helix bundle.
The ferroxidase center
As expected the ferroxidase center is coordinated by Glu18 and His54 as terminal ligands to Fe1, Glu94 and His130 as terminal ligands to Fe2, and Glu51 and Glu127 as bridging ligands (Figure 6-A). The presence of iron at the ferroxidase center is corroborated by strong peaks in the difference electron density map (Fo-Fc) as well as a strong anomalous signal from data acquired at 1.6314 Å. All 24 independent subunits exhibit anomalous peak intensity and B factors for Fe1 and Fe2 that are comparable to that displayed by heme-iron (Table 2 and Figure S2), suggesting nearly complete occupancy. The average distance between Fe1 and Fe2 is 4.00 Å with a range encompassing 3.96 to 4.04 Å. These distances, which are similar to those observed in the structures of Av Bfr (14, 28) and Dd Bfr (16) in their reduced form, are consistent with a di-Fe2+ oxidation state (9). Figure 6-B depicts a superposition of the empty (green) and filled (magenta) ferroxidase centers to illustrate that with the exception of His130 the side chains of ligands to the di-iron center undergo minimal structural reorganization upon binding iron. The side chain of His130, on the other hand, rotates into the ferroxidase center to coordinate an iron atom (Fe2). This observation suggests that the conformational flexibility of the His130 side chain is associated with binding of external Fe2+ to the ferroxidase center, where it is oxidized to Fe3+ and finally released into the internal cavity as the side chain of His130 adopts its conformation in the ferroxidase empty state. Hence, the conformational flexibility of the His130 side chain may be important in gating iron from the ferroxidase center into the core. In this context, it is noteworthy that residual difference electron density was observed near the side chain of His130. When the structure derived from mineralized and Fe soaked Pa BfrB are superimposed the residual difference electron density from the latter fits closely to the His130 side chain from the former, implying that His130 in the Fe soaked structure experiences two conformations, one in which it is coordinated to Fe2 (~ 70% occupancy) and one similar that seen in the structure devoid of iron (Figure 6-C). The approximate occupancy of the alternate His130 conformations was determined manually from various structural refinement cycles.
Figure 6.

Ferroxidase center of Fe soaked Pa BfrB. (A) Coordination of Fe ions at the ferroxidase center of Pa BfrB. (B) Overlay of the mineralized (green) and Fe soaked (magenta) Pa BfrB structures showing their ferroxidase centers. All residues adopt similar conformations with the exception of His130, which in the Fe-soaked structure is rotated toward the ferroxidase center, as indicated by the arrow, to facilitate binding of Fe2. (C) His130 from the Fe soaked structure (magenta) in its coordinative and non-coordinative conformations; the latter is nearly superimposed with the His130 side chain of the mineralized structure shown in green. The 2Fo-Fc map (grey mesh) is contoured at 1σ and the Fo-Fc omit map (blue mesh) at 3σ. The Fe2 atom is represented as an orange sphere.
Table 2.
Anomalous peak heights and B-factors for Fe atoms in the Fe soaked structure.
| Fe(out) | Fe1 | Fe2 | Fe(in) | Heme Fe | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Subunit | B (Å2) | Peak Heighta | B (Å2) | Peak Heighta | B (Å2) | Peak Heighta | B (Å2) | Peak Heighta | Heme # | B (Å2) | Peak Heighta |
| A | 26.6 | 11.7 | 24.4 | 11.2 | 37.8 | 10.3 | 25.8 | 12.9 | 1 | 22.4 | 12.1 |
| B | 32.9 | 8.1 | 25.9 | 12.4 | 35.0 | 11.1 | 25.4 | 12.5 | 2 | 22.5 | 12.7 |
| C | 33.4 | 9.0 | 23.6 | 11.8 | 34.0 | 11.1 | 21.0 | 13.5 | 3 | 21.4 | 12.8 |
| D | 37.5 | 8.0 | 26.4 | 12.2 | 35.4 | 9.2 | 24.4 | 12.9 | 4 | 22.2 | 14.1 |
| E | 36.7 | 8.1 | 24.7 | 14.0 | 35.2 | 9.7 | 21.7 | 12.2 | 5 | 22.6 | 12.1 |
| F | 32.1 | 7.9 | 27.1 | 11.9 | 33.2 | 9.1 | 24.5 | 12.8 | 6 | 26.4 | 12.5 |
| G | 37.4 | 9.4 | 25.0 | 12.2 | 35.5 | 10.1 | 22.7 | 12.9 | 7 | 25.8 | 12.5 |
| H | 39.5 | 8.2 | 27.4 | 11.3 | 36.3 | 8.7 | 25.7 | 11.8 | 8 | 23.6 | 13.0 |
| I | 35.4 | 9.0 | 27.3 | 13.1 | 38.2 | 8.6 | 25.3 | 11.7 | 9 | 23.5 | 13.3 |
| J | 43.2 | 8.6 | 24.3 | 11.2 | 36.8 | 8.7 | 21.3 | 14.6 | 10 | 21.2 | 12.9 |
| K | 31.9 | 10.7 | 24.0 | 14.3 | 31.8 | 10.7 | 24.5 | 14.3 | 11 | 26.1 | 12.8 |
| L | 32.6 | 9.0 | 26.6 | 12.6 | 34.1 | 10.3 | 24.5 | 13.6 | 12 | 20.7 | 12.1 |
| M | 42.7 | 7.5 | 28.6 | 11.0 | 35.1 | 9.4 | 24.6 | 13.2 | |||
| N | 39.3 | 7.3 | 29.7 | 11.9 | 40.4 | 8.7 | 28.4 | 11.4 | |||
| O | 45.2 | 7.6 | 31.1 | 11.3 | 40.0 | 8.1 | 24.2 | 12.7 | |||
| P | 40.3 | 8.0 | 28.3 | 10.5 | 35.4 | 9.2 | 29.3 | 12.2 | |||
| Q | 23.8 | 14.0 | 26.1 | 10.0 | 32.1 | 7.3 | 24.0 | 13.6 | |||
| R | 29.2 | 9.9 | 24.1 | 11.6 | 33.1 | 8.2 | 26.0 | 13.3 | |||
| S | 32.4 | 8.3 | 25.7 | 10.8 | 33.7 | 9.6 | 22.5 | 13.2 | |||
| T | 33.5 | 9.3 | 23.0 | 12.0 | 36.5 | 9.2 | 22.0 | 12.8 | |||
| U | 31.7 | 9.5 | 26.2 | 12.1 | 35.5 | 9.1 | 19.7 | 13.8 | |||
| V | 34.3 | 11.0 | 23.1 | 11.7 | 32.5 | 8.5 | 21.2 | 11.7 | |||
| W | 35.7 | 9.3 | 26.3 | 12.1 | 34.4 | 10.3 | 25.5 | 12.3 | |||
| X | 42.1 | 7.4 | 28.3 | 11.2 | 39.2 | 8.6 | 28.2 | 12.1 | |||
| Avg. b | 35.4 | 9.0 | 26.1 | 11.9 | 35.5 | 9.3 | 24.3 | 12.8 | Avg. | 23.2 | 12.7 |
Peak heights were determined from the anomalous Fourier difference electron density map from data collected at λ =1.6314 Å.
