

Abstract
Olmesartan medoxomil (OM) is a prodrug type angiotensin II type 1 receptor antagonist widely prescribed as an antihypertensive agent. Herein, we describe the identification and characterization of the OM bioactivating enzyme that hydrolyzes the prodrug and converts to its pharmacologically active metabolite olmesartan in human liver and intestine. The protein was purified from human liver cytosol by successive column chromatography and was identified by mass spectrometry to be a carboxymethylenebutenolidase (CMBL) homolog. Human CMBL, whose endogenous function has still not been reported, is a human homolog of Pseudomonas dienelactone hydrolase involved in the bacterial halocatechol degradation pathway. The ubiquitous expression of human CMBL gene transcript in various tissues was observed. The recombinant human CMBL expressed in mammalian cells was clearly shown to activate OM. By comparing the enzyme kinetics and chemical inhibition properties between the recombinant protein and human tissue preparations, CMBL was demonstrated to be the primary OM bioactivating enzyme in the liver and intestine. The recombinant CMBL also converted other prodrugs having the same ester structure as OM, faropenem medoxomil and lenampicillin, to their active metabolites. CMBL exhibited a unique sensitivity to chemical inhibitors, thus, being distinguishable from other known esterases. Site-directed mutagenesis on the putative active residue Cys132 of the recombinant CMBL caused a drastic reduction of the OM-hydrolyzing activity. We report for the first time that CMBL serves as a key enzyme in the bioactivation of OM, hydrolyzing the ester bond of the prodrug type xenobiotics.
Keywords: Enzymes, Metabolism/Drug, Protein/Purification, Site-directed Mutagenesis, Xenobiotics, CMBL, Activation, Esterase, Identification, Prodrug
Introduction
In any biological organisms, the hydrolysis of endogenous and exogenous compounds is catalyzed by an extremely large variety of enzymes collectively known as hydrolases that catalyze numerous hydrolytic reactions of various bonds (i.e. amides, lactams, peptides, esters, and lactones) and have an essential role in biological activity in multiple sites (1). In particular, enzymes that hydrolyze various types of ester bonds have been categorized as esterases and have been well investigated as key enzymes that are frequently used for the bioactivation of ester-based prodrugs.
Historically, many ester-based prodrugs have been developed with the aim of overcoming a number of barriers to drug-like properties (2–4). Esterases, which are involved in the prodrug bioactivation, as typically represented by carboxylesterases (5, 6), cholinesterases (7, 8), and paraoxonases (9), are widely distributed in the blood, liver, intestine, and many other biological fluids and tissues. Therefore, these esterases are frequently targeted as bioactivating enzymes for ester-based prodrugs (10, 11). For example, human carboxylesterase, which is listed as one of the most important enzymes involved in the bioactivation of various therapeutic prodrugs such as anti-tumor drugs and angiotensin-converting enzyme inhibitors, show ubiquitous tissue expression profiles with the highest levels in liver microsomes (5, 6, 12). Human paraoxonase 1/arylesterase (PON1),2 reported to be a bioactivating enzyme of the antibacterial agent prulifloxacin (9, 13), is localized predominantly in plasma where it is associated with high density lipoprotein and the liver microsomal fraction (9, 14). As a more recent example, the novel prodrug bioactivating enzyme valacyclovirase, which hydrolyzes the amino acid ester prodrugs valacyclovir and valcyclovir, was identified and characterized (15, 16). To date, as shown in this case, molecular-based studies have revealed many valuable aspects of these enzymes, such as their protein structure and catalytic site.
Olmesartan medoxomil (OM) is the most recently launched angiotensin receptor blocker (ARB) and is prescribed worldwide as monotherapy and in combination with a thiazide diuretic or a calcium channel blocker (17–19). As shown in Fig. 1, OM is one of the exemplary cases of bioavailability improvement by derivatization into (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl ester (medoxomil-ester) prodrug (17). In several clinical trials, no components other than the active de-esterified metabolite olmesartan were detected in plasma after the oral administration of radiolabeled OM (20). This suggests that the medoxomil-ester prodrug is rapidly and completely converted to its pharmacologically active form in the gastrointestinal mucosa, portal blood, and liver before it comes into systemic circulation. As previously reported, human plasma PON1 (20) and plasma albumin (21) are capable of hydrolyzing OM to olmesartan. In addition to the plasma esterases, in vitro studies on esterase(s) involved in the bioactivation of OM were carried out using human liver and intestinal preparations. These studies demonstrated that the enzyme characteristics in these tissues are clearly different from those of the plasma esterases. This interesting finding has led us to investigate the unknown hydrolase(s) responsible for the OM bioactivation in the liver and intestine.
FIGURE 1.

Bioactivation of olmesartan medoxomil. A, bioactivation of olmesartan medoxomil is shown. Hydrolysis librates its active metabolite olmesartan and generates a diketone. Another possible product, RNH-8097, was not detected in in vitro reaction mixture. B, shown is the estimated mechanism of the base-catalyzed activation of olmesartan medoxomil, supporting the postulated mechanism previously published for several medoxomil ester prodrugs of carboxylic acid (39, 40).
The identification and characterization of prodrug bioactivating enzymes are important because they directly determine the pharmacokinetic and pharmacodynamic properties of the drug entities, such as the onset of drug action and potency of therapeutic efficacy. In this report we present the successful purification and identification of the OM bioactivating hydrolase from human liver cytosol. Here, the identified protein, human carboxymethylenebutenolidase homolog (carboxymethylenebutenolidase-like protein (CMBL), GenBankTM accession number NP_620164.1) is a novel esterase that has not received much attention until now, in contrast to the well characterized prodrug bioactivating esterases mentioned above. Notably, no information has been reported for CMBL concerning its biological functions in any higher eukaryotes, including humans. We describe the enzymatic characteristics of CMBL, namely its substrate specificity and inhibition properties, tissue distribution of its gene products, and predicted catalytic triad of this novel esterase.
EXPERIMENTAL PROCEDURES
Chemicals
OM and olmesartan were synthesized in Daiichi Sankyo Co., Ltd. Lenampicillin hydrochloride was extracted from Varacillin tablets (Organon-Japan, Osaka, Japan). Faropenem medoxomil and faropenem were purchased from Toronto Research Chemicals Inc. (Ontario, Canada) and Hangzhou-Hetd Industry Co., Ltd. (Hangzhou, China), respectively. Ampicillin and eserine were purchased from Sigma. Bis-p-nitrophenylphosphate (BNPP) and p-chloromercuribenzoate (PCMB) were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Phenylmethylsulfonyl fluoride (PMSF), diisopropyl fluorophosphate (DFP) and EDTA were purchased from Wako Pure Chemical. (Osaka, Japan).
