

Abstract
Hepatic nuclear factor 1α (HNF1α) is a key regulator of development and function in pancreatic beta cells and is specifically involved in regulation of glycolysis and glucose-stimulated insulin secretion. Abnormal expression of HNF1α leads to development of MODY3 (maturity-onset diabetes of the young 3). We report that NK6 homeodomain 1 (NKX6.1) binds to a cis-regulatory element in the HNF1α promoter and is a major regulator of this gene in beta cells. We identified an NKX6.1 recognition sequence in the distal region of the HNF1α promoter and demonstrated specific binding of NKX6.1 in beta cells by electrophoretic mobility shift and chromatin immunoprecipitation assays. Site-directed mutagenesis of the NKX6.1 core-binding sequence eliminated NKX6.1-mediated activation and substantially decreased activity of the HNF1α promoter in beta cells. Overexpression or small interfering RNA-mediated knockdown of the Nkx6.1 gene resulted in increased or diminished HNF1α gene expression, respectively, in beta cells. We conclude that NKX6.1 is a novel regulator of HNF1α in pancreatic beta cells. This novel regulatory mechanism for HNF1α in beta cells may provide new molecular targets for the diagnosis of MODY3.
Keywords: Diseases/Diabetes, Gene/Promoters, Gene/Regulation, Gene/Transcription, Transcription/Regulation, Transcription/Tissue-specific Factors, HNF1α, MODY3, NKX6.1, Beta Cell Lines
Introduction
Hepatic nuclear factor 1α (HNF1α)2 is a key transcription factor involved in glucose-stimulated insulin secretion (GSIS) and is the major factor involved in most cases of maturity-onset diabetes of the young (MODY), which accounts for ∼1% of worldwide diabetes cases (1, 2). Precise cellular concentrations of HNF1α are required to maintain normal beta cell function, and both underexpression and overexpression have been shown to lead to diabetes (3–9).
HNF1α is an important gene involved in the developmental regulation of the liver, pancreas, kidneys, stomach, and intestines (10–14). This transcription factor is essential for control of mature cellular phenotype in these tissues. In pancreatic beta cells, HNF1α is involved in regulating the transcription of several genes that are involved in glycolysis and GSIS such as insulin (15), the Glut2 glucose transporter (16), pyruvate kinase (17), aldolase B (18), HNF3γ (19), and HNF4γ (19). Although the regulation of HNF1α has been well characterized in hepatocytes, its regulation in beta cells has not been studied in detail, and this gene is assumed to be regulated based on mechanistic studies from hepatocytes (20).
HNF1α has an HNF4α-binding site in its proximal promoter that has been shown to be sufficient for its activation in hepatocytes (21, 22), but evidence exists to suggest that the mechanism for regulation of HNF1α may different in beta cells. First, HNF1α is expressed as three isoforms (A, B, and C) that have tissue-specific distribution ratios (14). HNF1αA is the predominant form in hepatocytes, whereas HNF1αB is predominant in pancreatic islets. It has been shown that HNF1αB and HNF1αC have greater transactivation potential than HNF1αA (23). Second, the major regulator of this gene, HNF4α, is predominantly expressed from the P1 promoter in hepatocytes, whereas in the pancreas, only transcripts from the P2 promoter can be detected according to most reports (24–26). HNF4α transcripts expressed in the pancreas from the P2 promoter have a truncated N-terminal region containing the transactivation domain, and these isoforms have been shown to have lower transactivation potential compared with isoforms expressed from the P1 promoter (27–29). This suggests that these HNF4α isoforms expressed from the P2 promoter may be insufficient to fully activate HNF1α in the pancreas. Third, Huang et al. (30) have show that, although a −497-bp proximal HNF1α-luciferase reporter was activated in hepatocytes, it failed to be activated in rat insulinoma (INS1) beta cells, and this promoter contains the proximal HNF4α-binding site used for activation in hepatocytes. The aforementioned data suggest that HNF1α may utilize a different mechanism for gene transcription in beta cells than previously identified for hepatocytes.
NK6 homeodomain 1 (NKX6.1) is a homeodomain transcription factor involved in pancreatic differentiation and beta cell homeostasis (31). In mature human islets, it is exclusively expressed in beta cells (32) and is required for normal GSIS (33). Overexpression of NKX6.1 has been shown to increase GSIS in rat islets (34). It is also of interest that islets isolated from type 2 diabetic patients have altered NKX6.1 expression (35). However, the specific function of NKX6.1 in GSIS of mature beta cells remains elusive.