Average B-factors and peak heights for each Fe site
It is interesting to note that in the as-isolated and mineralized structures, the side chain of His130 adopts only the conformation corresponding to the non-coordinated form, indicating that the alternate conformation is populated only in the presence of iron in the ferroxidase center. Hence, it is plausible that the Fe soaked structure represents a snapshot where the ferroxidase center of Pa BfrB is freshly loaded with Fe2+ and where the His130 side chain has been caught in the process of returning to its coordinative position. These findings are in agreement with the observation of non-coordinative conformation of His130 in the crystal structure of Av Bfr, which led to the suggestion that His130 acts as a gate that allows the movement of iron from the ferroxidase center to the internal cavity (14). Moreover, the conformation of the His130 side chain in Av Bfr has also been shown to change from its coordinative to its noncoordinative position upon reduction with dithionite (28), an observation that is in agreement with the notion that the His130 side chain may act as a gate for the entry or iron from the ferroxidase center to the interior cavity.
With the exception of the as isolated Dd Bfr structure, which showed a ferroxidase center fully occupied with iron, Bfr structures from most organisms investigated, which contained metal ions at the ferroxidase center, i.e. iron, zinc or manganese, showed lower occupancy of Fe2 relative to Fe1 (14–16, 27, 28). In addition, when Dd Bfr was reduced with dithionite and then allowed to oxidize, the Fe2 site in the oxidized protein was nearly depleted (16). Hence, the relatively low occupancy of the Fe2 site in most Bfr structures investigated thus far is consistent with the idea of Fe2 translocation from the ferroxidase center to the internal cavity as part of the mechanism of iron uptake (14, 16, 28). Given that soaking mineralized Pa BfrB crystals in iron solution resulted in a structure with a fully occupied di-Fe2+ center, it was of interest to investigate whether oxidation of the protein in air would result in a ferroxidase center with iron at the Fe1 position and nearly depleted Fe2. To this end, crystals of mineralized Pa BfrB were soaked for 15 min in iron solution, then soaked for 15 min in crystallization solution before they were flash frozen for data collection. The structure obtained from these crystals reveals a ferroxidase center almost completely devoid of iron, where the side chains of the ferroxidase ligands attain conformations identical to those observed in the structure of the as-isolated and mineralized structures and the side chain of His130 is rotated to its non-coordinative position. Weak residual difference electron density can be observed in the ferroxidase center, which is likely due to low occupancy iron. In comparison, relatively larger peaks were observed in the difference electron density maps at the Fein sites that also display a weak anomalous signal. Refinement of Fe ions with full occupancy at these sites resulted in B-factors that were 2–3 times that of the average value for the model. Fe ions with ~0.5 occupancy were ultimately modeled at these Fein sites as determined manually from iterative refinement cycles. The relatively weak electron density and lengthened non-bonded contacts between Fein and His153/His155 at these sites relative to the Fe soaked structure, most likely reflects the dynamic motions likely resulting in incorporation of Fein in the core mineral. Likewise, higher B-factors relative to the overall model were observed for the potassium ions in the 4-fold pores which refined with 0.5 occupancy. These findings are so far unique to Pa BfrB and indicate that the di-iron center in this protein is relatively labile. This in turn suggests that the instability of the di-iron center may be a property that permits Pa BfrB to efficiently uptake iron via the ferroxidase center. This idea was explored further with the aid of further structural and kinetic information; results from these investigations are described below.
The ferroxidase pore
Inspecting the outer surface of the Pa BrfB structure reveals a pore that likely serves as a conduit of Fe2+ from the surface to the ferroxidase center and upon oxidation to Fe3+, from the ferroxidase center to the interior cavity. Figure 7-A shows a semi-transparent surface representation (magenta) of the mineralized Pa BfrB structure (apo ferroxidase) delineating the section of the pore leading from the exterior protein surface to the ferroxidase center. The walls are lined by the backbone and side chain of Asn17, the side chain of Ile20, Leu93, the hydrophobic component of the Lys96 side chain and by the backbone of Ala97. The carboxyl oxygen of Glu51 and Glu94, a terminal and bridging ligands in the ferroxidase center, respectively can be observed at the bottom of the pore. For comparison, an identical view of the Fe soaked structure (holo-ferroxidase) is presented in Figure 7-B, where the iron ions have been omitted for clarity. It is worth noting that the conformation of the His130 side chain in the mineralized structure does not obstruct the pore, which we term the “gate-open” conformation (Figure 7-A). In contrast, the conformation of the His130 chain in the Fe soaked structure adopts its “gate closed” conformation, allowing it to function as a cap at the bottom of the pore and as ligand to Fe2. The view of Figure 7-C, which is identical to that in 7-B, except that the ferroxidase iron ions have been included (orange spheres), shows an unobstructed view of Fe2 at the bottom of the pore, where it is coordinated by capping ligands Glu94 (above) and His130 (below) and bridged to Fe1 by Glu51 and Glu127, visible by virtue of the semi-transparent surface rendering. The picture emerging from this analysis suggests that Fe2+ enters the ferroxidase center via the pore illustrated in Figure 7, where it is coordinated by the ferroxidase ligands, including His130; the latter moves from its “gate-open” to its “gate-closed” conformation, likely triggered by small conformational changes that occur to accommodate a di-Fe2+ center. Oxidation to a di-Fe3+ moiety likely leads to a shorter internuclear distance (9) and perhaps subtle ligand rearrangement needed to accommodate a bridging oxo or hydroxo ligand, which in turn may signal His130 to move to its “gate-open” conformation, thus allowing Fe3+ to move into the internal cavity. It is also interesting to note that in addition to the large conformational change of the His130 side chain there is a more subtle, but likely important conformational change in the side chain of Asp50. Whereas this side chain does not obstruct the pore in the “gate-open” conformation (Figure 7-A), it clearly aids in capping the bottom of the pore in the “gate-closed” conformation of Figure 7-C, where it can be seen below Fe1. Hence, opening and closing the gate for the translocation of Fe3+ from the ferroxidase center to the interior cavity of Pa BfrB likely involves a concerted motion of the D50 and His130 side chains.