Human plasma was prepared from fresh blood samples collected from healthy subjects under a protocol approved by the Institutional Human Ethical Committee. Human liver and intestinal subcellular fractions (cytosol and microsomes) were purchased from Tissue Transformation Technologies (Edison, NJ) or XenoTech LLC (Lenexa, KS). Human liver cytosolic fraction for purification of OM hydrolase was purchased from Human and Animal Bridging Research Organization (Tokyo, Japan).
OM Bioactivation and Chemical Inhibition Properties in Human Tissue Preparations
The OM-hydrolyzing activity in human liver or intestinal subcellular fractions (cytosol and microsomes) was measured in 10 mm potassium phosphate buffer (pH 7.4, incubation volume of 0.25 ml) at 37 °C. The protein concentrations were as follows: liver and intestinal cytosols, 0.05 mg protein/ml; liver microsomes, 0.5 mg protein/ml; intestinal microsomes, 0.25 mg protein/ml. The effect of esterase inhibitors was investigated using BNPP, DFP, PCMB, eserine, PMSF (1000 μm each), and EDTA (500 μm) by incubating liver, intestinal cytosols (0.2 mg protein/ml each), or diluted plasma (2.5 mg protein/ml) in the incubation volume of 0.25 ml and OM as a substrate (250 μm) with each inhibitor for 5 min. The concentration of the active metabolite olmesartan was determined by an HPLC system (SLC-10A vp system, Shimadzu, Kyoto, Japan) with a reversed-phase C18 column (YMC-Pack ODS-A A-312, C-18, 5 μm, 6.0 inner diameter (ID) × 150 mm, YMC, Kyoto, Japan) and UV detection at 254 nm. The peaks of olmesartan and OM were observed at the retention times of 5.0 and 15.2 min, respectively, with a mobile phase of 40% acetonitrile containing 1% PIC-A (Waters, Milford, MA) at a flow rate of 1.0 ml/min. The enzymatic activity was expressed as a metabolite formation rate (v0; nmol/min/mg of protein) based on the production of olmesartan, which was subtracted from that in the buffer control as non-enzymatic hydrolysis.
Purification of Human CMBL from Human Liver Cytosol
All column purification steps were performed using a fast protein liquid chromatography system (Amersham Biosciences) with monitoring absorbance at 280 nm. Human liver cytosolic fraction was added to four volumes of 20 mm potassium phosphate buffer (pH 7.4) containing 1.7 m ammonium sulfate followed by centrifugation at 19,000 × g for 10 min. The supernatant was loaded onto a hydrophobic interaction column (HiPrep 16/10 Octyl FF, Amersham Biosciences) and eluted with a linear gradient of 1.7–0 m ammonium sulfate in 20 mm potassium phosphate buffer (pH 7.4). The active fractions were purified on an ion exchange column (DEAE-Sepharose Fast Flow column, Amersham Biosciences) with a linear gradient of 0–500 mm NaCl in 20 mm potassium phosphate buffer (pH 7.4). The active fractions were loaded onto a gel filtration column (Hi Prep 16/60 Sephacryl S-200 column, Amersham Biosciences) with 20 mm potassium phosphate buffer (pH 7.4). Between the serial column purification steps, the active fractions were concentrated by an ultrafiltration method using Ultracel Amicon YM10 (Millipore, Billerica, MA). A portion of the active fractions from the last column purification was loaded onto SDS-polyacrylamide gel (10–20% gradient gel, Bio-Rad) under reduced conditions, and the gel was stained with SYPRO Ruby (Molecular Probes) and scanned using a Molecular Imager FX (Bio-Rad). The active metabolite olmesartan was monitored as the OM-hydrolyzing activity by the HPLC method described above. The protein concentration was determined by a modified Lowry method using a DC Protein Assay (Bio-Rad) with bovine serum albumin as a standard.
To identify the protein, the bands on the SDS-polyacrylamide gel were excised and underwent in-gel tryptic digestion (modified trypsin, Promega), then the resultant peptides were subjected to a reverse-phase liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis as reported previously (22) with a slight modification (see supplemental Method 1). The tandem mass spectra were searched against the GenBankTM non-redundant protein data base using the Mascot program (Matrix Science, London, UK).
Molecular Cloning of Human CMBL
Human CMBL gene was amplified by PCR from the human liver and skeletal muscle cDNA library using forward and reverse primers (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGGCTAACGAAGCTTATCCTTGTCC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTACATGTACTTGTTCAGCCACTCAATTAA-3′, respectively). The resulting PCR product was cloned into pDONR221 entry vector by the BP reaction, and the sequences were confirmed by DNA sequencing.
Expression of Human CMBL in Escherichia coli and Mutant Generation
The open reading frame of the full-length human CMBL in the entry vector was inserted by the LR reaction into the corresponding site of the expression vector pDEST-T7/lac, which was constructed in-house. The resulting plasmid was used as the template for PCR. A QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to create two single mutants in human CMBL (C132A or C132S) in this study. The forward and reverse primers used for the mutagenesis were shown as follows: 5′-GGCATCGTGGGATTCGCCTGGGGTGGAACTGCTGTCC-3′ and 5′-GTTCCACCCCAGGCGAATCCCACGATGCCAATTTTC-3′ for the mutant C132A and 5′-CATCGTGGGATTCAGCTGGGGTGGAACTGCTGTCC-3′ and 5′-CCACCCCAGCTGAATCCCACGATGCCAATTTTC-3′ for the mutant C132S, respectively. To confirm the desired mutation and verify the absence of unintended mutations, the constructs were sequenced. Each plasmid for the native CMBL, and the mutants was transformed into E. coli BL21(DE3) competent cells (Novagen, Madison, WI), which were then grown in MagicMedia E. coli expression medium (Invitrogen) containing ampicillin (100 μg/ml). The template protein and two mutants were expressed as N-terminal His6-tagged proteins and purified using a column packed with His-select nickel affinity gel (Sigma). From the eluted recombinant proteins the tag was cleaved by biotinylated thrombin (Novagen) treatment. The resulting protein solutions were stored at 4 °C until use. The protein concentration was determined using a Bradford protein assay kit (Bio-Rad) with bovine serum albumin as a standard.
Expression of Human CMBL in Mammalian Cell Line
To obtain the destination vector pFLAG-CMV-2GW, reading frame B of a Gateway cassette (Invitrogen) was inserted into pFLAG-CMV-2 (Sigma). The converted destination vector was confirmed by DNA sequencing. The open reading frame of the full-length human CMBL was subcloned into the expression vector pFLAG-CMV-2GW, which encodes human CMBL with an N-terminal FLAG tag. The expression of the recombinant protein of human CMBL was performed using mammalian Freestyle 293-F cells (Invitrogen) according to the manufacturer's instructions. As a negative control, pFLAG-CMV-2GW destination vector was transfected into the cells. The transfected cells were cultured for 72 h in Freestyle 293 Expression Medium (Invitrogen) and lysed with Cellytic-M (Sigma) containing a protease inhibitor mixture (complete, Roche Applied Science). The cell extract was centrifuged, and the cell lysate supernatant was used as an enzyme source for the following in vitro metabolic reactions. The protein concentrations of human CMBL- and control vector- transfected cell lysate were determined by the Lowry method using a DC protein assay (Bio-Rad) with bovine serum albumin as a standard and were stored at −80 °C until use.
