

Abstract
The Arabidopsis thaliana genome contains two genes encoding the mitochondrial NAD-malic enzyme (NAD-ME), NAD-ME1 (At2g13560) and NAD-ME2 (At4g00570). The characterization of recombinant NAD-ME1 and -2 indicated that both enzymes assemble as active homodimers; however, a heterodimeric enzyme (NAD-MEH) can also be detected by electrophoretic studies. To analyze the metabolic contribution of each enzymatic entity, NAD-MEH was obtained by a co-expression-based recombinant approach, and its kinetic and regulatory properties were analyzed. The three NAD-MEs show similar kinetic properties, although they differ in the regulation by several metabolic effectors. In this regard, whereas fumarate activates NAD-ME1 and CoA activates NAD-ME2, both compounds act synergistically on NAD-MEH activity. The characterization of two chimeric enzymes between NAD-ME1 and -2 allowed specific domains of the primary structure, which are involved in the differential allosteric regulation, to be identified. NAD-ME1 and -2 subunits showed a distinct pattern of accumulation in the separate components of the floral organ. In sepals, the NAD-ME1 subunit is present at a slightly higher proportion than the NAD-ME2 subunit, and thus, NAD-MEH and NAD-ME1 act in concert in this tissue. On the other hand, NAD-ME2 is the only isoform present in anthers. In view of the different properties of NAD-ME1, -2, and -H, we suggest that mitochondrial NAD-ME activity may be regulated by varying native association in vivo, rendering enzymatic entities with distinct allosteric regulation to fulfill specific roles. The presence of three different NAD-ME entities, which originate by alternative associations of two subunits, is suggested to be a novel phenomenon unique to plant mitochondria.
Keywords: Allosteric Regulation, Enzyme Structure, Mitochondrial Metabolism, Protein Assembly, Protein Chimeras, Malic Enzyme
Introduction
Malic enzyme (ME)2 decarboxylates malate to pyruvate and CO2 in the presence of a divalent metal ion using NAD or NADP as cofactor. MEs can be classified in three different groups. The first group includes NADP-MEs (NADP-ME, EC 1.1.1.40), which are widely distributed in animals, plants, and microorganisms; this type uses NADP as cofactor and can decarboxylate oxaloacetate (OAA). The second group (NAD-ME, EC 1.1.1.38) is composed of enzymes found in Ascaris suum and bacteria, which use NAD and can decarboxylate OAA in addition to malate. Third category is plant NAD-MEs, which are not able to decarboxylate OAA (NAD-ME, EC 1.1.1.39).
MEs are widely distributed in nature, and the products of their reaction participate in many biosynthetic pathways and in respiratory metabolism. In plants NADP-MEs are localized to both plastids and cytosol (1, 2), whereas NAD-MEs are found in mitochondria (3). Apart from being involved in C4-photosynthesis and Crassulacean acid metabolism, non-photosynthetic roles have been proposed for NADP- and NAD-ME, including plant defense responses (4–6), tolerance to osmotic stress (7), lipid and lignin biosynthesis (8–10), control of cytosolic pH (11), and malate respiration (12).
NADP-MEs, and non-plant NAD-MEs are homooligomeric proteins. The tetramer is the most common form, but higher and lower structural conformations have also been reported (2, 13–16). On the other hand, plant mitochondrial NAD-MEs are composed of two dissimilar subunits (α and β) at a 1:1 molar radio (17). Depending on the source of the enzyme, pH, and l-malate concentration, plant NAD-MEs assemble as heterodimers, heterotetramers, or heterooctamers (17–19). However, NAD-MEs purified from some C4 plants, e.g. Eleusine coracana, Panicum dichotomiflorum, and Amaranthus tricolor, are octamers composed of identical subunits (20).
In Arabidopsis thaliana, two genes encoding NAD-MEs, At2g13560 (NAD-ME1) and At4g00570 (NAD-ME2), show expression in all mature organs (12). The products of both genes, NAD-ME1 and NAD-ME2, share about 65% sequence identity and are immunological different (12). Recombinant NAD-ME1 and -2 are active homodimers with similar kinetic parameters (12). Arabidopsis insertion mutants defective in each NAD-ME gene exhibit residual NAD-ME activity, indicating that NAD-ME1 and -2 are functional as homodimers in vivo (12). Moreover, electrophoretic studies revealed that NAD-ME1 and -2 can also associate to form a heterodimeric enzyme both in vivo and in vitro (12).
Previous kinetic analyses of plant NAD-ME have been limited to enzymes that were purified from plant tissues, where the α and β subunits always co-purified (17–19). Currently, the available resources and characteristics of Arabidopsis as a model plant organism allow us to achieve a more complete understanding of the different properties of the separated NAD-ME oligomers. Here, we present the first biochemical characterization of a plant heteromeric NAD-ME (NAD-MEH) obtained by the recombinant technology and a co-expression-based approach. The metabolic regulation of the purified NAD-MEH was investigated and compared with that obtained with the isolated NAD-ME1 and -2 homodimers. In addition, the analysis of chimeric proteins between NAD-ME1 and -2 allowed assignment of the differential regulatory properties to specific regions of the primary structure. Finally, differential relative protein accumulation patterns suggest that NAD-ME activity may also be modulated by alternative associations of the subunits in some organs of Arabidopsis.
EXPERIMENTAL PROCEDURES
Heterologous Expression and Purification of Arabidopsis NAD-ME1, -2, and -H
For the co-expression of NAD-ME1 and -2, the cDNA fragment corresponding to the mature NAD-ME2 (12) was cloned in the pET29a vector using the BamHI and SalI sites. The pET29-NAD-ME2 construction expresses the mature NAD-ME2 without any fusion vector-coded sequence because the cloned cDNA has a stop codon in their 5′ end. BL21(DE3) Escherichia coli cells were simultaneously transformed with pET29-NAD-ME2 and pET32-NAD-ME1, which expresses NAD-ME1 as a His-tag fusion protein (12). The cells containing both vectors were selected on LB-agar plates supplemented with 100 μg/ml ampicillin (pET32 selection agent) and 30 μg/ml kanamycin (pET29 selection agent). The co-transformed cells were grown in LB medium until the culture reached an A600 of 0.6. The inductor lactose (1% w/v) was added, and the cells were cultured for a further 16 h at 16 °C. The cells were then harvested by centrifugation and resuspended in Buffer A (20 mm Tris-HCl, pH 7.9, 5 mm imidazole, and 2 mm phenylmethylsulfonyl fluoride), sonicated, and centrifuged for 10 min at 7000 × g at 4 °C. The supernatant was loaded onto a nickel-nitrilotriacetic acid column previously equilibrated with Buffer A. The co-expressed proteins were eluted with Buffer A containing 200 mm imidazole. Purified NAD-MEH was treated with enterokinase to remove the His tag fusion in NAD-ME1 and stored as previously described for the separated recombinant proteins (12). The expression and purification of NAD-ME1 and NAD-ME2 fusion proteins by pET32-NAD-ME1 and pET32-NAD-ME2 vectors were performed as previously described (12). The amino-terminal sequences encoded for by the expression vectors were removed using enterokinase, and the mature proteins were stored as previously described (12). The typical protein yield of NAD-ME1 or -2 was nearly 1.5 mg/200 ml of bacterial culture. For NAD-MEH, the typical protein yield was lower, nearly 0.5 mg/200 ml of bacterial culture.