In this work, we hypothesized that a distinct regulatory mechanism for HNF1α gene expression exists in pancreatic beta cells. Here, we report a novel finding that NKX6.1 is a key regulator of HNF1α expression in beta cells, which may provide insight into understanding the regulation of GSIS in beta cells.
EXPERIMENTAL PROCEDURES
Cell Culture
NIH 3T3 mouse fibroblast cells, rodent insulinoma cell lines (βTC3, NIT1, and INS1), and Huh7 human hepatocarcinoma cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 0.1% kanamycin in a 37 °C incubator with 100% humidity and 5% CO2.
Plasmid Construction
For HNF1α-luciferase, 2772 bp of the mouse HNF1α proximal promoter was cloned into a pGL3 vector (Promega) using the EcoRI and BglII restriction sites. The pGL-TK expression vector was purchased from Promega. The pcDNA3 vector was purchased from Invitrogen. For pCMV-PDX1, the human PDX1 expression plasmid (pCMV6-XL5) was purchased from OriGene. The human NKX6.1 expression plasmid (pBAT12) was a generous gift from Dr. Michael German (University of California, San Francisco, CA). The mouse Ngn3 expression plasmid was a generous gift from Dr. Marko Horb (Institute de Recherches Cliniques de Montréal, Montréal, Quebec, Canada). Mouse Ngn3 cDNA was cloned into the pcDNA3 vector using the BamHI restriction site. Human MafA cDNA was cloned into the pTYF lentiviral vector cassette under the control of the elongation factor 1α promoter using the BamHI and SpeI restriction sites. Mouse NeuroD1 cDNA was cloned into the pcDNA3.1CT-GFP-TOPO vector. The Xenopus Pax6 expression plasmid (a generous gift from Dr. Marko Horb) was derived by inserting Pax6 cDNA into the pcDNA3 vector using the HindIII and XhoI restriction sites. The human HNF1α expression plasmid was a generous gift from Dr. Michael German. Human HNF1α cDNA was cloned into the pCMV-Sport6 vector. The human HNF1β and mouse HNF4α expression plasmids (pCMV-Sport6) were purchased from Open Biosystems. Mouse Pbx1 was cloned into the pcDNA3 vector using the BamHI and XbaI restriction sites.
Transient Transfection and Luciferase Assays
Cells were cultured as indicated above and transfected with 0.1–1.0 μg of DNA (as indicated) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. 0.02 μg of pTK-Luc plasmid was used as a transfection control in all experiments. Cell lysates were harvested and measured 24 h post-transfection using the Dual Luciferase reporter kit (Promega) according to the manufacturer's protocol except that only 50 μl of each substrate reagent was used (as optimized by our laboratory) to read samples using a Lumat LB 9507 luminometer (Berthold Technologies). All luciferase assays were done in triplicate.
Site-directed Mutagenesis
Primers were designed (IDT Technologies) to induce a block mutation of 4–6 bp as indicated at the core sequence, inducing a restriction enzyme site for easy confirmation of mutation. Primer sequences were as follows: Nkx6.1, 5′-GGACCTGTTCCTCGAGGAAATGTGACACTTTAC-3′ (forward) and 5′-GTAAAGTGTCACATTTCCTCGAGGAACAGGTCC-3′ (reverse); and HNF4α, 5′-CTTGCAAGGCTGAAGTCCGGCCGTCAGTCCCTTCCTAAGCGCAC-3′ (forward) and 5′-GCTTAGGAAGGGACTGACGGCCGGACTTCAGCCTTGCAAGTGCAG-3′ (reverse).
Mutations were induced using PCR on the relevant plasmid with Pfu polymerase. PCR products were incubated for 1 h with the DpnI (New England Biolabs) methylation-sensitive restriction enzyme to remove template plasmid and transformed into competent Escherichia coli. Positive colonies were inoculated in LB medium (MP Biomedicals), and plasmid was purified using a Qiagen plasmid purification maxikit.