Figure 7.

Stereo view of the ferroxidase pore viewed from the protein exterior. The semi-transparent surface representation (magenta) is constituted by residues lining the pore wall (Asn17, Ile20, Leu93, Lys93 and Ala97). (A) The “gate-open” conformation of the Asp50 and His130 side chains (observed in the as isolated and mineralized Pa BfrB structures) allows a nearly unobstructed view of the interior cavity through the pore. (B) The “gate-closed” conformation of Asp50 and His130 (observed in the Fe-soaked structure) obstruct the bottom of the pore and poise H130 to coordinate Fe2. The ferroxidase iron is not shown for clarity. (C) View identical as in (B) but showing the ferroxidase iron ions as orange spheres. The side chain is His130 is below Fe2, which is located at the bottom of the pore. Fe1 in the interior can be seen through the semi-transparent surface.
A similar pore has been observed on the external surface of Ec (13) and Dd (16) Bfr and hypothesized to be an entry route for Fe2+ ion to the ferroxidase center. Macedo and coworkers described the ferroxidase pore in Dd Bfr and hypothesized that upon oxidation of the di-Fe2+ center the relative low occupancy of the Fe2 site is suggestive of Fe3+ moving into the internal Bfr cavity (16, 29). These authors speculated that translocation of Fe3+ from the ferroxidase center into the internal cavity of Dd Bfr would require a concerted conformational change of the His59 and Glu131 side chains, which would gate open a path for ferroxidase iron to move into the interior of the bacterioferritin. In this context, it is noteworthy that the structures of Pa BfrB with the His130 side chain in their gate open and gate closed conformations (Figure 7) provide for the first time direct experimental observation of a pore leading from the protein surface to the ferroxidase center, and upon concerted motion of D50 and H130, a pore for the translocation of ferroxidase iron from the ferroxidase center to the interior cavity of Pa BfrB. These structural observations strongly suggest that the ferroxidase center in Pa BfrB acts in a dual role or catalytic center and pore for the incorporation of iron into the internal cavity, where it is hydrolyzed and incorporated into the core mineral. Additional observations supporting this notion are presented later in this report.
The three- and four-fold pores in Pa BfrB
The core of the Pa-BfrB structure is connected to the protein exterior by eight three-fold pores and by six four-fold pores, illustrated in Figure 2. In the eukaryotic ferritins structural studies have uncovered the presence of divalent cations in the three-fold pores which are lined with highly conserved negatively charged residues (30). Replacement of these residues has resulted in decreased rates of iron uptake which has led to the suggestion that iron traffics in and out of eukaryotic bacterioferritins via the three-fold pores (30–32). In addition, localized unfolding at or near the three-fold pores has been shown to accelerate the process of iron release from eukaryotic ferritins, observations that are in agreement with the idea that iron traffics in and out of these molecules via the three-fold pores (5, 33, 34). In comparison, the role of three-fold channels in the bacterioferritins, which are lined with both positively and negatively charged residues, is less clear. Figure 8-A depicts a three-fold pore in the mineralized Pa BfrB structure viewed from the interior cavity: at the top, the entrance to the channel from outside is formed by three Glu109 side chains which form salt bridges with three Arg117 side chains and cap the pore access. By comparison, the three-fold pore in the structure of Fe soaked Pa BfrB shows a similar architecture of the most external layer lining the three-fold pore, where the electrostatic interactions between the side chains of Glu109 and Arg117 cap the pore entrance. In the interior, however, the situation presents itself differently; the side chains corresponding to Lys121 are likely disordered because there is no electron density beyond the beta carbon (Figure 8-B). However, residual electron density was observed in the three-fold pore which was modeled as either sulfate or phosphate. Additional refinement indicated that the residual electron density likely corresponds to a sulfate ion in the pore; SO42- is abundantly present in the soaking solution which consists of 50 mM FeSO4, and in the crystallization solution (see Experimental Procedures) which contains 200 mM Li2SO4. The presence of sulfate in three fold pores only in the Fe soaked Pa BfrB structure makes it tempting to hypothesize that the three-fold pores may serve as conduits for the traffic of anions; additional work is clearly necessary to test this hypothesis. In this frame, it is noteworthy that concomitant traffic of iron and phosphate in and out of the internal cavity is important in bacterioferritins because it is known that the anion is an integral component of the core mineral and may also be involved in nucleating and catalyzing its growth (35, 36). Consequently, it is attractive to postulate that the phosphate needed to support the growth of a mineral core in Pa BfrB enters the bacterioferritin via the three-fold pores.
Figure 8.

(A) Stereo view of a three-fold channel in the mineralized Pa BfrB structure, viewed from the interior cavity. The top of the pore is capped by the side chains of Glu109 and Asn117 (O is in red and N in blue). Below the pore becomes wider and is lined by the side chains of Lys121 and Asp118, which also undergo stabilizing electrostatic interactions. (B) Identical view of a three-fold channel in the Fe-soaked structure. The top of the pore is still capped by the interactions of Glu109 and Asn117. The interior however shows the presence of sulfate (magenta) and disorder in the side chains of Lys21. (C) View of a four fold pore where a potassium ion (yellow sphere) is coordinated by Gln151 and Asn148. The potassium ion in the four fold pore is present in as isolated, mineralized, Fe-soaked and double-soaked structures.