Construction and Purification of Chimeric NAD-ME1q and -2q
For the generation of NAD-ME chimeric protein NAD-ME1q, a PCR reaction was conducted using as template the NAD-ME2 full-length cDNA and the following primer pairs: NAD-ME2F (5′-GGATCCTGCATCGTCCACAAGCGT-3′) and NAD-ME2PstI-HindIII (5′-CTGCAGCAGCGACATACATGACAAGCTTTCCAA-3′). The cloned product was treated with the restriction enzymes BamHI and HindIII, and the purified fragment was ligated to the fragment pET32-NAD-ME1 treated with the same endonucleases (Fig. 4B). For the generation of the chimeric protein NAD-ME2q, the amplification product obtained as indicated above was treated with the restriction enzymes BamHI and PstI, and the purified fragment was ligated to the fragment pET29-NAD-ME2 treated with the same endonucleases. The ligation product obtained has a cDNA of NAD-ME2 with a HindIII site at the position 530, not present in NAD-ME2 sequence. This construction was treated with HindIII and SalI, and the fragment obtained was purified and ligated to the vector pET32-NAD-ME1 digested with HindIII and SalI (Fig. 4B). The inserts of the chimeric constructs (pET32-NAD-ME1q and pET32-NAD-ME2q) were sequenced to verify correct swapping of the corresponding fragments and assume that no mistakes were introduced during the subcloning procedures. BL21(DE3) E. coli was transformed with the pET32 plasmid containing the chimeric NAD-MEs. The induction and purification of NAD-ME1q and NAD-ME-2q were performed as previously described (12). The amino-terminal sequences encoded by the expression vectors were removed using enterokinase, and the mature proteins were stored as previously described (12).
FIGURE 4.

Chimeric NAD-MEs constructed and analyzed in the present work. A, sequence alignment of the amino-terminal end of NAD-ME1 and -2 is shown. The sequences correspond to the first 200 amino acids of the mature NAD-ME1 and -2 obtained after eliminating the predicted mitochondrial targeting peptide (ARAMEMNON). The region with the most significant differences is underlined. The amino acid residues homologous to the residues involved in fumarate activation of human NAD(P)-ME and A. suum NAD-ME are indicated in light gray (33, 41, 42). The arrow indicates the site where the sequences of NAD- ME1 and -2 were exchanged for the generation of the chimeric proteins NAD-ME1q and -2q. B, shown are the regulatory properties of NAD-ME1q and -2q. The restriction sites HindIII and PstI of the parental enzymes (NAD-ME1 and NAD-ME2) were used to construct the reciprocal chimeric enzymes. The modulation of activity by CoA, acetyl-CoA, fumarate, and succinate of the parental and chimeric NAD-MEs are indicated on the right. A, activated; I, inhibited; N, no effect. C, shown are the postulated regions in the NAD-ME1 and -2 primary structure involved in allosteric activity modulation. Postulated CoA/acetyl CoA and fumarate/succinate binding sites in the parental NAD-ME sequences are indicated.
Gel Filtration Chromatography
The molecular mass of NAD-MEH was evaluated by gel filtration chromatography on a fast protein liquid chromatography system using a Superdex 200 10/300 GL column (Amersham Biosciences). The column was equilibrated with 25 mm Tris-HCl, pH 7.5, or with 50 mm Mes-NaOH, pH 6.5, and calibrated using molecular mass standards. The sample, and the standards were applied separately in a final volume of 50 μl at a constant flow rate of 0.5 ml/min.
Enzyme Activity Measurements
The enzymatic activity was determined spectrophotometrically using a standard reaction mixture containing 50 mm Hepes (pH 6.4 or 6.6 depending on the enzyme), 10 mm MnCl2, 4 mm NAD, and 10 mm l-malate in a final volume of 0.5 ml. The reaction was started by the addition of l-malate. Initial velocity studies were performed by varying the concentration of one of the substrates around its Km value while keeping the other substrate concentration at saturating levels. All kinetic parameters were calculated at least in triplicate using free concentrations of all substrates (21). Data were fitted to the Michaelis-Menten equation. In the case of sigmoidal kinetics, initial rates were fitted to the Hill equation by nonlinear regression. When testing different compounds as possible inhibitors or activators of the enzymatic activity, NAD-ME activity was measured in the presence of 0.5 or 2 mm concentrations of each effector and non-saturating concentrations of malate (Km l-malate value of each enzyme, Table 1).
TABLE 1.
Kinetic properties of recombinant Arabidopsis NAD-ME1, -2, and -H
The indicated values are the average of at least three different experiment with no more than 5% S.D. among them.
| pH optimum | kcat | Km NAD | kcat/Km NAD | Kml-malate | kcat/Kml-malate | |
|---|---|---|---|---|---|---|
| s−1 | mm | mm | ||||
| NAD-ME1a | 6.4 | 31.1 | 0.50 | 60.2 | 3.0b (1.9) | 10.3 |
| NAD-ME2a | 6.6 | 44.1 | 0.50 | 88.2 | 3.0 | 14.7 |
| NAD-MEH | 6.5 | 39.0 | 0.55 | 67.0 | 2.7b (2.0) | 14.2 |
| NAD-ME1q | 6.6 | 32.1 | 0.52 | 61.7 | 0.2 | 160.5 |
| NAD-ME2q | 6.4 | 11.4 | 0.58 | 19.6 | 4.0a (2.1) | 2.9 |
a Values previously obtained (12) are included for comparison.
b S0.5; the calculated Hill coefficient (nH) is indicated in parentheses.
The apparent activation constant (A50) values were obtained by varying the concentration of activator while keeping the NAD concentrations at saturating levels and the l-malate at non-saturating concentrations. Data were fitted, by nonlinear regression to Equation 1,
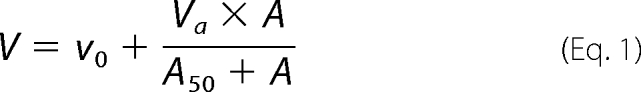 |
where v0 is the rate in absence of activator; Va is the maximum activated rate, A is the concentration of activator, and A50 is the concentration of activator that gives the 50% Va.
In the case of the activation by CoA for NAD-ME2 and NAD-MEH, the studies involved varying of concentration of a substrate while keeping the other substrate concentration fixed and at saturating levels and with varying concentrations of CoA. For NAD-ME2, the resulting data were fitted to Equation 2,
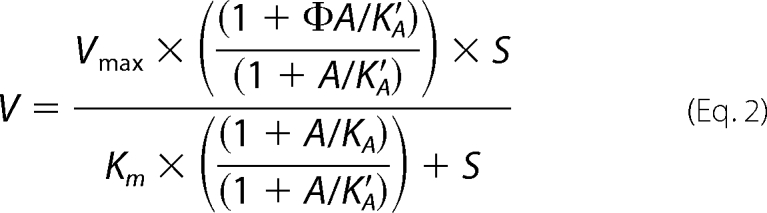 |
where V is the activity measured at a specific l-malate concentration (S), Vmax is the maximum activity, KA and KA′ are the dissociation constants of the activator (A) for the enzyme free and for the enzyme plus malate, respectively, and Φ is the ratio between k2 and k1. This last value (Φ) should be high than 1, as the activity when the allosteric site is occupied is high than when it is empty.