OM Bioactivation by Recombinant CMBL in E. coli System
The OM-hydrolyzing activity of the recombinant CMBL overexpressed in E. coli system was measured in 100 mm HEPES buffer (pH 7.4, incubation volume of 0.2 ml) at 37 °C containing OM as a substrate (10 μm) and the tag-purified protein (0.015 mg of protein/ml). The concentration of the active metabolite olmesartan was determined by an LC-MS/MS system consisting of Prominence LC-20A (Shimadzu) and API 3200 (Applied Biosystems). Olmesartan was separated with a reversed-phase C18 column (Atlantis T3, S-5 μm, 2.1 mm ID × 150 mm, Waters) and a mobile phase of 64% methanol containing 0.2% formic acid at a flow rate of 0.2 ml/min and was determined by monitoring the ion transition of m/z 447 to 207 with multiple reaction monitoring in the positive electrospray ionization mode. The enzymatic activity was expressed as a metabolite formation rate (v0; nmol/min/mg of protein) based on the production of olmesartan for the reaction by the recombinant protein, which was subtracted from that in the HEPES buffer as nonenzymatic hydrolysis.
OM Bioactivation by Recombinant CMBL in Mammalian Cell Lysate and Chemical Inhibition Properties
The OM-hydrolyzing activity of the CMBL-transfected mammalian cell lysate (0.05 mg of protein/ml) was measured in 100 mm HEPES buffer (pH 7.4, incubation volume of 0.2 ml) at 37 °C containing OM as a substrate (50 μm). The concentration of the active metabolite olmesartan was determined by an HPLC system (SLC-10A vp system, Shimadzu) with a reversed-phase C18 column (SunFire C18, S-5 μm, 4.6 mm ID × 150 mm, Waters) and UV detection at 254 nm. The peaks of olmesartan and OM were observed at the retention times of 2.8 and 5.3 min, respectively, with a mobile phase of 32% acetonitrile containing 0.1% trifluoroacetic acid at a flow rate of 1.0 ml/min. The enzymatic activity was expressed as a metabolite formation rate (v0; nmol/min/mg of protein) based on the production of olmesartan for the reaction by the recombinant protein, which was subtracted from that of the vector control as nonenzymatic hydrolysis. The effect of esterase inhibitors was investigated using the CMBL-transfected mammalian cell lysate as an enzyme source. OM as a substrate (50 μm) and each inhibitor, BNPP, DFP, PCMB, eserine, PMSF (10, 100, and 1000 μm), or EDTA (50, 500, and 5000 μm) were incubated with the preincubated cell lysate at 0.05 mg of protein/ml for 5 min.
Prodrug Bioactivation by Recombinant CMBL
The enzymatic activities hydrolyzing the medoxomil-ester type prodrugs, namely faropenem medoxomil and lenampicillin, were investigated. Each substrate was incubated in 100 mm HEPES buffer (pH 7.4, incubation volume of 0.2 ml) at 37 °C with the human CMBL-transfected mammalian cell lysate as an enzyme source at the protein concentrations of 0.1 and 0.4 mg of protein/ ml for faropenem medoxomil and lenampicillin, respectively. The concentration of faropenem, the active metabolite of faropenem medoxomil, was determined by an HPLC system (SLC-10A vp system, Shimadzu) with a reversed-phase C18 column (SunFire C18, S-5 μm, 4.6 mm ID × 150 mm, Waters) and UV detection at 254 nm. The peaks of the metabolite and substrate were observed at the retention times of 2.9 and 9.1 min, respectively, with a mobile phase of 32% acetonitrile containing 0.1% trifluoroacetic acid at a flow rate of 1.0 ml/min. The concentration of ampicillin, the active metabolite of lenampicillin, was determined by an LC-MS/MS system consisting of Alliance 2795 (Waters) and Micromass Quattro LC (Micromass). Ampicillin was separated with a reversed-phase C18 column (XBridge C18, S-5 μm, 2.1 mm ID × 150 mm, Waters) and a mobile phase of 21% acetonitrile containing 0.1% formic acid at a flow rate of 0.2 ml/min and was determined using the previously reported method by positive electrospray ionization mass spectrometry (23). The enzymatic activity was expressed as an active metabolite formation rate (v0; nmol/min/mg of protein) based on the production of active moiety, which was subtracted from that of the vector control as nonenzymatic hydrolysis.
Kinetic Analysis
The kinetic constant (Km) and maximum velocity (Vmax) for OM hydrolysis by the recombinant CMBL expressed in the E. coli system and the mammalian cells, and in human liver or intestinal cytosols and microsomes were calculated from the metabolite formation rate (v0) at substrate concentrations [S] ranging from 7.81 to 1000 μm. The kinetic parameters were estimated using WinNonlin Professional (Version 1.4.1, Pharsight, CA) by a nonlinear least square regression analysis fitting to the Michaelis-Menten equation, v0 = Vmax × [S]/(Km + [S]). For the kinetic analysis of the other prodrugs, faropenem medoxomil and lenampicillin, the following substrate concentration ranges were selected: 7.81–1000 μm and 0.781–100 μm for faropenem medoxomil and lenampicillin, respectively.
Expression Analysis of Human CMBL Transcript
Human total RNA isolated from 20 different tissues was purchased from Clontech (Mountain View, CA), and the cDNA was synthesized from 1 μg of the total RNA using an Omniscript RT kit (Qiagen) for reverse transcriptase (RT)-PCR. Glyceraldehyde-3-phosphate dehydrogenase was used as a reference gene. A TaqMan gene expression assay for human CMBL (Hs00540853_m1) and Pre-Developed TaqMan assay reagent human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were purchased from Applied Biosystems. Quantitative RT-PCR was performed on a 7500 real-time PCR system (Applied Biosystems) using a QuantiTech Probe PCR kit (Qiagen). The cycle threshold (Ct) values were obtained using Applied Biosystems 7500 system SDS software (Version 1.3). The data were normalized by setting the average placenta expression value to 1.
Western Blotting Analysis of Human CMBL
The following sample proteins were subjected to Western blot analysis of human CMBL: 10 ng of FLAG tag-purified recombinant CMBL and 400 ng each of CMBL-transfected cell lysate, human liver, intestinal cytosolic and microsomal fractions, and human plasma. The sample proteins were separated by SDS-PAGE using 12.5% sodium dodecyl sulfate-polyacrylamide gel (Ready Gels J 12.5%, Bio-Rad) and were transferred electrophoretically onto a polyvinylidene difluoride membrane (Immun-Blot polyvinylidene difluoride membrane, 0.2 μm, Bio-Rad). The native CMBL proteins expressed in the human tissue preparations were detected with affinity-purified rabbit polyclonal IgG against a human CMBL peptide (Immuno-Biological Laboratories, Takasaki, Japan, 1:5,000 dilution) as a primary antibody and ECL anti-rabbit IgG horseradish peroxidase-linked, from donkey (Amersham Biosciences, 1:10,000 dilution) as a secondary antibody. These immunoblots were visualized by chemiluminescence with an ECL Advance Western blotting detection kit (Amersham Biosciences).