Figure 8-C depicts a four-fold pore in the structure of mineralized BfrB, which shows a potassium ion coordinated by the side chains of four Asn148 and four Gln151 residues. The possibility that the metal ion in the four-fold pore is iron was eliminated by the negligible anomalous difference density at this position in a data set collected at 1.6314 Å. In addition, residual negative and positive difference density was observed when Fe ions or water molecules, respectively, were modeled at these sites. The structure obtained from crystals soaked in Fe2+ solution also contains potassium ions in equivalent positions of the four-fold pore. In fact, analysis of data sets acquired at 1.6314 Å obtained from these crystals add strong corroborating evidence for the absence of iron in the four-fold pore due to the absence of anomalous difference density at this position, which contrasts with the strong anomalous signal observed from the heme-iron, ferroxidase iron, as well as from Fein and Feout (see Table 2). As was the case with data obtained from crystals not soaked in iron solution, a model in which potassium ions are coordinated by the side chains of Asn148 and Gln151 did not yield negative difference density. The presence of potassium is simple to explain since the metal ion is abundantly present in the protein storage buffer, which consists of 100 mM potassium phosphate. The presence of cations in the four-fold pore, also coordinated by Asn148 and Gln151, has been observed in two independent structures of Av Bfr, one containing iron (28) and the other Ba2+ (14). These observations led to the hypothesis that iron may enter or leave the protein via these pores. In contrast, the four-fold pores in the structures of Ec, Ms and Dd Bfr are devoid of metal ions (12, 13, 15, 16), which is perhaps a reflection of the different architecture of the pores in the distinct Bfrs: In Pa and Av Bfr the four-fold pores are built in two layers, each formed by Asn148 (outer) and Gln151 (inner), whereas in Ec, Ms and Dd Bfr the pores are built from three layers; these are Asn148 (outer) Glu15 (central) and Arg155 (inner) in Ec Bfr and equivalent residues in Ms and Dd Bfr. It is therefore possible that the ubiquitous presence of metal ions in the four-fold pores of Pa and Av BfrB structure, which share the same architecture, is indicative of a role for iron trafficking in these two and perhaps in other not yet studied bacterioferritins with similar four-fold pore structures.
Iron bound to the internal (Fein) and external (Feout) surfaces
A striking feature of the mineralized Fe soak Pa BfrB structure is the presence of two additional iron sites, one located in the exterior surface, Fe(out), and one on the internal surface, protruding into the internal cavity Fe(in). The position of Fe(in) in the Pa BfrB structure is shown in Figure 9-A which shows a portion of the molecule viewed from the interior cavity, depicting a four-fold pore (subunits E, J, Q, V), its associated K+ ion (green), and subunits I, F, R and U, which pack against the four-fold pore-forming subunits to form the so called B pores (16), four of which can be readily seen in the CPK rendering of Figure 9-B. The figures also illustrate the position of the associated Fein ions, as well as the ferroxidase-iron ions, which are shown as orange spheres. It is interesting that there are four Fein ions surrounding each four-fold pore, each coordinated by His153 and His155 from different subunits and water molecules complete the octahedral coordination (Figure 9-C and Figure S3-A).
Figure 9.

(A) A four fold-pore and neighboring B pores in the Fe-soaked Pa BfrB structure viewed from the interior cavity. The iron and potassium ions are represented by orange and green spheres, respectively. (B) An identical view in CPK rendering, which facilitates visualization of the B pores and of the four Fe(in) ions surrounding the four-fold pore. (C) Zoomed-in view illustrating side chains that may form possible routes for moving iron ions from the ferroxidase pore, four-fold pore or B pores to their Fe(in) position.
Analysis of the structure suggests at least three possibilities for potential iron entry paths leading to iron at this location, (a) entry via the ferroxidase pore, (b) entry via the four-fold pores, and (c) entry via the B pores. These are described below and illustrated in Figure 9-C: In the case of iron accessing the internal cavity via the ferroxidase pore, a conformational change of the His130 side chain to its “gate open” conformation allows Fe3+ to move into the interior, where it may interact with Glu47 and subsequently with His46, His43 and or Tyr133 before it moves to its observed position in the structure. It is noteworthy that the side chain of His46 also occupies two conformations, which in the content of this discussion may be related to the transport of Fe3+ from the ferroxidase center to the Fe(in) position. Iron ions entering through the four-fold pore (presumably Fe2+) may be transported to their position in the structure via interactions with Arg39 and His55 from different subunits; side chains from these residues and equivalent residues in other subunits that interact to structure the four-fold pore constitute the widest section of the wall lining the pore. Finally, iron entering the cavity via the B pores may be guided to its position in the interior cavity via interactions with Asp34, Thr136 and Asp132, which form a potential path of coordinative contacts linking the B pores with the location of Fein. In a recent report it has been shown that Ec Bfr can also mineralize iron in the crystal state, leading to the observation of an iron in the inner surface of the protein (12). The location of this iron ion, however, is different from that observed for Fe(in) in Pa BfrB; the Ec Bfr internal iron is almost directly below the ferroxidase center where it is coordinated by Asp50 and His46.
Soaking crystals of mineralized Pa BfrB in iron also allowed observation of an unprecedented iron site on the surface of the protein. These iron ions bound to the outside surface (Feout) are coordinated by His10, His153 and three waters (Figure 10 and Figure S3-B), and are located in relative proximity to the ferroxidase pore yet distant from the four-fold and B pores. Their presence in the crystal structure with near full occupancy (see Table 2) suggests a moderate to high affinity of the Pa BfrB surface for iron, which may be useful for capturing iron and directing it to an internalization channel. It is interesting to consider this idea in the context of Figure 10, which depicts residues lining the ferroxidase pore in CPK rendering and hydrophilic side chains connecting Feout with the pore in stick rendering. The side chain of Lys96, which shapes part of the pore wall, forms a salt bridge with Asp100, suggesting the possibility that Fe2+ approaching the pore from its Fe(out) position induces a conformational change in the Asp100 side chain that breaks the salt bridge, which in turn facilitates a change in conformation of Lys96 that grants the Fe2+ ion ready access to the pore. Clearly, the potential role played by residues involved directly in binding Feout, as well as that played by residues intervening between the Feout and the ferroxidase pore will have to be probed systematically to elucidate their influence in iron capture and iron internalization by Pa BfrB.
Figure 10.
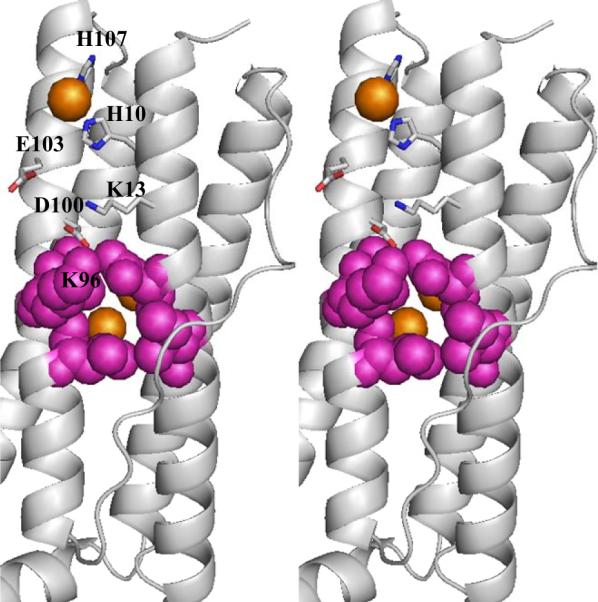
Stereo view of a single subunit showing the location of Fe(out) on the external surface of the Fe-soaked structure, where it is coordinated by His10, His107 and three water molecules (the latter are not shown here for clarity, but can be seen in Figure S3). The view also shows the side chains of residues that may be involved in transporting the Fe(out) to the ferroxidase pore where a disruption of the salt bridge between D100 and K96 facilitates the entry of iron to the pore. Iron ions are in orange, nitrogen atoms are in blue, oxygen atoms in red and residues lining the external outermost layer of the pore are in magenta and in CPK rendering.