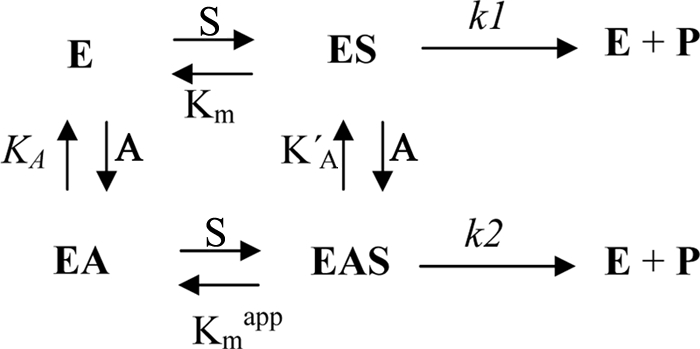
The model of activation that fit best to the kinetic data obtained is shown as Scheme 1, where the activator (CoA) can bind to the free enzyme and to the enzyme complexed with the substrates.
SCHEME 1.
The decarboxylation of OAA was monitored by measuring the disappearance of OAA at 260 nm (ϵ260 nm = 850 m−1 cm−1) in an assay medium containing 50 mm Mes-NaOH, pH 5.5, 1 mm OAA, and 10 mm MnCl2 in a final volume of 0.25 ml. The reported velocities were corrected for the background rate resulting from the nonenzymatic OAA decarboxylation catalyzed by the divalent metal ion.
One unit is defined as the amount of enzyme that catalyzes the formation of 1 μmol of NADH min−1 under the specified conditions. Protein concentration was determined by the method of Sedmak and Grossberg (22) using bovine serum albumin as standard.
Protein Crude Extract Preparations
Different organs (leaf, stem, flowers, and roots) of 6-week-old Arabidopsis were ground in N2, and the resulting powder was suspended in a buffer containing 50 mm Mes-NaOH, pH 6.5, 5 mm MnCl2, 1 mm EDTA, 10 mm 2-mercaptoethanol, 0.05% Triton X-100, 20% glycerol, and 1 mm phenylmethylsulfonyl fluoride). The homogenates were clarified by centrifugation and subjected to electrophoresis. Homogenates enriched in mitochondrial fraction from leaves were prepared by rounds of centrifugation, as previously described (12).
PAGE and Western Blot Analysis
Denaturing PAGE (SDS-PAGE) was performed in 8% (w/v) or 10% (w/v) polyacrylamide gels according to Laemmli (23). Proteins were visualized with Coomassie Blue or electroblotted onto a nitrocellulose membrane for immunoblotting. Antibodies against Arabidopsis NAD-ME1 or -2 were used for detection (12). When indicated, a mixture of purified antibodies against NAD-ME1 (1:500) and -2 (1:200) in a 1:1 relation were used. Bound antibodies were visualized by linking to alkaline phosphatase-conjugated goat anti-rabbit IgG according to the manufacturer's instructions (Sigma). Alkaline phosphatase activity was detected colorimetrically. Quantification of the intensity of the bands was conducted by image analysis software in at least three independent blots.
Native PAGE was performed using a 6% (w/v) acrylamide separating gel. Electrophoresis was run at 150 V at 10 °C. Gels were electroblotted onto a nitrocellulose membrane and subjected to Western blot analysis.
Circular Dichroism (CD) Spectra
CD spectra were made with a Jasco J-810 spectropolarimeter using 0.1-cm path length cell and averaging 10 repetitive scans between 250 and 200 nm. Typically, 50 μg of the wild type or chimeric NAD-ME in phosphate buffer (20 mm NaPi, pH 6.5) were used for each assay. Mean residue ellipticity (Φ) was obtained by the equation.
in which 111.42 was used as MMRW (the mean amino acid residue weight), d is the cell path in cm, and c is the concentration of the protein in mg/ml.
RESULTS
Heterologous Co-expression of Arabidopsis NAD-ME1 and -2 and Purification of NAD-MEH
To obtain the heteromeric Arabidopsis NAD-ME (NAD-MEH), NAD-ME1 fused to a His tag and NAD-ME2 without the His tag were co-expressed in E. coli. After induction of the expression, the bacterial extracts showed the presence of both proteins with the expected molecular masses of 80 kDa in the case of NAD-ME1 fusion protein and 58 kDa in the case of NAD-ME2 (Fig. 1A, lane 1). NAD-ME1 and -2 co-eluted after elution with imidazole (Fig. 1A, lane 3). To rule out the possibility of a nonspecific interaction of NAD-ME2 with the affinity column, E. coli expressing only NAD-ME2 without the His tag was similarly treated (Fig. 1A, lanes 5–7). In this case, NAD-ME2 mature protein was detected in bacterial extracts (Fig. 1A, lane 5) and the washing fractions (Fig. 1A, lane 6), but it was absent in the fraction eluted from the column after imidazole treatment (Fig. 1A, lane 7), indicating its failure to bind to the column due to the lack of the His tag. Moreover, specific antibodies against NAD-ME1 or -2 reacted with the respective proteins, indicating that the lower band that co-eluted with NAD-ME1 corresponds to NAD-ME2 and was not a partial proteolysis of NAD-ME1 (Fig. 1B). Densitometric analysis of the bands obtained after SDS-PAGE of the eluted fraction indicated that NAD-ME1 and -2 co-purify in an equimolar ratio (Fig. 1B, lane 1). These results indicate that the co-elution of NAD-ME1 and -2 is due to specific protein-protein interactions resulting in an heteromeric assembly (1:1) of NAD-ME1 and -2 (NAD-MEH).
FIGURE 1.

Purification of recombinant NAD-MEH by co-expression of NAD-ME1 and NAD-ME2. A, purification steps from E. coli BL21 cells co-transformed with pET32-NAD-ME1 and pET29-NAD-ME2 (lanes 1–3) or E. coli BL21 cells transformed with pET29-NAD-ME2 (lanes 5–7) were analyzed by Western blot after SDS-PAGE using a mixture of antibodies against NAD-ME1 and NAD-ME2. Lanes 1 and 5, 20 μg of E. coli crude extract after induction; lanes 2 and 6, last nickel-nitrilotriacetic acid column fraction wash; lanes 3 and 7, elute fraction with 200 mm imidazole. Purified NAD-ME1 fusion protein (3 μg) was loaded in lane 4. B, shown is Coomassie Blue-stained SDS-PAGE (lane 1, 10 μg) of purified NAD-MEH after enterokinase digestion. The same protein was analyzed by Western blot using antibodies against NAD-ME1 (Lane 2, 2 μg) or against NAD-ME2 (Lane 3, 2 μg). MM, molecular weight marker. C, shown is a Western blot analysis after native-PAGE of recombinant NAD-MEs using a mixture of antibodies against NAD-ME1 and NAD-ME2. Approximately 5 μg of NAD-ME1, -2, and -H were loaded. A mitochondrial leaf crude extract (mle, 30 μg) was also loaded on the gel. Molecular weight markers (MM) were run in parallel and stained with Coomassie blue.