RESULTS
OM Bioactivation and Chemical Inhibition Properties in Human Tissue Preparations
In both human liver and intestinal cytosols, OM was substantially converted into the active metabolite, olmesartan, in a single-enzyme Michaelis-Menten kinetics manner as shown in Fig. 2, A and B, with Km values of 160 and 193 μm (Table 1), respectively. The activities in both liver and intestinal cytosolic fractions were partially inhibited by 1000 μm BNPP, a carboxylesterase inhibitor, and were strongly inhibited by 1000 μm PCMB, a free thiol modifier, whereas they were not inhibited by the addition of the following esterase inhibitors: 1000 μm DFP and PMSF, irreversible serine hydrolase inhibitors; 1000 μm eserine, choline esterase inhibitor; 500 μm EDTA, divalent cation chelator inhibiting metallohydrolases (Fig. 2C). The chemical inhibition patterns observed in liver and intestinal cytosols were quite similar between these two tissues but were different from that in human plasma, where the activity was almost completely inhibited by PCMB and EDTA. These findings indicate that the major enzymes with OM-hydrolyzing activity in human liver and intestine would be an identical or a similar subtype of enzymes, but they could be clearly distinguished from PON1, which has been reported to be the major OM-bioactivating enzyme in human plasma (20). The OM-hydrolyzing activity was much higher in the cytosolic fractions than that in the microsomal fractions prepared from human liver and intestine (Fig. 2D and Table 1). Therefore, human liver cytosol was used as the starting material for purification of our target OM hydrolase by successive column chromatography.
FIGURE 2.

Enzymatic OM hydrolysis in human tissue preparations. A and B, kinetic analysis of OM hydrolysis in human liver and intestinal cytosols, respectively, is shown. Data points represent the means ± S.D. of triplicate determinations. The solid line is the best fit by nonlinear least squares regression to the Michaelis-Menten equation. The respective Eadie-Hofstee plot is presented in the inset. C, shown is the inhibitory effect of known esterase inhibitors on OM-hydrolyzing activity in human plasma, liver, and intestinal cytosols. Each inhibitor concentration was as follows: BNPP, DFP, PCMB, eserine, and PMSF, 1000 μm; EDTA, 500 μm. Data represent the means of duplicate determinations. D, shown is OM-hydrolyzing activity in human liver and intestinal preparations. Data represent the means ± S.D. of triplicate determinations.
TABLE 1.
Kinetic parameters of hydrolysis of medoxomil prodrugs
To estimate the kinetic parameters, data were fitted to the Michaelis-Menten equation by nonlinear least squares regression. The estimated parameters are expressed as the means ± S.E. of triplicate determinations. “Recombinant CMBL” represents lysate supernatant of Freestyle 293-F cell-transfected human CMBL as an enzyme source.
| Substrate | Enzyme source | Km | Vmax | Vmax/Km |
|---|---|---|---|---|
| μm | nmol/min/mg protein | ml/min/mg protein | ||
| Olmesartan medoxomil | Recombinant CMBL | 170 ± 18 | 24.6 ± 0.9 | 0.145 |
| Liver cytosol | 160 ± 19 | 19.7 ± 0.8 | 0.123 | |
| Intestinal cytosol | 193 ± 24 | 45.1 ± 2.0 | 0.234 | |
| Liver microsomes | 481 ± 23 | 15.0 ± 0.3 | 0.0312 | |
| Intestinal microsomes | 236 ± 10 | 6.76 ± 0.11 | 0.0286 | |
| Faropenem medoxomil | Recombinant CMBL | 283 ± 32 | 16.4 ± 0.8 | 0.0580 |
| Lenampicillin | Recombinant CMBL | 63.4 ± 4.7 | 4.00 ± 0.15 | 0.0631 |
Purification and Identification of CMBL
The OM hydrolase was purified from human liver cytosol through successive three-step column chromatography. After the last purification step, although multiple bands with different molecular masses were observed on SDS-polyacrylamide gel, bands of 30 kDa correlated well with the OM-hydrolyzing activity as shown in Fig. 3. The protein of this 30-kDa band was identified as human carboxymethylenebutenolidase homolog (CMBL, GenBankTM accession number NP_620164.1) by mass spectrometry. The identified peptide sequences are shown in Fig. 4A. The predicted molecular mass of CMBL deduced from its amino acid sequence (245 amino acids) was 28 kDa. A BLAST search against GenBankTM databases revealed that human, mouse, and rat CMBL genes consist of five exons and are mapped onto 5p15.2, 15B2, and 2q22, respectively. Mouse and rat CMBL proteins share 82 and 84% homology with human protein, respectively (Fig. 4A). A Pfam search (24) revealed that the CMBL proteins have only one potential functional domain of the dienelactone hydrolase family (Pfam: PF01738), which is a member of the α/β hydrolase-fold family (25), with a high score (E value: 2.8 × 10−21, 3.8 × 10−26, and 4.9 × 10−26 for human, mouse, and rat CMBL, respectively), supporting that we had purified CMBL from human liver cytosol as the protein with the OM-hydrolyzing activity. Notably, CMBL was the only protein having the dienelactone hydrolase domain in the human genome found in a Pfam search. A BLAST search demonstrated no human proteins sharing more than 30% homology with full-length human CMBL or dienelactone hydrolase domain sequences (data not shown).
FIGURE 3.

Identification of bands corresponding to OM-hydrolyzing activity. Protein with OM-hydrolyzing activity was purified from human liver cytosol by successive column chromatography. A, fractions of the last purification step were subjected to SDS-PAGE, and the gel was stained with a fluorescent dye (SYPRO Ruby, Bio-Rad). Arrowheads indicate 30-kDa bands correlating with OM-hydrolyzing activity. B, the OM-hydrolyzing activity in the fractions determined by HPLC and the intensities of the bands shown as arrows in A are indicated by the solid line and bars, respectively.
FIGURE 4.