The ferroxidase pore is an iron entry port in Pa BfrB
As indicated previously, the mineralized Pa BfrB structure does not contain iron, despite the fact that the crystals were grown from Pa-BfrB mineralized with ~600 iron atoms. Soaking the crystals in iron solution incorporates iron into the ferroxidase center as well as in specific sites at the internal (Fein) and external (Feout) surfaces of the protein. These findings, together with the observation of a ferroxidase pore leading from the external surface to the ferroxidase center, which by virtue of the flexibility built in the His130 side chain, opens a path for internalization of ferroxidase iron into the Pa BfrB cavity, suggest that the ferroxidase center in Pa BfrB is a transit stop in the route to iron internalization. Additional structural evidence supporting this idea was sought by soaking crystals of mineralized Pa BfrB for 15 min in a solution of FeSO4, followed by soaking them for 15 min in crystallization solution, prior to acquiring X-ray diffraction data. The resulting structure is devoid of iron and shows an empty ferroxidase center with structural features identical those observed in the structure obtained prior to soaking in iron solution, including 100% occupancy of the “gate-open” conformation of His130. These observations confirm that “gate-open” is the preferred conformation of the His130 side chain in the empty ferroxidase center and support the notion that the ferroxidase center acts as a pore for the internalization of iron. The latter was studied further with the aid of experiments aimed at probing the uptake of iron by Pa-BfrB in solution. Results from these experiments are discussed below.
It has been shown that iron uptake by Ec Bfr involves a least three kinetically resolved phases: The binding of Fe2+ ions at the ferroxidase center (phase 1), followed by their oxidation to form what is most likely a μ-oxo-bridged di-Fe3+ moiety at each ferroxidase center (phase 2) and finally growth of the iron core (phase 3). Phase 1, detected by a small shift of the Soret band is the fastest, with t½ ~ 5 ms, whereas phases 2 and 3, typically monitored near 340 nm, last approximately ~15 sec and ~15 min, respectively (7). It is also important to stress that in experiments conducted with Ec Bfr the change in absorbance near 340 nm (ΔA340) resulting from phase 2 saturates after ~ 50 iron ions (two iron ions per ferroxidase center) have been added per Ec Bfr molecule, an observation that has been interpreted to indicate that a stable di-iron ferroxidase center is formed per Bfr subunit during this phase. Addition of more iron causes only relatively slow increases in ΔA340, i.e. only phase 3 kinetics are observed (1, 7). These observations together with the absence of an obvious structural conduit for the translocation of ferroxidase iron to the core were interpreted to indicate that the ferroxidase center of Ec Bfr does not act as a pore for iron uptake. Rather, once the ferroxidase center is formed, Fe2+ is taken in via pores in the Bfr structure and oxidized by transferring electrons to the ferroxidase iron, which in turn reduce molecular O2 (12).
The uptake of Fe2+ by as-isolated Pa BfrB in solution was initially studied by adding aliquots containing 50 Fe2+ ions per Pa BfrB, while monitoring the response with the aid of a conventional diode array spectrophotometer. The experiments were conducted at pH 6.5 and 30 °C to compare the observations with those made in the study of Ec Bfr (1, 7). The resulting observations, discussed below, are strikingly different from those typically observed with Ec Bfr: Addition of 50 Fe2+ results in a rapid increase of the absorbance near 300 nm (plotted at 320 nm in Figure 11), which rapidly reaches a plateau and remains constant thereafter. Note that the magnitude of the change in absorbance at 320 nm (ΔA320) following the addition of each aliquot of Fe2+ remains nearly constant throughout the experiment. This indicates that the ferroxidase center in Pa BfrB is indeed labile and that once all Fe2+ ions are oxidized and internalized, the ferroxidase center is vacated, an observation that is consistent with the fact that the ferroxidase center in the as isolated, mineralized and double soak structures is vacant.
Figure 11.
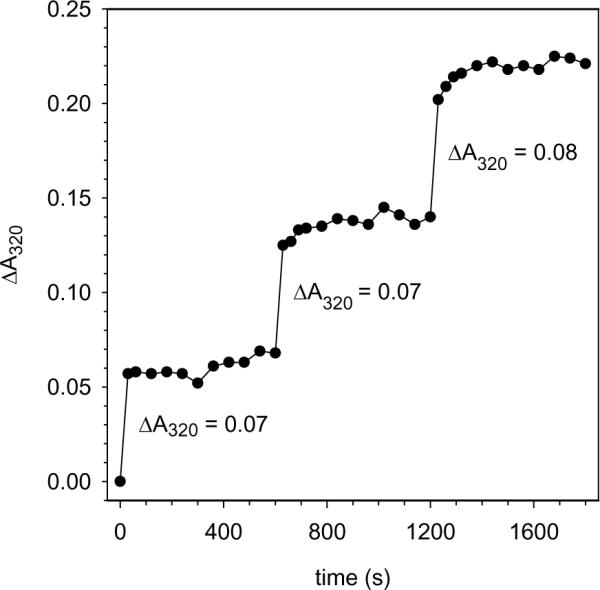
Iron uptake in solution: Change in ΔA320 upon subsequent additions of Fe2+ aliquots (50 Fe2+ ions per BfrB molecule) to a solution of as isolated Pa BfrB (0.8 μM) in 100 mM MES buffer, 100 mM KCl, pH 6.5. The uptake of iron was monitored with the aid of a conventional diode array spectrophotometer by acquiring spectra before the addition of an iron aliquot and every 30 seconds for 2 minutes; thereafter spectra were acquired every one minute for eight additional minutes. The responses illustrate a rapid rise in A320 which rapidly reaches a steady value after the addition of each aliquot, suggesting that the ferroxidase center is completely vacated at the end of each plateau.