Structural Properties of NAD-MEH
The purified recombinant NAD-MEH was analyzed by Western blot after native PAGE using a mixture of antibodies against NAD-ME1 and -2. NAD-MEH showed an immunoreactive band with similar mobility to the major immunoreactive band observed in Arabidopsis leaf mitochondrial extracts (Fig. 1C). Previous work demonstrated that this band is composed of both NAD-ME1 and -2, and it reacts with antibodies against NAD-ME1 or -2 used separately (12). Apart from this band, mitochondrial extracts also show bands corresponding to NAD-ME1 and -2 homodimers (Fig. 1C).
The native molecular mass of NAD-MEH determined by size exclusion chromatography was 125 ± 10 kDa. Thus, NAD-MEH is a dimer composed of NAD-ME1 and -2 in a 1:1 ratio. By the same technique, the native molecular mass of NAD-ME1 and -2 was previously estimated (120.0 and 117.5 kDa, respectively (12)).
Kinetic Properties of Arabidopsis NAD-ME1, NAD-ME2, and NAD-MEH
A kinetic characterization of the recombinant NAD-MEH was performed, and the results were compared with those obtained with NAD-ME1 and -2 homodimers. NAD-MEH has a pH activity optimum of 6.5 (Table 1), a value similar to those of NAD-ME1 and -2 (12). The kcat and the Km values for NAD of NAD-MEH were also similar to those reported for the homodimers (Table 1). On the other hand, NAD-MEH displayed a non-hyperbolic kinetic behavior with respect to malate, showing sigmoidal kinetics (nH = 2). This kinetic response was also observed for NAD-ME1 but not for NAD-ME2 (Table 1 (12)). Despite the different kinetics that were observed with respect to malate, the three enzymes showed similar affinity toward this substrate (Table 1). Hence, the three Arabidopsis NAD-MEs display similar catalytic efficiency (kcat/Km) for both NAD and malate (Table 1). Finally, as in the case of NAD-ME1 and -2, NAD-MEH was unable to decarboxylate OAA.
Regulatory Properties of Arabidopsis NAD-ME1, NAD-ME2, and NAD-MEH
Several intermediates of glycolysis and the tricarboxylic acid (TCA) cycle were tested as possible effectors of the individual NAD-ME activities. The results indicated that each NAD-ME responds differentially to the effectors tested (Fig. 2). Interestingly, succinate and fumarate showed opposite effects on the activity of the homodimers (Fig. 2). Although NAD-ME1 was strongly activated by these organic acids, NAD-ME2 was inhibited. The activation effect of succinate and fumarate was also observed in the case of NAD-MEH (Fig. 2). On the other hand, although PEP and FBP were the strongest activators of NAD-ME2 and -H (nearly 400% activation with 2 mm PEP or FBP), they did not modify the activity of NAD-ME1 (Fig. 2). The three NAD-MEs were activated by 2-ketoglutarate, whereas OAA activated NAD-ME1 and -H but did not modify the activity of NAD-ME2 (Fig. 2). NAD-ME2 and -H were activated more than 2-fold in the presence of CoA or acetyl-CoA, whereas the activity of NAD-ME1 was not significantly modified (Fig. 2). Finally, phosphorylated nucleosides (AMP and ATP) did not significantly modify the activity of the NAD-MEs even at high concentrations (Fig. 2).
FIGURE 2.

Regulatory properties of recombinant NAD-ME1, -2, and -H. NAD-ME1, -2, and -H activities were measured at the optimum pH in the absence or presence of each effector. l-Malate concentration was kept at the Km value for each enzyme (Table 1). The results represent the % of activity in the presence of each effector in relation to the activity measured in the absence of the metabolites (100%). Assays were done at least by triplicate, and error bars indicate S.D. Bars with dark gray and parallel lines, significant inhibition (less than 70% residual activity). Bars with light gray and crossed lines, significant activation (more than 140%).
The apparent activation constant (A50, activator concentration that gives 50% of activation) was estimated in the case of FBP, PEP, and fumarate (Table 2). The results showed that NAD-MEH is more sensitive with respect to FBP and PEP than NAD-ME2, exhibiting a 5.2- and 4.8-fold decrease in the A50 value, respectively (Table 2). NAD-MEH displays a similar apparent affinity (A50 values) for fumarate as NAD-ME1 (Table 2).
TABLE 2.
Apparent activation constant (A50) for FBP, PEP, fumarate, and CoA
The indicated values are the average of the values obtained from at least three different data sets with no more than 5% S.D. among them. −, no activation was observed (Fig. 2).
| FBP | PEP | Fumarate | CoA | |
|---|---|---|---|---|
| μm | mm | mm | μm | |
| NAD-ME1 | − | − | 1.1 | − |
| NAD-ME2 | 115 | 0.48 | − | 15.0 |
| NAD-MEH | 22.0 | 0.10 | 0.84 | 16.1 |
CoA Activation of NAD-ME2 and NAD-MEH
CoA is a typical activator of plant NAD-MEs, and it is present at high levels in mitochondria (24). Thus, the activation of NAD-ME2 and NAD-MEH by CoA was further analyzed. In the presence of 50 μm CoA, NAD-ME2 and NAD-MEH optimum pH was shifted to a more alkaline value (pH 6.8; Tables 1 and 3). In addition, in the presence of CoA, the NAD saturation curves of NAD-ME2 and NAD-MEH did not significantly differ from the ones obtained in the absence of CoA (not shown), but the kinetic behavior of both enzymes with l-malate was dependent on the CoA concentration (Fig. 3). The CoA activation effect was examined by varying l-malate concentration at several fixed concentrations of the activator. In the case of NAD-ME2, the analysis of the hyperbolic malate saturation kinetics obtained in the presence of CoA indicated that the activator increased both the maximal catalytic rate and the affinity toward malate (Fig. 3A). In the presence of 50 μm CoA, a 1.6-fold increase in the kcat value (Fig. 3A) and a 15-fold decrease in the Km for l-malate were observed in comparison to the enzyme without activator (Fig. 3C, Tables 1 and 3). Hence, the catalytic efficiency (kcat/Km l-malate) was increased by 24-fold in the presence of 50 μm CoA (Tables 1 and 3). The kinetic data obtained for the activation by CoA of NAD-ME2 fitted to an equation corresponding to a mixed activation mode (Equation 2) gave KA and KA′ values of 17.2 and 2.2 μm, respectively. The apparent activation constant (A50) for CoA was 15.0 μm (Table 2).
TABLE 3.
Kinetic parameters of NAD-ME2, -H, and -1q in the presence of 50 μm CoA
The indicated values are the average of at least three different experiment with no more than 5% S.D.
| 50 μm CoA |
||||
|---|---|---|---|---|
| pH | Kml-malate | kcat | kcat/Km malate | |
| mm | s−1 | |||
| NAD-ME2 | 6.8 | 0.20 | 69.1 | 345 |
| NAD-MEH | 6.8 | 0.80a | 40.6 | 51 |
| NAD-ME1q | 6.8 | 0.06 | 45.0 | 750 |
a S0.5; nH = 1.3.