Amino acid sequences of CMBL and identification of the active residue. A, the human, rat, and mouse CMBL (GenBankTM accession number: NP_620164.1, NP_001008770.1, and NP_853619.1, respectively) are aligned. Asterisks and dots, respectively, indicate identical and homologous residues between the three sequences. Arrowheads and bold letters indicate the putative catalytic triad of human CMBL, Cys132, Asp179, and His212, predicted based on the crystal structure of dienelactone hydrolase from Pseudomonas sp. B13 (34) (PDB code 1DIN, gi: 1827808) as a template with the aid of ModBase (26). Underlined letters indicate the dienlactone hydrolase family domain (pfam01738). Shadowing indicates the identified sequences by LC-MS/MS analysis of the purified protein from human liver cytosol. B and C, shown is OM-hydrolyzing activity of recombinant CMBL proteins overexpressed in E. coli system, the wild-type (WT) and two mutant constructs with each of an amino acid substitution (C132A or C132S) on the putative active site, Cys132. B and C show the activities at 10 μm of the substrate concentration and the kinetic analysis, respectively. Data represent the means ± S.D. of triplicate determinations. The solid line in C is the best fit by nonlinear least squares regression to the Michaelis-Menten equation. The Km value in the wild-type construct was estimated to be 116 μm.
Mutant Generation and Identification of the Active Residue of CMBL
The wild-type construct of human CMBL with His6 tag expressed in E. coli system was used as the template for generating mutants. Two mutant proteins with each amino acid substitution (C132A or C132S) on the putative active site Cys132, as predicted with the aid of ModBase (26), were obtained (see Fig. 4A and “Discussion”). All the overexpressed proteins were purified using the affinity tag, and the tag was cleaved from the proteins by thrombin treatment. SDS-PAGE analysis indicated the high levels of purity and complete tag cleavage (supplemental Fig. S1).
In the results from the OM-hydrolyzing activity measurement at a low substrate concentration (10 μm), C132A was catalytically disabled and retained ∼3% of the wild-type activity, as shown in Fig. 4B. In contrast, C132S displayed a measurable but considerably low activity, ∼30% that of the wild type. Furthermore, as shown in Fig. 4C, the activity in both mutants was almost completely abolished at higher substrate concentrations (up to 1000 μm) but not in the wild-type construct (Km = 116 μm). These data indicate that the OM-hydrolyzing activity of CMBL exists in the Cys132 residue.
Expression of Human CMBL in Mammalian Cell Line and Enzyme Characterization
To compare the enzyme characteristics between a recombinant protein and the native protein in human tissue preparations, the expression vector for human CMBL was transfected into Freestyle 293-F cells, a mammalian cell line derived from human embryonic kidney cells (HEK-293 cells) that is supposed to express proteins with post-translational modifications more closely than bacterial expression systems, and the OM-hydrolyzing activity by the recombinant protein was measured. Significant OM-hydrolyzing activity was observed in the cell lysate of the human CMBL-transfectant compared with that of vector-transfectant (Fig. 5A). This result convinced us that our purified protein from human liver cytosol was CMBL, which could work as an OM-bioactivating enzyme.
FIGURE 5.

Enzyme characterization of recombinant CMBL overexpressed in mammalian cell line. A, OM-hydrolyzing activity of recombinant CMBL expressed in Freestyle 293-F cells is shown. The cell lysate supernatant was used as an enzyme source for the in vitro metabolic reactions. Data represent the means ± S.D. of triplicate determinations. B, kinetic analysis of OM hydrolysis by recombinant CMBL is shown. Data points represent the means ± S.D. of triplicate determinations. The solid line is the best fit by nonlinear least squares regression to the Michaelis-Menten equation. The respective Eadie-Hofstee plot is presented in the inset. C, the inhibitory effect of known esterase inhibitors on OM-hydrolyzing activity of recombinant CMBL is shown.
In the kinetic analysis of OM-hydrolyzing activity by the recombinant CMBL, OM was hydrolyzed and converted to the pharmacologically active olmesartan in a simple Michaelis-Menten kinetics manner (Fig. 5B). As summarized in Table 1, the Km value in the recombinant CMBL was 170 μm, and it was quite consistent with those in the liver and intestinal cytosols. The microsomal fractions, where typical drug-metabolizing enzymes such as cytochrome P450 members and carboxylesterases are expressed, showed metabolic intrinsic clearance (Vmax/Km) considerably lower than those of the recombinant human CMBL and the liver and intestinal cytosols. This result indicates less importance of the microsomal hydrolases than the cytosolic hydrolases in bioactivating OM.
We also investigated the effects of chemical inhibitors on the OM-hydrolyzing activity by the recombinant CMBL and compared them with those in human liver and intestinal cytosols. The human CMBL activity was almost completely inhibited by 1000 μm PCMB and was partially inhibited by 1000 μm BNPP by ∼50% (Fig. 5C). The complete inhibition by the free thiol modifier PCMB indicates that human CMBL would be a member of cysteine hydrolases. In contrast, no inhibition was observed by the addition of 1000 μm DFP, PMSF, eserine, and 5000 μm EDTA. This inhibition pattern in the recombinant CMBL is quite consistent with those in human liver and intestinal cytosols (Fig. 2C), where only PCMB and BNPP had a meaningful inhibitory effect on the OM-hydrolyzing activity.
Gene Transcript and Protein Expression of Human CMBL
The tissue distribution of mRNA encoding CMBL was examined with quantitative RT-PCR. Human CMBL mRNA was widely distributed in all the 20 human tissues tested, and the highest level of expression was observed in the liver, which is the most important site for xenobiotic metabolism, followed by the kidney, small intestine, and colon (Fig. 6).
FIGURE 6.

Expression of CMBL gene transcript in human tissues. The relative expression of mRNA from multiple human tissues was measured by quantitative RT-PCR using glyceraldehyde-3-phosphate dehydrogenase as a reference gene and normalized to the placenta expression.
Western blot analysis demonstrated that CMBL protein is definitely expressed in human liver and intestine (Fig. 7), as indicated by the expression of the transcript. The protein expression was localized in cytosolic fractions rather than in microsomal fractions in these two tissues, consistent with the higher subcellular distribution of the OM-hydrolyzing activity in the cytosolic fraction than in the microsomal fraction in both the liver and intestine (Fig. 2D). In contrast, CMBL was not stained in the human plasma, indicating that CMBL clearly differs from the plasma OM-bioactivating enzyme, PON1, which is mainly distributed to the plasma (9).
FIGURE 7.