Given that iron uptake occurs rapidly, additional experiments were conducted with the aid of a stopped-flow apparatus. In these experiments Pa BfrB containing solutions were mixed with Fe2+ containing solutions in different proportions (30, 50, 100, 200 and 300 Fe2+ ions per BrfB), while monitoring the mineralization process by following the time-dependent growth of the absorbance at 320 nm. The results, summarized in Figure 12-A, show that when the iron load is 100 Fe2+/BfrB or less, two phases can be observed, a fast phase that lasts less than 10 sec and a slower phase, which becomes progressively faster as the iron load is increased, such that when the iron load is 200 and 300 Fe2+ ions/BfrB the two phases are no longer resolved. It is interesting that when the iron load is 30 or 50 Fe2+ ions, the increase in ΔA320 corresponding to the fast phase is followed by a decrease in absorbance before the onset of the slower phase; this is seen more clearly in Figure 12-B, which shows only the progress curves obtained upon addition of 30 and 50 Fe2+ ions. It is likely that the initial rapid increase in ΔA320 (fast phase) corresponds to the oxidation of the di-Fe2+ center to form a di-Fe3+ center that is likely μ-oxo (or μ-hydroxo) bridged. The subsequent decrease in absorbance may correspond to migration of Fe3+ ions to the internal cavity where mineralization (slow phase) takes place; as the iron load increases the decrease in absorbance is obscured by the earlier onset and larger amplitude of the slower phase. Hence, the decrease in absorbance observed at the low iron loads appears to be consistent with the notion derived from the structural studies indicating that the ferroxidase center is vacated when Fe3+ is translocated to the interior cavity.
Figure 12.

(A) Progress curves obtained with the aid of a stopped flow apparatus after the addition of 30, 50, 100, 200 and 300 Fe2+ ions per Pa BfrB molecule in 100 mM MES buffer, 100 mM KCl, pH 6.5 and 30 °C. Protein concentration is 1.0 μM (after mixing) and the path length is 1.0 cm. (B) Progress curves corresponding to the addition of 50 and 100 Fe2+ ions in (A) are shown to more clearly illustrate the decrease in absorbance that follows the fast phase. (C) A plot of the amplitude obtained at the end of 200 s for the progress curves in (A). (D) A plot of the amplitude of the fast phase as a function of Fe2+ load; the amplitudes were obtained from fitting the first 5 s of the progress curves in (A), obtained after the addition of 30, 50 and 100 Fe2+ ions, to a mono exponential expression.
At the higher Fe2+ loads (200 and 300) the iron uptake process is virtually complete in ~ 100 s, which contrasts with the ~ 800 s necessary to complete the uptake of similar loads of iron by Ec Bfr, at the same pH and temperature (7). As indicated above, at the lower Fe2+ loads the iron uptake by Pa BfrB also contrasts with the uptake by Ec Bfr in that a slow phase is observed, which likely corresponds to iron mineralization in the interior cavity. By comparison, the progress curves obtained with Ec Bfr at similar loads only show the fast phase because iron in the ferroxidase center is not translocated into the interior cavity (7). The mineralization reaction in Pa BfrB at a load of 30 Fe2+ ions appears to proceed relatively slowly, as can be inferred from the fact that the slow phase does not seem to approach completion at 200 s (Figure 12-B). This can also be seen in Figure 12-C, where the amplitude obtained at the end of 200 s has been plotted as a function of iron load; the magnitude of the amplitude corresponding to 30 iron atoms is lower than may be anticipated from the trend of amplitudes obtained at the higher Fe2+ loads. The slow mineralization observed with a load of 30 Fe2+ ions may be a consequence of slow nucleation due to the relatively small number of available Fe3+ ions. As the number of available Fe3+ ions increases (higher loads) the rate of mineralization increases such that at the higher loads there is no clear distinction between the two phases. The initial 5 seconds of the progress curves (fast phase) corresponding to Fe2+ loads of 30, 50 and 100 were fitted to a mono exponential function to obtain the amplitudes and rate constant corresponding to the fast phase. A plot of the fast phase amplitude as a function of iron load (Figure 12-D) shows that the fast phase does not saturate at an iron load of 50 Fe2+ ions, as is the case with the equivalent phase observed with Ec Bfr, which has been interpreted to indicate that the ferroxidase center in Ec Br is stable and does not regenerate (7, 12). The value of the rate constant obtained for the fast phase of Pa BfrB is 0.67 s−1 ± 0.04 (pH 6.5, 30 °C, and protein concentration 1.0 μM), which appears to be faster than the rate constant obtained for the equivalent phase of Ec Bfr (0.17 s−1 ± 0.05) obtained at the same pH and temperature (7, 37), although direct comparison is not possible because the rate constant for Ec Bfr was obtained with protein concentration of 0.5 μM.
Taken together, the observations made in solution phase suggest that the ferroxidase center of Pa BfrB is labile, unlike that of Ec Bfr, and therefore support the notion derived from the structural investigations that the ferroxidase center of Pa BfrB functions in a dual role of substrate oxidizing center and pore for iron internalization. Elucidation of the mechanism of iron uptake will require detailed kinetic experiments similar to those carried out with the eukaryotic ferritins (38, 39) and Ec Bfr (1, 7, 8).
The picture emerging from the structural and kinetic studies reported herein points to the idea that iron trafficking in and out of bacterioferritins does not take place via the same routes, despite the similar architecture of the molecules, including the binding of 12 heme macrocycles. As has been shown previously with Ec Bfr (1, 7, 8, 12), the entry of iron into the interior cavity, likely occurs via one or more of the pores in the structure, but not via the ferroxidase center. Mineralization of internalized iron requires formation of a stable ferroxidase center, which functions nearly exclusively as a catalytic site that relays electrons released from the oxidation of internalized Fe2+ to molecular O2. Structural evidence of Ec Bfr iron uptake in crystalo did not show a possible route for the access of iron from the ferroxidase center to the internal cavity (12), which is in agreement with the idea derived from kinetic studies that the ferroxidase center in Ec Bfr is stable. In comparison, the oxidation and entry of iron into Pa BfrB appears to take place via the ferroxidase pore. This mechanism appears to impart the protein with a higher efficiency to capture, oxidize and store iron.
It is also interesting to consider that the genetic regulation and individual role of the ferritins, bacterioferritins and Dps molecules coexisting in an organism such as Ec or Pa are likely related to the bacterial lifestyle and its ecological niche (40). Hence, it is possible that the mechanisms of iron uptake and release exhibited by the distinct ferritin-like proteins are tuned to meet the particular conditions causing their biosynthesis. In the case of Pa-BfrB it has been recently demonstrated that iron release requires the binding of apo-bacterioferritin associated ferredoxin (Pa Bfd), which accelerates iron release from the core of Pa BfrB by enabling heme mediation of electron transfer from the surface to the core iron mineral (18). Low-iron conditions down-regulate the Pa BfrB gene (41), while at the same time strongly up-regulate the Pa Bfd gene (42), thus the Bfd BfrB interactions are important to ensure that iron stored in Pa BfrB is efficiently mobilized from the core of BfrB during iron starvation conditions (18), likely enabling the bacterium to survive while other mechanisms of iron uptake procure the nutrient from extracellular sources.