FIGURE 3.

Effect of CoA on NAD-ME2 (A) and NAD-MEH (B) activity as a function of the malate concentration is shown. Assays were done at 4 mm NAD and 10 mm MnCl2 in the presence of the indicated CoA concentration. The Km for l-malate of NAD-ME2 (C) and S0.5 for malate of NAD-ME-H (D) as a function of CoA concentration is indicated as an inset in each graph. The estimated kcat and Km or S0.5 for l-malate of each enzyme in the presence of 50 μm CoA are indicated in Table 3.
In the case of NAD-MEH, the increase in CoA concentration resulted in a decrease in the nH value, with a concomitant decrease in the S0.5 for l-malate and no significant modification of the kcat (Fig. 3, B and D). Thus, the catalytic efficiency of NAD-MEH was increased by 3.6-fold in the presence of 50 μm CoA (Table 1 and Table 3). The A50 value for CoA was similar to that of NAD-ME2 (16.1 μm, Table 2). No further variations in the kinetic parameters of NAD-ME2 and NAD-MEH were observed with CoA concentration higher than 50 μm (Fig. 3, C and D).
Synergistic Activation of NAD-MEH by Fumarate and CoA
The analysis presented above indicated that the individual NAD-MEs showed differential activation by several metabolites (Fig. 2). Among the different metabolites tested, fumarate was the strongest activator of NAD-ME1, whereas low concentrations of CoA activated NAD-ME2 (Fig. 2, Table 2). As fumarate and CoA increased the activity of NAD-MEH (Fig. 2, Table 2), it was important to analyze if these compounds are able to simultaneously modify the activity of NAD-MEH. The results obtained showed that the A50 for CoA decreases at increasing fumarate concentration, e.g. in the presence of 5 mm fumarate, the A50 for CoA was 4.5 μm, a value more than 3-fold lower than that obtained in the absence of fumarate (16.1 μm, Table 2). The converse experiment in which the A50 for fumarate was estimated in the presence of different CoA concentrations also demonstrated the synergistic effect of both activators on the NAD-MEH activity (not shown). In the presence of CoA (0.1 mm), the A50 for fumarate was 0.30 mm, a value nearly 3-fold lower than that obtained without CoA (0.84 mm, Table 2).
Construction, Expression, and Characterization of Chimeric NAD-MEs
At the amino acid level, Arabidopsis NAD-ME1 and -2 share 65% identity, exhibiting the most sequence divergence at the amino-terminal end (Fig. 4A). To determine whether this segment of the primary structure is responsible for the differences in regulatory properties, two chimeras (NAD-ME1q and NAD-ME2q) were constructed by interchanging the first 176 amino residues between NAD-ME1 and -2 (Fig. 4B). Both chimeric proteins were expressed in E. coli and purified to homogeneity. NAD-ME1q and -2q showed enzymatic activity and, thus, were kinetically and structurally characterized. The comparison of CD spectra of all chimeric and parental proteins did not indicate substantial changes (not shown), suggesting that the construction of the chimeric proteins did not result in a loss of overall structural integrity.
After native electrophoresis, purified NAD-ME1q presented a similar mobility to that of NAD-ME2, whereas NAD-ME2q mobility was similar to that of NAD-ME1 (supplemental Fig. 1). These results indicate that, as in the case of the parental proteins, the chimeric proteins assemble as dimers, and the amino-terminal segment of each NAD-ME homodimer is responsible for the differential mobility of NAD-ME1 and -2.
The kinetic characterization of NAD-ME1q and -2q indicated that the kcat value of NAD-ME1q was similar to the one obtained for NAD-ME1 (Table 1). However, the kcat value of NAD-ME2q was 4-fold lower than that of NAD-ME2 (Table 1). Both chimeric proteins showed Km NAD values similar to those of the parental enzymes (Table 1). Nevertheless, although NAD-ME2q affinity for l-malate was slightly lower than that of NAD-ME1 and -2, NAD-ME1q affinity for l-malate was 15-fold higher than that of the parental enzymes (Table 1). Moreover, the kinetic behavior of NAD-ME1q with respect to malate was hyperbolic, whereas NAD-ME2q presented sigmoidicity (nH = 2.1, Table 1).
The effect of CoA, acetyl CoA, fumarate, and succinate on the enzymatic activity of the chimeras was analyzed and compared with the results obtained with the parental enzymes. As in the case of NAD-ME2, NAD-ME1q was activated by CoA and acetyl-CoA (Fig. 4B). On the other hand, NAD-ME2q activity was not modified by these effectors, as in the case of NAD-ME1 (Fig. 4B). Moreover, although NAD-ME2q was activated by fumarate and succinate, NAD-ME1q activity was not modified by these compounds, which were strong activators of NAD-ME1 and inhibitors of NAD-ME2 (Figs. 4B and 2). The estimated A50 value for fumarate of NAD-ME2q (0.88 mm) as well as the A50 value for CoA of NAD-ME1q (24.5 μm) was similar to those of the respective parental enzymes (Table 2), indicating that the binding sites for these metabolites were conserved in the chimeric enzymes.
The CoA activation effect on NAD-ME1q was further analyzed, and the kinetic parameters were estimated in the presence of 50 μm CoA (Table 3). The results obtained show that, in the presence of CoA, the apparent Km l-malate value of NAD-ME1q decreased 3-fold, whereas the kcat value increased 1.4-fold (Tables 1 and 3). Finally, as in the case of NAD-ME2 and -H, the pH optimum of NAD-ME1q moved to a more alkaline value in the presence of CoA (Tables 1 and 3).
Relative Organ-specific Accumulation of NAD-ME1 and -2
Previous work indicated that the transcripts of NAD-ME1 and -2 can be detected in leaf, stem, flower, and root (12). Moreover, the comparison of the abundance of each transcript was very similar in all mature organs (12). However, as the level of protein accumulation might not directly parallel the level of transcript, Western blot analysis of crude extracts from different organs was performed using a mixture of specific antibodies against NAD-ME1 and -2. To quantify the amount of immunoreactive protein detected in each organ, NAD-MEH was used as control, as it is composed of NAD-ME1 and -2 in a 1:1 protein:protein ratio (Fig. 1). The relative expression of NAD-ME1 and -2 was determined by densitometric analysis of several gels using different crude extracts (Fig. 5A). The results indicated that the relative abundance of NAD-ME2 to -1 in inflorescences differ significantly from the control (1:1) in that NAD-ME2 protein accumulation was higher than that of NAD-ME1 (Fig. 5A). In leaf, stem, and root, the relative abundance of NAD-ME1 and -2 was not significantly different from the control (Fig. 5A).
FIGURE 5.