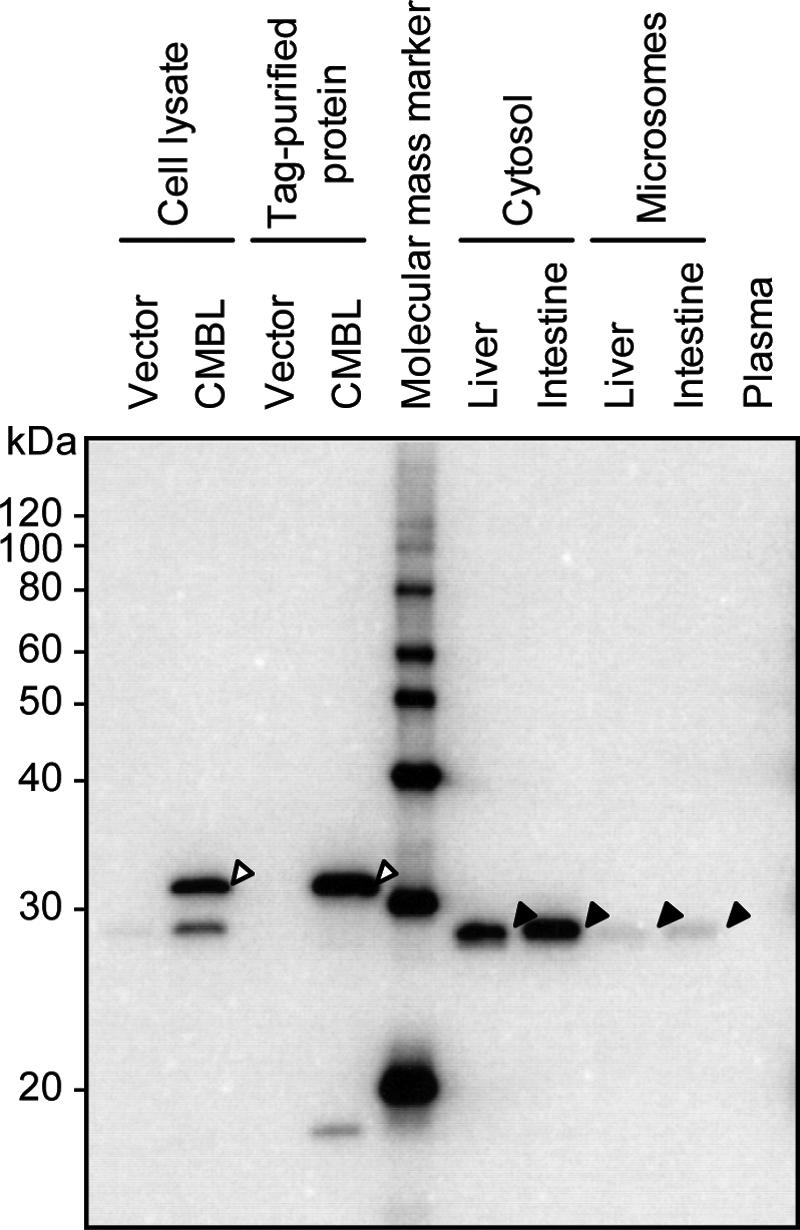
Protein expression of native CMBL proteins in human tissues. Protein expression of native CMBL in human liver, intestine, and plasma was analyzed by Western blot. The SDS-PAGE-separated proteins were transferred electrophoretically onto a polyvinylidene difluoride membrane and stained by polyclonal anti-CMBL antibody. Open and closed arrowheads indicate the recombinant CMBL-FLAG fusion protein- and the native CMBL bands, respectively.
Enzyme Kinetics of Prodrug Bioactivation by Recombinant Human CMBL
In addition to OM, recombinant human CMBL expressed in mammalian cells also catalyzed the hydrolytic activation of other known medoxomil-ester prodrugs: the β-lactam antibiotic faropenem medoxomil and lenampicillin. These prodrugs with a medoxomil ester pro-moiety were substantially hydrolyzed by the recombinant CMBL and exhibited simple Michaelis-Menten kinetics (Fig. 8). As shown in Table 1, the human CMBL exhibited higher affinity for lenampicillin than that for OM (Km = 63.4 versus 170 μm, respectively), whereas it had lower affinity for faropenem medoxomil (Km = 283 μm). The intrinsic metabolic clearance calculated as the Vmax/Km for the hydrolysis of the two hydrophilic β-lactam antibiotics were relatively low (0.0580 and 0.0631 ml/min/mg of protein, respectively) compared with that of OM (0.145 ml/min/mg of protein) with moderate lipophilicity. Interestingly, prulifloxacin (Fig. 8C), in which the medoxomil moiety is linked directly to the nitrogen atom in the piperazinyl group of its active metabolite ulifloxacin, was not enzymatically hydrolyzed by the recombinant human CMBL (supplemental Table S1).
FIGURE 8.

Kinetic analysis of medoxomil-prodrug hydrolysis by recombinant CMBL overexpressed in mammalian cell line. Enzyme kinetics of the reactions with chemical structures of faropenem medoxomil and lenampicillin are shown in A and B, respectively. Data points represent the means ± S.D. of triplicate determinations. The solid line is the best fit by nonlinear least-squares regression to the Michaelis-Menten equation. The respective Eadie-Hofstee plot is presented in the inset. C, chemical structure of prulifloxacin.
DISCUSSION
In this work we have identified human CMBL as a bioactivating hydrolase of the prodrug-type ARB, OM. Furthermore, we demonstrated that CMBL catalyzed the hydrolytic activation of the other two medoxomil-type prodrugs, faropenem medoxomil (27) and lenampicillin (28), whose bioactivating enzyme has not been revealed so far. To the best of our knowledge this is the first paper that reported the biological function of this unfamiliar protein. Successive column purification, protein separation by SDS-PAGE, and protein identification by mass spectrometry allowed us to successfully identify CMBL from human liver cytosol. Surprisingly, CMBL is a protein whose biological and enzymatic functions have remained unrevealed not only in humans but also in other higher eukaryotes. Only one report regarding protein-protein interactions including human CMBL by large-scale mapping has been previously published (29), although no further information on human CMBL function has been reported. Notably, human CMBL exhibited a unique sensitivity to esterase inhibitors and ubiquitous tissue expression, which are distinguishable from other known prodrug activating esterases such as carboxylesterases, cholinesterases, and paraoxonases. A protein data base search showed that various eukaryotic homologs of CMBL have been found. Highly conserved amino acid sequences with more than 80% homologies among several mammals including mice and rats were observed, reflecting the functional importance of these proteins. Furthermore, these animal models could provide further opportunities to define the physiological role of this uncharacterized protein in the future. Significant sequence homologies were also found in hypothetical protein LOC100126103 from Danio rerio (GenBankTM accession number NP_001103302.1, 64% homology). Moderate homology was detected with a dienelactone hydrolase family protein AT1G35420 from Arabidopsis thaliana (GenBankTM accession number NP_564458.1, 32% homology) and a hypothetical protein Os05g0429500 from Oryza sativa (GenBankTM accession number NP_001055619.1, 29% homology). So far the homology search suggests that CMBL is a relatively unique enzyme in eukaryotes with unknown endogenous functions that may be evolutionarily conserved among various eukaryotes.