Supplementary Material
ACKNOWLEDGMENTS
Use of the IMCA-CAT beamline 17-BM at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
This work was supported by a grant from the National Institute of Health, GM-50503 (M.R.) and by use of the KU COBRE Center in Protein Structure and Function, Protein Structure Laboratory (NIH grant RR-017708).
Footnotes
Coordinates and crystallographic structure factors for the distinct Pa BfrB structures have been deposited in the protein data bank under accession codes 3ISF (as isolated), 3IS7 (mineralized, non-soaked), 3IS8 (mineralized, soaked in FeSO4), and 3ISE (mineralized, soaked in FeSO4 and then in crystallization solution).
SUPPORTING INFORMATION AVAILABLE Amino acid sequence alignment of several bacterioferritins, single subunit of Fe soak Pa BfrB showing anomalous difference map and microenvironment of Fe(in) and Fe(out). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Yang X, Le Brun NE, Thomson AJ, Moore GR, Chasteen ND. The Iron Oxidation and Hydrolysis Chemistry of Escherichia coli Bacterioferritin. Biochemistry. 2000;39:4915–4923. doi: 10.1021/bi992631f. [DOI] [PubMed] [Google Scholar]
- 2.Bunker J, Lowry T, Davis G, Zhang B, Brosnahan D, Lindsay S, Costen R, Choi S, Arosio P, Watt GD. Kinetic Studies of Iron Deposition Catalyzed by Recombinant Human Liver Heavy and Light Ferritins and Azotobacter vinelandii Bacterioferritin using O2 and H2O2 as Oxidants. Biophys. Chem. 2005;114:235–244. doi: 10.1016/j.bpc.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Zhao G, Bou-Abdallah F, Arosio P, Levi S, Janus-Chandler C, Chasteen ND. Multiple Pathways for Mineral Core Formation in Mammalian Apoferritin. The Role of Hydrogen Peroxide. Biochemistry. 2003;42:3142–3150. doi: 10.1021/bi027357v. [DOI] [PubMed] [Google Scholar]
- 4.Lewin A, Moore GR, Le Brun NE. Formation of Protein-Coated Iron Minerals. Dalton Trans. 2005:3597–3610. doi: 10.1039/b506071k. [DOI] [PubMed] [Google Scholar]
- 5.Liu X, Theil EC. Ferritins: Dynamic Management of Biological Iron and Oxygen Chemistry. Acc. Chem. Res. 2005;38:167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 6.Levi S, Luzzago A, Cesareni G, Cozzi A, Franceschinelli F, Albertini A, Arosio P. Mechanism of Ferritin Iron Uptake: Activity of the H-chain and Deletion Mapping of the Ferro-oxidase Site. J. Biol. Chem. 1988;263:18086–18092. [PubMed] [Google Scholar]
- 7.Le Brun NE, Wilson MT, Andrews SC, Guest JR, Harrison PM, Thomson AJ, Moore GR. Kinetic and Structural Characterization of an Intermediate in the Biomineralization of Bacterioferritin. FEBS Lett. 1993;333:197–202. doi: 10.1016/0014-5793(93)80404-i. [DOI] [PubMed] [Google Scholar]
- 8.Baaghil S, Lewin A, Moore GR, Le Brun NE. Core Formation in Escherichia coli Bacterioferritin Requires a Functional Ferroxidase Center. Biochemistry. 2003;42:14047–14056. doi: 10.1021/bi035253u. [DOI] [PubMed] [Google Scholar]
- 9.Kurtz DM. Stuctural Similarity and Functional Diversity of Diiron-Oxo Proteins. J. Biol. Inorg. Chem. 1997;2:159–167. [Google Scholar]
- 10.Chasteen ND, Theil EC. Iron Binding by Horse Spleen Apoferritin. J Biol Chem. 1982;257:7672–7677. [PubMed] [Google Scholar]
- 11.Le Brun NE, Andrews SC, Guest JR, Harrison PM, Moore GR, Thomson AJ. Identification of the Ferroxidase Centre of Escherichia coli Bacterioferritin. Biochem J. 1995;312:385–392. [PMC free article] [PubMed] [Google Scholar]
- 12.Crow A, Lawson TL, Lewin A, Moore GR, Le Brun NE. Structural Basis for Iron Mineralization by Bacterioferritin. J. Am. Chem. Soc. 2009;131:6808–6813. doi: 10.1021/ja8093444. [DOI] [PubMed] [Google Scholar]
- 13.Frolow F, Kalb AJ, Yariv J. Structure of a Unique Twofold Symmetric Haem-Binding Site. Nat. Struct. Biol. 1994;1 doi: 10.1038/nsb0794-453. [DOI] [PubMed] [Google Scholar]
- 14.Liu H-L, Zhou H-N, Xing W-M, Zhao J-F, Li S-X, Huang J-F, Bi R-C. 2.6 Å Resolution Crystal Structure of the Bacterioferritin from Azotobacter vinelandii. FEBS Lett. 2004;573:93–98. doi: 10.1016/j.febslet.2004.07.054. [DOI] [PubMed] [Google Scholar]
- 15.Janowski R, Auerbach-Nevo T, Weiss MS. Bacterioferritin from Mycobacterium smegmatis Contains Zinc in its Di-Nuclear Site. Prot. Sci. 2007;17:1138–1150. doi: 10.1110/ps.034819.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macedo S, Romão CV, Mitchell E, Matias PM, Liu MY, Xavier AV, LeGall J, Teixeira M, Lindley P, Carrondo MA. The Nature of the Di-Iron Site in the Bacterioferritin from Desulfovibrio desulfuricans. Nat. Struct. Biol. 2003;10:285–290. doi: 10.1038/nsb909. [DOI] [PubMed] [Google Scholar]
- 17.Cobessi D, Huang LS, Ban M, Pon NG, Daldal F, Berry EA. The 2.6 Å Resolution Structure of Rhodobacter capsulatus Bacterioferritin with Metal-Free Dinuclear Site and Heme Iron in Crystallographic “Special Position”. Acta. Cryst. 2002;D58:29–38. doi: 10.1107/s0907444901017267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weeratunga S, Gee CE, Lovell S, Zeng Y, Woodin CL, Rivera M. Binding of Pseudomonas aeruginosa Apobacterioferritin-Associated Ferredoxin to Bacterioferritin B Promotes Heme Mediation of Electron Delivery and Mobilization of Core Mineral Iron. Biochemistry. 2009;48:7420–7431. doi: 10.1021/bi900561a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Vagin A, Teplyakov A. MOLREP: an Automated Program for Molecular Replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Carson M. Ribbons. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]