SDS- and native-PAGE of extracts of Arabidopsis organs analyzed by Western blot. A, shown is a Western blot of crude extracts of different Arabidopsis organs after SDS-PAGE. To assess the relative NAD-ME1 and -2 immunoreactivity toward the antibodies, 50 ng of NAD-MEH, which has a 1:1 protein:protein relationship between NAD-ME1 and -2, was loaded in the first lane (NAD-MEH). Fifty μg of total soluble protein from leaf (L), stem (S), flower (F), and root (R) were loaded. Molecular masses of the immunoreactive bands are shown on the right (kDa). The assay was performed using a mixture of specific antibodies against NAD-ME1 and -2. The relative quantification of the immunoreactive bands of NAD-ME1 and -2 for each line is shown in the upper graph. Standard deviations of the densitometric analysis among at least three different Western blots are shown, and the asterisk indicates a significant different relative level of expression (p < 0.05). B, shown is a Western blot of the separated components of Arabidopsis flowers after SDS-PAGE. Fifty μg of total soluble protein from leaf (L), flower (F), gynoecium (G), sepal (Sp), filament (Fi), and anther (A) were loaded. Molecular masses of the immunoreactive bands are shown on the right (kDa). The assay was performed using a mixture of specific antibodies against NAD-ME1 and -2. C, shown is a Western blot of the separated components of Arabidopsis flowers after Native-PAGE. Fifty μg of total soluble protein from flower (F), gynoecium (G), sepal (Sp), filament (Fi), and anther (A) were loaded. The assay was performed using a mixture of specific antibodies against NAD-ME1 and -2. Molecular mass markers (MM) were run in parallel and stained with Coomassie Blue. The mobility of purified NAD-ME1, -2, and -H in native gels (Fig. 1C) is indicated on the right.
To further analyze NAD-ME1 and -2 protein accumulation in inflorescences, Western blot analysis was performed in the separated parts of the flowers (gynoecium, sepals, filaments, and anthers). The densitometric analysis of the immunoreactive bands obtained indicated that the relative level of NAD-ME1 and -2 in gynoecium and filaments did not differ from the one estimated in leaves (not shown). However, NAD-ME1 was prevalent in sepals, whereas only NAD-ME2 was found in anthers (Fig. 5B). In addition, crude extracts from sepals, gynoecium, filaments and anthers were analyzed by Western blot after native PAGE (Fig. 5C). Whole flowers, gynoecium, and filaments showed three immunoreactive bands with mobilities corresponding to NAD-ME1, -2, and -H (Figs. 5C and 1C). On the other hand, in sepals, two immunoreactive bands corresponding to NAD-MEH and NAD-ME1 were detected (Fig. 5C), whereas in anthers only a band corresponding to NAD-ME2 was found (Fig. 5C).
Transgenic Arabidopsis lines expressing the GUS reporter gene (12) were used to analyze the activity of NAD-ME1 and -2 promoters in inflorescences. In mature flowers, NAD-ME1 is expressed in the filaments, vasculature of sepals, and apical part of the gynoecium (supplemental Fig. 2). In developing flowers, a weak activity could be observed in the sepals. GUS expression driven by the NAD-ME2 promoter was found in the filaments, apical part of the gynoecium, and mature pollen grains (supplemental Fig. 2). It should be noted that the differential expression pattern observed is consistent with AtGenExpress data from the Genevestigator microarray data base (25). Thus, the different NAD-ME1 and -2 protein levels detected (Fig. 5) are due to differential expression of the corresponding transcripts, although regulation at the posttranscriptional level cannot be ruled out.
DISCUSSION
Reconstitution of an Active NAD-MEH through a Co-expression-based Approach
Previous studies indicated a heteromeric association between NAD-ME1 and -2 in Arabidopsis mitochondria (12). However, the homodimeric forms of NAD-ME1 and -2 were also active in vitro and in vivo (12). To compare the properties of the different native associations between Arabidopsis NAD-ME1 and -2, the heteromeric Arabidopsis NAD-ME (NAD-MEH) was successfully obtained by a recombinant approach (Fig. 1). NAD-MEH was active as a dimer composed of NAD-ME1 and -2 in a 1:1 ratio. The maximum catalytic rate and the affinity toward the substrates were very similar among the three native associations (Table 1). Native molecular masses assessed by gel filtration chromatography were very similar and in all cases compatible with a dimer. Nevertheless, differential migration in native gels was observed for the three NAD-MEs (Fig. 1C), which may indicate a different net charge of the oligomeric forms.
NAD-ME1, -2, and -H Are Differentially Regulated by Key Metabolites
The very distinct regulatory patterns obtained for NAD-ME1, -2, and -H (Fig. 2) suggest different metabolic contribution of each NAD-ME in Arabidopsis. Fumarate, succinate and OAA behaved as activators of NAD-ME1 and NAD-MEH (Fig. 2). Because of the structural similarity of these organic acids, the activation observed could be a result of the existence of a unique allosteric site in the NAD-ME1 subunit. The low A50 values of fumarate of NAD-ME1 and -H (nearly 1 mm, Table 2) and the high levels of this compound found in Arabidopsis leaves (26) suggest that the regulation of NAD-ME activity by this organic acid should have physiological significance in vivo. Fumarate and l-malate can be used to replenish the TCA cycle pool and as a carbon source for mitochondrial respiration. During the day, Arabidopsis leaves accumulate great amounts of these organic acids, which are metabolized during the following night (26). Total NAD-ME activity is enhanced at the end of the night period due to a higher transcriptional activity of NAD-ME1 and -2 genes (12). By the onset of darkness, when the fumarate levels are high, the allosteric activation of NAD-ME1 and -H by this organic acid would further increase the NAD-ME activity in addition to the transcriptional up-regulation. In addition, fumarate, succinate and OAA levels increase by cold stress (27). Low temperatures reduce the availability of Pi and adenylates, and thus, a lower supply of pyruvate to the mitochondria via the pyruvate kinase reaction is expected (28). However, a respiratory homeostasis has been reported in plants stressed by low temperature (29). Thus, in these situations increased concentrations of organic acids would stimulate the supply of pyruvate by increasing mitochondrial NAD-ME activity. Interestingly, fumarate and succinate behave as inhibitors of NAD-ME2 (Fig. 2). These compounds could inhibit the activity of this enzyme by competing with the substrate l-malate in the active site as a result of structural homology.
The strong effect of the products of the two irreversible reactions of glycolysis, FBP and PEP, on the enzymatic activity of NAD-ME2 and -H (Fig. 2) is consistent with the hypothesis that NAD-ME would supply pyruvate when pyruvate kinase is inhibited. NAD-MEH showed the highest sensitivity to FBP and PEP (Table 2), suggesting that this enzyme could be involved in such function in vivo. In this regard, plant mitochondria can import PEP from the cytosol by a PEP/H+ symporter or a PEP/ATP antiporter (30). Moreover, the PEP concentration in plant cells is in the micromolar range (31), and it increases by conditions that limit the pyruvate kinase reaction, e.g. by cold stress or low phosphate (28).