CMBL was first identified in Pseudomonas sp.B13 (30), with its gene was encoded on a plasmid. This bacterial enzyme, also called dienelactone hydrolase, has been well investigated as the third enzyme of the halocatechol degradation pathway in a complex set of catabolic reactions used by bacteria for the utilization of aromatic compounds and catalyzes the hydrolysis of both (E) and (Z) dienelactone (4-carboxymethylene-but-2-ene-4-olide) to maleyl acetate (2-oxo-but-1,3-diene-1,4-dicarboxylate) (31). This bacterial CMBL is a 25.5-kDa monomer that belongs to the α/β hydrolase fold family (32) that is shared by several enzymes which apparently diverged from a common ancestor with various different catalytic functions. It was reported that the bacterial CMBL has a catalytic triad consisting of a nucleophilic cysteine (Cys123), a histidine (His202), and an aspartic acid (Asp171) (33–36), like all the α/β hydrolase fold family enzymes, which contain a conserved nucleophile (serine/cysteine)-histidine-acid catalytic triad. Thus, we chose the characterized crystal structure of dienelactone hydrolase from Pseudomonas sp. B13 (34) (PDB 1DIN, gi:1827808) as a template to predict a catalytic triad in human CMBL with the aid of ModBase (26). As a result of this prediction, a putative catalytic triad of human CMBL composed of Cys132-Asp179-His212 (Fig. 4A) was indicated. Interestingly, this catalytic triad was completely conserved in a comparison of 10 eukaryotic CMBL homologs, even in plants, by a multiple sequence alignment. Furthermore, the OM-hydrolyzing activity was completely abolished by the free thiol modifier PCMB as was the dienelactone hydrolase activity in the bacterial CMBL (30), suggesting that human CMBL is a member of cysteine hydrolases with an active cysteine residue, most probably Cys132, in the active center. In fact, site-directed mutants expressed in the E. coli system with an amino acid substitution on the putative active residue Cys132 into alanine or serine catalyzed the OM hydrolysis with drastically reduced rates (Fig. 4, B and C) compared with the wild type. These results confirmed that the Cys132 residue is catalytically quite important in the human CMBL, as similarly demonstrated for the active residue Cys123 in the bacterial CMBL (37).
The Km value for OM in the recombinant CMBL produced in mammalian cells agreed well with those in human liver and intestinal cytosols (Table 1), strongly suggesting a predominant contribution of this enzyme to the OM hydrolysis in these tissues. Importantly, Western blot analysis (Fig. 7) clearly showed that CMBL protein certainly exists in human liver and intestine. Moreover, as a result of a gel filtration assay of the human liver cytosol (supplemental Fig. S2), the OM-hydrolyzing activity, eluted in a single peak, correlated well with CMBL protein distribution in the corresponding fractions, suggesting a predominant contribution of this enzyme to the reaction. However, further experiments by a specific chemical inhibitor, a neutralizing antibody, or immunodepletion are necessary to conclude that the CMBL is the responsible enzyme for the OM bioactivation in the liver and intestine. For the orally administered prodrug, the first pass bioactivation may occur in the intestine or liver before it reaches the systemic circulation. In the case of OM, rapid hydrolytic bioactivation catalyzed by intestinal and liver CMBL is thought to predominantly contribute to the quick onset of drug action. In addition, as previously reported, plasma esterases PON1 (20) and albumin (21) play a significant role as OM bioactivating enzymes in portal blood in the first pass metabolism as well as in circulating blood. In fact, no components other than olmesartan were detected in plasma after oral administration of radiolabeled OM in healthy volunteers (20). Also, in rats, no prodrug was detected in either the portal or circulating blood (data not shown). Taken together, after oral administration of OM, the multiple bioactivating enzymes in multiple sites in humans in vivo are considered to cause the rapid and complete drug action of this prodrug, allowing us to disregard the genetic polymorphism in the bioactivating enzymes, which may cause an extremely varied production of the pharmacologically active metabolite.
Using the recombinant human CMBL preparations, we examined the hydrolysis of the other medoxomil prodrugs, faropenem medoxomil, lenampicillin, and prulifloxacin, into their respective active metabolites and found that the prodrugs with medoxomil moiety being linked to the oxygen atom (faropenem medoxomil and lenampicillin) are readily hydrolyzed by CMBL (Fig. 8), whereas prulifloxacin, where the medoxomil moiety being linked to the nitrogen atom, was not hydrolyzed at all (supplemental Table S1). PON1 has been shown to hydrolyze prulifloxacin and OM (13, 20), indicating that the substrate recognition between CMBL and PON1 partially overlap. In additional experiments we confirmed that the recombinant CMBL does not hydrolyze typical esterase substrates, namely p-nitrophenyl acetate, phenyl acetate, and acetylthiocholine iodide as substrates of carboxylesterases, PON1, and cholinesterases, respectively (supplemental Fig. S3), nor candesartan cilexetil, another prodrug type ARB that contains an extended double ester chain and is bioactivated by carboxylesterases (supplemental Fig. S4) (38). Although only limited information on the substrate specificity of CMBL is available at present, CMBL quite likely prefers cyclic esters as a substrate over non-cyclic esters. In fact, bacterial CMBL has been reported to hydrolyze simple esters and amides such as p-nitrophenyl acetate and trans-cinnamoyl imidazole with a low catalytic activity (37), whereas it hydrolyzed cyclic esters with quite high activity. Like other historically investigated esterases whose true physiological substrates remain unknown, much more research is needed to further define the endogenous substrates and to elucidate the physiological functions of CMBL.
In conclusion, we have purified the OM-bioactivating enzyme from human liver and identified it as CMBL. This is the first report that CMBL is involved in the metabolism of xenobiotics in humans, whereas further research is needed to determine the physiological functions of CMBL proteins, which are evolutionarily conserved from bacteria to mammals, including humans.
Supplementary Material
Acknowledgments
We gratefully acknowledge Tatsuya Inoue, Kenji Nakamaru, Shinya Tokuhiro, and Takako Kimura for expert help with informatics. We also express our appreciation to Osamu Ubukata and Kaori Yoshida for experimental suggestions and contributions to this work as well as Masakatsu Kotsuma for many helpful discussions.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Method 1, Table S1, and Figs. S1–S4.
- PON1
- paraoxonase1/arylesterase
- OM
- olmesartan medoxomil
- CMBL
- carboxymethylenebutenolidase homolog
- ARB
- angiotensin receptor blocker
- BNPP
- Bis-p-nitrophenylphosphate
- PCMB
- p-chloromercuribenzoate
- PMSF
- phenylmethylsulfonyl fluoride
- DFP
- diisopropylfluoro-phosphate
- LC-MS/MS
- reverse-phase liquid chromatography with tandem mass spectrometry
- RT
- reverse transcriptase
- HPLC
- high performance liquid chromatography
- ID
- inner diameter.