- 24.Potterton L, McNicholas S, Krissinel E, Grubber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 Molecular-Graphics Project. Acta Crystallogr D Biol Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 25.Lovell SC, Davis IW, Arendall W. B. r., de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DS. Structure Validation by C-Alpha Geometry: Phi, Psi and C-Beta Deviation. Proteins. 2003;30:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 26.Yariv J, Kalb AJ, Sperling R, Bauminger ER, S.G. C, Ofer S. The Composition and Structure of Bacterioferritin of Escherichia coli. Biochem J. 1981;197:171–175. doi: 10.1042/bj1970171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eerde A, Wolterink-van Loo S, Van der Oost J, Dijkstra BW. Fortuitous Structure Determination of `As-Isolated' Escherichia coli Bacterioferritin in a Novel Crystal Form. Acta. Cryst. 2006;F62:1061–1066. doi: 10.1107/S1744309106039583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swartz L, Kuchinskas M, Li H, Poulos TL, Lanzilotta WN. Redox-Dependent Structural Changes in the Azotobacter vinelandii Bacterioferritin: New Insights into the Ferroxidase and Iron Transport Mechanism. Biochemistry. 2006;45:4421–4428. doi: 10.1021/bi060146w. [DOI] [PubMed] [Google Scholar]
- 29.Carrondo MA. Ferritins, Iron Uptake and Storage from the Bacterioferritin Viewpoint. EMBO J. 2003;22:1959–1968. doi: 10.1093/emboj/cdg215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hempstead PD, Yewdall SJ, Fernie AR, Lawson DM, Artymiuk PJ, Rice DW, Ford GC, Harrison PM. Comparison of the Three-Dimensional Structures of Recombinant Human H and Horse L Ferritins at High Resolution. J. Mol. Biol. 1997;268:424–448. doi: 10.1006/jmbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 31.Levi S, Santambrogio P, Corsi B, Cozzi A, Arosio P. Evidence that Residues Exposed on the Three-Fold Channels Have Active Roles in the Mechanism of Ferritin Iron Incorporation. Biochem J. 1996;317:467–473. doi: 10.1042/bj3170467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Treffry A, Bauminger ER, Hechel D, Hodson NW, Nowik I, Yewdall SJ, Harrison PM. Defining the Roles of the Threefold channels in Iron Uptake, Iron Oxidation and Iron-Core Formation in Ferritin: A Study Aided by Site-Directed Mutagenesis. Biochem J. 1993;296:721–728. doi: 10.1042/bj2960721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takagi H, Shi D, Ha Y, Allewell NM, Theil EC. Localized Unfolding at the Junction of Three Ferritin Subunits. J. Biol. Chem. 1998;273:18685–18618–18688. doi: 10.1074/jbc.273.30.18685. [DOI] [PubMed] [Google Scholar]
- 34.Jin W, Takagi H, Pancorbo B, Theil EC. “Opening” the Ferritin Pore for Iron Release by Mutation of Conserved Amino Acids at Interhelix and Loop Sites. Biochemistry. 2001;40:7525–7532. doi: 10.1021/bi002509c. [DOI] [PubMed] [Google Scholar]
- 35.Mann S, Williams JM, Treffry A, Harrison PM. Reconstituted and Native Iron-Cores of Bacterioferritin and Ferritin. J. Mol. Biol. 1987;198:405–416. doi: 10.1016/0022-2836(87)90290-7. [DOI] [PubMed] [Google Scholar]
- 36.Watt GD, Frankel RB, Jacobs D, Huang H. Fe2+ and Phosphate Interactions in Bacterial Ferritin from Azotobacter vinelandii. Biochemistry. 1992;31:5672–5679. doi: 10.1021/bi00139a035. [DOI] [PubMed] [Google Scholar]
- 37.Andrews SC, Le Brun NE, Barynin V, Thomson AJ, Moore GR, Guest JR, Harrison PM. Site-Directed Replacement of the Coaxial Heme Ligands of Bacterioferritin Generates Heme-free Variants. J Biol Chem. 1995;270:23268–23274. doi: 10.1074/jbc.270.40.23268. [DOI] [PubMed] [Google Scholar]
- 38.Yang X, Chen-Barrett Y, Arosio P, Chasteen ND. Reaction Paths of Iron Oxidation and Hydrolysis in Horse Spleen and Recombinant Human Ferritins. Biochemistry. 1998;37:9743–9750. doi: 10.1021/bi973128a. [DOI] [PubMed] [Google Scholar]
- 39.Bou-Abdallah F, Zhao G, Mayne HR, Arosio P, Chasteen ND. Origin of the Unusual Kinetics of Iron Deposition in Human H-Chain Ferritin. J. Am. Chem. Soc. 2005;127:3885–3893. doi: 10.1021/ja044355k. [DOI] [PubMed] [Google Scholar]
- 40.Smith JL. The Physiological Role of Ferritin-Like Compounds in Bacteria. Crit. Rev. Microbiol. 2004;30:173–185. doi: 10.1080/10408410490435151. [DOI] [PubMed] [Google Scholar]
- 41.Palma M, Worgall S, Quadri LEN. Transcriptome Analysis of the Pseudomonas aeruginosa Response to Iron. Arch. Microbiol. 2003;180:374–379. doi: 10.1007/s00203-003-0602-z. [DOI] [PubMed] [Google Scholar]
- 42.Ochsner UA, Wilderman PJ, Vasil AI, Vasil ML. GeneChip Expression Analysis of the Iron Starvation Response in Pseudomonas Aeruginosa: Identification of Novel Pyoverdine Biosynthesis Genes. Mol. Microbiol. 2002;45:1277–1287. doi: 10.1046/j.1365-2958.2002.03084.x. [DOI] [PubMed] [Google Scholar]
- 43.Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JA, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, Thomas CD, Shaw WV, Harrison PM. Solving the Structure of Human H Ferritin by Genetically Engineering Intermolecular Crystal Contacts. Nature. 1991;349:541–544. doi: 10.1038/349541a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.