CoA is a typical activator of plant NAD-ME that has been shown to activate all enzymes characterized so far (24). In contrast to this, the results of this study clearly show that the activity of NAD-ME1 is not modulated by CoA, whereas the catalytic efficiency of NAD-ME2 was drastically increased by this compound (Tables 1 and 3). The kinetic data suggest a mixed mechanism of activation of NAD-ME2 with modification of the maximal catalytic activity and the affinity toward l-malate (Fig. 3, Table 3). On the other hand, the catalytic efficiency of NAD-MEH showed a moderate increment due to an increased affinity toward l-malate. The KA (and KA′) and A50 values for CoA (Table 2) of NAD-ME2 and NAD-MEH are in the micromolar range, and thus, it is possible that both enzymatic activities are subjected to metabolic control in vivo. Moreover, CoA and fumarate showed a synergistic activating effect on NAD-MEH because each metabolite increases the affinity of the enzyme for the other metabolite, as was observed for the purified NAD-ME from cauliflower (32). In addition, this work shows that fumarate and CoA bind to different subunits on the heterodimer surface. Thus, the binding of one of the effectors may produce changes in the heterodimer interface that are transmitted to the allosteric site for the other activator.
The mitochondrial NAD-ME enzyme occupies a key position in mitochondrial carbon metabolism, providing a means whereby organic acids can be partitioned between replenishment of mitochondrial pools and complete oxidation (Fig. 6A). As such, it would not be surprising that NAD-ME represents an important regulatory site for the control of mitochondrial carbon flux. The different NAD-ME entities, originated by alternatives assembly of two proteins, could be a mechanism to respond to differential allosteric regulation, rendering isoforms best suited to fulfill specific roles or to act under specific metabolic situations (Fig. 6B). Furthermore, the kinetic behavior with respect to l-malate is also modified by alternative associations. In this regard, the strong activation by fumarate and succinate and its sigmoidal kinetics to l-malate renders NAD-ME1 best suited for physiological situations where organic acids are the main substrates for mitochondrial respiration (Fig. 6A). On the other hand, NAD-ME2 responds to the glycolytic intermediate PEP and CoA (Fig. 6A), suggesting a concerted function with phosphoenolpyruvate carboxylase under situations where the pyruvate supply to the TCA cycle by the pyruvate kinase reaction is diminished. Finally, NAD-MEH is a form that can integrate the several forms of regulation and respond synergistically to activators of NAD-ME1 or NAD-ME2.
FIGURE 6.

A, the scheme shows the most relevant metabolites regulating NAD-ME1 or NAD-ME2 activity. NAD-ME1 and -2 are differentially activated by intermediates of the TCA cycle (fumarate, succinate, and OAA), the glycolytic pathway (FBP and PEP), and CoA. B, shown is postulated regulation of NAD-ME activity in Arabidopsis by differential native association. The three native associations of NAD-ME (NAD-ME1, -2, and -H) display different kinetic responses to malate and differential regulation by metabolites. The most relevant differential properties among the oligomers are indicated.
The Amino-terminal Region of NAD-ME1 and -2 Is Critical for Activation by C4 Organic Acids and CoA
NAD-ME1 and -2 significantly diverge at the amino-terminal end where the differences mostly reflect changes of charge (Fig. 4A). The analysis of the recombinant chimeras NAD-ME1q and -2q indicated that the amino-terminal regions of NAD-ME1 and -2 are associated with differences in the regulatory properties observed in these proteins; that is, activation by CoA and the C4-organic acids fumarate and succinate (Fig. 4, B and C).
As fumarate activates both NAD-ME2q and NAD-ME1 (Fig. 4B), it is concluded that an allosteric site responsible for such activation is present in the amino-terminal region of NAD-ME1 (Fig. 4C). This segment of NAD-ME1 possesses homologous residues to those of the human NAD(P)-ME that are involved in fumarate binding at an allosteric site: Arg-67, Arg-91, and Asp-102 (Fig. 4A) (33). However, these residues are also present in NAD-ME2, which is inhibited by this metabolite. Thus, there may be additional factors controlling the binding capacity and response to fumarate in the plant enzyme. In addition, the activation of NAD-ME2q by succinate further supports the hypothesis of an allosteric site for organic acids at the amino-terminal region of NAD-ME1 (Fig. 4C). However, further studies are necessary to establish the existence of this site.
On the other hand, NAD-ME1q was activated by CoA and acetyl CoA but not by the C4-organic acids fumarate and succinate (Fig. 4B). Thus, residues of the amino-terminal region of NAD-ME2 are involved in the regulation by CoA and acetyl-CoA (Fig. 4, B and 4C), probably by binding at the same allosteric site. Notable it the high affinity for malate of this chimeric enzyme, which displays a nearly 15-fold decrease in the Km value for malate with respect to the parental enzymes (Table 1). This increase in affinity may be due to a particular arrangement of the amino acid residues in the malate binding site of NAD-ME1q by participating residues from the both segments that were swapped. Finally, the fact that the kinetic behavior with respect to l-malate was hyperbolic for NAD-ME1q and sigmoidal for NAD-ME2q suggests that some amino acids residues of the amino-terminal end of the NAD-MEs would also be implicated in the l-malate binding.
Is the Formation of Alternative Oligomeric Forms a Fine-tuning Mechanism to Regulate NAD-ME Activity in Arabidopsis Mitochondria?
During the evolution of vascular plants, NAD-ME1 and -2 likely arose through gene duplication and subsequently evolved unique regulatory features. Although NAD-ME1 and -2 genes show a similar pattern of expression in mature organs of Arabidopsis (12), it cannot be ruled out that the proteins accumulate at different levels due to differences in stability or because the proteins are produced at different rates. Such changes in the protein ratio can influence the formation of the different NAD-ME native associations. In this regard, in mid-vein tissues of Arabidopsis, NAD-ME functions as a heterodimer, and NAD-ME2 can form an active homodimer, at least when NAD-ME1 is not present (34). In contrast, NAD-ME1 gene product is unable to form functional homodimers in cells of the mid-vein (34).
In this work, we show that NAD-ME1 and -2 proteins accumulate at different levels in the separate parts of Arabidopsis inflorescences (Fig. 5B). In this respect, NAD-ME1 is present at higher proportions than NAD-ME2 in sepals (Fig. 5B), and thus, NAD-MEH and NAD-ME1 can act in concert in this tissue (Fig. 5C). On the other hand, NAD-ME2 homodimer is dominant in anthers (Fig. 5, B and C). As NAD-ME2 responds mostly to CoA and intermediates of the glycolytic pathway but not to intermediates of the TCA cycle (Fig. 6), it is possible that in anthers, where mitochondrial respiration is highly active, NAD-ME activity could be linked to the rate of glycolysis.
In plants, multisubunit composition and changes in oligomeric assembly, depending on the tissue and the metabolic situation, are properties of important glycolytic and TCA cycle enzymes such as pyruvate kinase, NAD-isocitrate dehydrogenase, and PPi-dependent phosphofructokinase and phosphoenolpyruvate carboxylase (35–39). These characteristics represent a mechanism of allosteric regulation of enzymes of central metabolic pathways (40). The presence of three different NAD-MEs originating by alternative associations of NAD-ME1 and -2 may be a novel phenomenon unique to plant mitochondria.