REFERENCES
- 1.Testa B., Mayer J. M. (2003) Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry, and Enzymology, 1st Ed., pp. 1–3, Wiley-VCH, Zürich, Switzerland [Google Scholar]
- 2.Ettmayer P., Amidon G. L., Clement B., Testa B. (2004) J. Med. Chem. 47, 2393–2404 [DOI] [PubMed] [Google Scholar]
- 3.Beaumont K., Webster R., Gardner I., Dack K. (2003) Curr. Drug Metab. 4, 461–485 [DOI] [PubMed] [Google Scholar]
- 4.Testa B. (2004) Biochem. Pharmacol. 68, 2097–2106 [DOI] [PubMed] [Google Scholar]
- 5.Satoh T., Hosokawa M. (2006) Chem. Biol. Interact. 162, 195–211 [DOI] [PubMed] [Google Scholar]
- 6.Hosokawa M. (2008) Molecules 13, 412–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor P. (1991) J. Biol. Chem. 266, 4025–4028 [PubMed] [Google Scholar]
- 8.Taylor P., Radić Z. (1994) Annu. Rev. Pharmacol. Toxicol. 34, 281–320 [DOI] [PubMed] [Google Scholar]
- 9.Draganov D. I., La Du B. N. (2004) Naunyn-Schmiedebergs Arch. Pharmacol. 369, 78–88 [DOI] [PubMed] [Google Scholar]
- 10.Liederer B. M., Borchardt R. T. (2006) J. Pharm. Sci. 95, 1177–1195 [DOI] [PubMed] [Google Scholar]
- 11.Satoh T. (2005) Toxicol. Appl. Pharmacol. 207, 11–18 [DOI] [PubMed] [Google Scholar]
- 12.Imai T. (2006) Drug Metab. Pharmacokinet. 21, 173–185 [DOI] [PubMed] [Google Scholar]
- 13.Tougou K., Nakamura A., Watanabe S., Okuyama Y., Morino A. (1998) Drug Metab. Dispos. 26, 355–359 [PubMed] [Google Scholar]
- 14.James R. W., Deakin S. P. (2004) Free Radic. Biol. Med. 37, 1986–1994 [DOI] [PubMed] [Google Scholar]
- 15.Kim I., Chu X. Y., Kim S., Provoda C. J., Lee K. D., Amidon G. L. (2003) J. Biol. Chem. 278, 25348–25356 [DOI] [PubMed] [Google Scholar]
- 16.Lai L., Xu Z., Zhou J., Lee K. D., Amidon G. L. (2008) J. Biol. Chem. 283, 9318–9327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott L. J., McCormack P. L. (2008) Drugs 68, 1239–1272 [DOI] [PubMed] [Google Scholar]
- 18.Chrysant S. G. (2008) Drugs Today 44, 443–453 [DOI] [PubMed] [Google Scholar]
- 19.Redon J., Fabia M. J. (2009) J. Renin Angiotensin Aldosterone Syst. 10, 147–156 [DOI] [PubMed] [Google Scholar]
- 20.Laeis P., Puchler K., Kirch W. (2001) J. Hypertens. Suppl. 19, S21–S32 [DOI] [PubMed] [Google Scholar]
- 21.Ma S. F., Anraku M., Iwao Y., Yamasaki K., Kragh-Hansen U., Yamaotsu N., Hirono S., Ikeda T., Otagiri M. (2005) Drug Metab. Dispos. 33, 1911–1919 [DOI] [PubMed] [Google Scholar]
- 22.Kubota K., Wakabayashi K., Matsuoka T. (2003) Proteomics 3, 616–626 [DOI] [PubMed] [Google Scholar]
- 23.Reyns T., Cherlet M., De Baere S., De Backer P., Croubels S. (2008) J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 861, 108–116 [DOI] [PubMed] [Google Scholar]
- 24.Finn R. D., Tate J., Mistry J., Coggill P. C., Sammut S. J., Hotz H. R., Ceric G., Forslund K., Eddy S. R., Sonnhammer E. L., Bateman A. (2008) Nucleic Acids Res. 36, D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carr P. D., Ollis D. L. (2009) Protein Pept. Lett. 16, 1137–1148 [DOI] [PubMed] [Google Scholar]
- 26.Pieper U., Eswar N., Davis F. P., Braberg H., Madhusudhan M. S., Rossi A., Marti-Renom M., Karchin R., Webb B. M., Eramian D., Shen M. Y., Kelly L., Melo F., Sali A. (2006) Nucleic Acids Res. 34, D291–D295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schurek K. N., Wiebe R., Karlowsky J. A., Rubinstein E., Hoban D. J., Zhanel G. G. (2007) Expert Rev. Anti Infect. Ther. 5, 185–198 [DOI] [PubMed] [Google Scholar]
- 28.Saito A., Nakashima M. (1986) Antimicrob. Agents Chemother. 29, 948–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ewing R. M., Chu P., Elisma F., Li H., Taylor P., Climie S., McBroom-Cerajewski L., Robinson M. D., O'Connor L., Li M., Taylor R., Dharsee M., Ho Y., Heilbut A., Moore L., Zhang S., Ornatsky O., Bukhman Y. V., Ethier M., Sheng Y., Vasilescu J., Abu-Farha M., Lambert J. P., Duewel H. S., Stewart, Kuehl B., Hogue K., Colwill K., Gladwish K., Muskat B., Kinach R., Adams S. L., Moran M. F., Morin G. B., Topaloglou T., Figeys D. (2007) Mol. Syst. Biol. 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ngai K. L., Schlömann M., Knackmuss H. J., Ornston L. N. (1987) J. Bacteriol. 169, 699–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt E., Knackmuss H. J. (1980) Biochem. J. 192, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ollis D. L., Cheah E., Cygler M., Dijkstra B., Frolow F., Franken S. M., Harel M., Remington S. J., Silman I., Schrag J. (1992) Protein Eng. 5, 197–211 [DOI] [PubMed] [Google Scholar]
- 33.Pathak D., Ngai K. L., Ollis D. (1988) J. Mol. Biol. 204, 435–445 [DOI] [PubMed] [Google Scholar]
- 34.Pathak D., Ollis D. (1990) J. Mol. Biol. 214, 497–525 [DOI] [PubMed] [Google Scholar]
- 35.Cheah E., Ashley G. W., Gary J., Ollis D. (1993) Proteins 16, 64–78 [DOI] [PubMed] [Google Scholar]
- 36.Beveridge A. J., Ollis D. L. (1995) Protein Eng. 8, 135–142 [DOI] [PubMed] [Google Scholar]
- 37.Pathak D., Ashley G., Ollis D. (1991) Proteins 9, 267–279 [DOI] [PubMed] [Google Scholar]
- 38.Easthope S. E., Jarvis B. (2002) Drugs 62, 1253–1287 [DOI] [PubMed] [Google Scholar]
- 39.Saari W. S., Halczenko W., Cochran D. W., Dobrinska M. R., Vincek W. C., Titus D. C., Gaul S. L., Sweet C. S. (1984) J. Med. Chem. 27, 713–717 [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto F., Ikeda S., Tsukamoto G. (1984) Chem. Pharm. Bull. 32, 2241–2248 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.