Supplementary Material
This work was supported by the Agencia de Promocion Cientifica y Tecnologica (PICT Grant 32233) and the Consejo Nacional de Investigaciones Científicas y Técnicas (Argentina) and the Deutsche Forschungsgemeinschaft (Germany).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- ME
- malic enzyme
- NAD-ME
- NAD-dependent ME
- FBP
- fructose 1,6-biphosphate
- PEP
- phosphoenolpyruvate
- OAA
- oxaloacetate
- Mes
- 4-morpholineethanesulfonic acid
- TCA
- tricarboxylic acid
- CoA
- coenzyme A.
REFERENCES
- 1.Drincovich M. F., Casati P., Andreo C. S. (2001) FEBS Lett. 490, 1–6 [DOI] [PubMed] [Google Scholar]
- 2.Gerrard Wheeler M. C., Tronconi M. A., Drincovich M. F., Andreo C. S., Flügge U. I., Maurino V. G. (2005) Plant Physiol. 139, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winning B. M., Bourguignon J., Leaver C. J. (1994) J. Biol. Chem. 269, 4780–4786 [PubMed] [Google Scholar]
- 4.Casati P., Drincovich M. F., Edwards G., Andreo C. S. (1999) Photosynth. Res. 61, 99–105 [Google Scholar]
- 5.Maurino V. G., Saigo M., Andreo C. S., Drincovich M. F. (2001) Plant Mol. Biol. 45, 409–420 [DOI] [PubMed] [Google Scholar]
- 6.Müller G. L., Drincovich M. F., Andreo C. S., Lara M. V. (2008) Plant Cell Physiol. 49, 469–480 [DOI] [PubMed] [Google Scholar]
- 7.Liu S., Cheng Y., Zhang X., Guan Q., Nishiuchi S., Hase K., Takano T. (2007) Plant Mol. Biol. 64, 49–58 [DOI] [PubMed] [Google Scholar]
- 8.Smith R. G., Gauthier D. A., Dennis D. T., Turpin D. H. (1992) Plant Physiol. 98, 1233–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eastmond P. J., Dennis D. T., Rawsthorne S. (1997) Plant Physiol. 114, 851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casati P., Spampinato C., Andreo C. S. (1997) Plant Cell Physiol. 38, 928–934 [Google Scholar]
- 11.Martinoia E., Rentsch D. (1994) Annu. Rev. Plant Physiol. Plant Mol. Biol. 45, 447–467 [Google Scholar]
- 12.Tronconi M. A., Fahnenstich H., Gerrard Weehler M. C., Andreo C. S., Flügge U. I., Drincovich M. F., Maurino V. G. (2008) Plant Physiol. 146, 1540–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagel W. O., Sauer L. A. (1982) J. Biol. Chem. 257, 12405–12411 [PubMed] [Google Scholar]
- 14.Moreadith R. W., Lehninger A. L. (1984) J. Biol. Chem. 259, 6222–6227 [PubMed] [Google Scholar]
- 15.Davisson V. J., Schulz A. R. (1985) Biochem. J. 225, 335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saigo M., Bologna F. P., Maurino V. G, Detarsio E., Andreo C. S., Drincovich M. F. (2004) Plant Mol. Biol. 55, 97–107 [DOI] [PubMed] [Google Scholar]
- 17.Willeford K. O., Wedding R. T. (1987) J. Biol. Chem. 262, 8423–8429 [PubMed] [Google Scholar]
- 18.Grover S. D., Wedding R. T. (1982) Plant Physiol. 70, 1169–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long J. J., Wang J. L., Berry J. O. (1994) J. Biol. Chem. 269, 2827–2833 [PubMed] [Google Scholar]
- 20.Murata T., Ohsugi R., Matsuoka M., Nakamoto H. (1989) Plant Physiol. 89, 316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Detarsio E., Andreo C. S., Drincovich M. F. (2004) Biochem. J. 382, 1025–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sedmak J. J., Grossberg S. E. (1977) Anal. Biochem. 79, 544–552 [DOI] [PubMed] [Google Scholar]
- 23.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 24.Wedding R. T. (1989) Plant Physiol. 90, 367–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmermann P., Hirsch-Hoffmann M., Hennig L., Gruissem W. (2004) Plant Physiol. 136, 2621–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fahnenstich H., Saigo M., Niessen M., Zanor M. I., Andreo C. S., Fernie A. R., Drincovich M. F., Flügge U. I., Maurino V. G. (2007) Plant Physiol. 145, 640–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan F., Kopka J., Haskell D. W., Zhao W., Schiller K. C., Gatzke N., Sung D. Y., Guy C. L. (2004) Plant Physiol. 136, 4159–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plaxton W. C., Podesta F. E. (2006) Crit. Rev. Plant Sci. 25, 159–198 [Google Scholar]
- 29.Kurimoto K., Millar A. H., Lambers H., Day D. A., Noguchi K. (2004) Plant Cell Physiol. 45, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 30.de Bari L., Valenti D., Pizzuto R., Atlante A., Passarella S. (2007) Biochim. Biophys. Acta 1767, 281–294 [DOI] [PubMed] [Google Scholar]
- 31.Farré E. M., Tiessen A., Roessner U., Geigenberger P., Trethewey R. N., Willmitzer L. (2001) Plant Physiol. 127, 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Canellas P. F., Wedding R. T. (1984) Arch. Biochem. Biophys. 229, 414–425 [DOI] [PubMed] [Google Scholar]
- 33.Yang Z., Lanks C. W., Tong L. (2002) Structure 10, 951–960 [DOI] [PubMed] [Google Scholar]
- 34.Brown N. J, Palmer B. G., Stanley S., Hajaji H., Janacek S. H., Astley H. M., Parsley K., Kajala K., Quick W. P., Trenkamp S., Fernie A. R, Maurino V. G., Hibberd J. M. (2010) Plant J. 61, 122–133 [DOI] [PubMed] [Google Scholar]
- 35.Kapri R., Dahan E., Zehavi U., Goren R., Sadka A. (2000) Acta Horticult. 535, 113–118 [Google Scholar]
- 36.Suzuki J., Mutton M. A., Ferro M. I., Lemos M. V., Pizauro J. M., Mutton M. J., Di Mauro S. M. (2003) Genet. Mol. Res. 2, 376–382 [PubMed] [Google Scholar]
- 37.Lemaitre T., Hodges M. (2006) Plant Cell Physiol. 47, 634–643 [DOI] [PubMed] [Google Scholar]
- 38.Andre C., Froehlich J. E., Moll M. R., Benning C. (2007) Plant Cell 19, 2006–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Leary B., Rao S. K., Kim J., Plaxton W. C. (2009) J. Biol. Chem. 284, 24797–24805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rontein D., Dieuaide-Noubhani M., Dufourc E. J., Raymond P., Rolin D. (2002) J. Biol. Chem. 277, 43948–43960 [DOI] [PubMed] [Google Scholar]
- 41.Karsten W. E., Pais J. E., Rao G. S., Harris B. G., Cook P. F. (2003) Biochemistry 42, 9712–9721 [DOI] [PubMed] [Google Scholar]
- 42.Hung H. C., Kuo M. W., Chang G. G., Liu G. Y. (2005) Biochem. J. 392, 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.