Electrophoretic Mobility Shift Assay (EMSA)
Biotin-labeled probes were designed (IDT Technologies) spanning the cis-regulatory element identified. Sequences were as follows: 5′-GAAGGATGGACCTGTTCCTAATGGAAATGTGACACTTTA-3′ (forward) and 5′-TAAAGTGTCACATTTCCATTAGGAACAGGTCCATCCTTC-3′ (reverse). Nuclear lysate from beta cell lines (INS1, βTC3, and NIT1) was prepared using the NE-PER nuclear extraction reagent (Pierce) according to the manufacturer's protocol. INS1 cells were a generous gift from Dr. Christopher Newgard (Duke University Medical Center, Durham, NC). Anti-NKX6.1 polyclonal antibodies (Santa Cruz Biotechnology) were used. Binding reactions were performed using the LightShift chemiluminescent EMSA kit (Promega) according to the manufacturer's protocol. Binding reactions were resolved by PAGE using a 7.5% Tris-HCl-polyacrylamide gel (Bio-Rad). Complexes were detected with the chemiluminescent nucleic acid detection module (Pierce) according to the manufacturer's protocol.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed using the ChIP assay kit (Millipore) according to the manufacturer's protocol. Beta cells cultured in 10-cm culture dishes were used both with and without transfection as indicated. Specific polyclonal antibodies to NKX6.1 or HNF4α (Santa Cruz Biotechnology) were used for immunoprecipitation. Following DNA isolation, sequences were evaluated by PCR using primers (IDT Technologies) flanking the respective cis-regulatory elements as indicated. Amplification primer sequences were as follows: Nkx6.1, 5′-CCCATCCAGGATGAAGTGAG-3′ (forward) and 5′-GACAAGGAGTTCTGGGCTAG-3′ (reverse); and HNF4α, 5′-TCACTCCCAATTGCAAGCCATG-3′ (forward) and 5′-TGCTGCTCTGTTTACATTGG-3′ (reverse).
Gene Expression and Quantitative Reverse Transcription (RT)-PCR
Cells were cultured as described above and transfected with DNA or small interfering RNA (siRNA) as indicated using Lipofectamine 2000 reagent according to the manufacturer's protocol. Nkx6.1-specific siRNA and control siRNA were purchased from Santa Cruz Biotechnology. Total RNA was extracted from cells using TRIzol reagent (Invitrogen), and RT-PCR and real-time RT-PCR were performed as described previously (36).3
Statistical Analysis
Statistical analysis was carried out using the two-sample Student's t test. A p value <0.01 was considered significant. All indicated significant values are in comparison with controls.
RESULTS
HNF1α Employs a Distinct Regulatory Element in Beta Cells
Regulation of the HNF1α promoter has been extensively studied in hepatocytes. Transcription is regulated through a TATA-like box (−21 to −15) and proximal HNF4α-binding site (−47) (21, 22, 37). The HNF1α −497-bp promoter (containing the HNF4α-binding site) is fully active in hepatocytes but not in INS1 rat insulinoma cells (30), providing evidence that its regulation may be controlled by alternative elements in other tissue types. To test this hypothesis, we cloned a 2772-bp region of the mouse HNF1α proximal promoter upstream of the firefly luciferase gene in a pGL3 vector to study its regulation in pancreatic beta cells. This 2772-bp HNF1α promoter has full activity in both beta cells and hepatic cells (Fig. 1, A and B). We created several truncated HNF1α promoter constructs by 5′-deletion analysis (Fig. 1A) using restriction enzymes and determined that the major regulation of HNF1α in beta cells is conferred through a more distal regulatory element (−2772 to −1820). Loss of this regulatory region significantly diminished transactivation of the HNF1α reporter in beta cells (Fig. 1A) but not in hepatic cells (Fig. 1B). NIH 3T3 cells, in which the HNF1α promoter is inactive, served as a control. This result shows that there is indeed a distinct regulatory region employed by beta cells for the regulation of HNF1α.
FIGURE 1.

Tissue-specific regulation of HNF1α. A and B, deletion analysis of the HNF1α promoter. Cells (3T3, βTC3, and Huh7) were transfected with various lengths of HNF1α promoter-luciferase (Luc) plasmids (1 μg) as indicated. Luciferase values are relative to the full-length constructs, which were arbitrarily set to 1.0. For the transactivation control, the shortest construct value for 3T3 and βTC3 cells was set as equal. Braces indicates the beta cell-specific cis-response element. C, luciferase activity of the mutant HNF4α-binding site HNF1α promoter. βTC3 cells were transfected with 1 μg of the indicated normal (open box) and mutant HNF4α-binding site (closed box) HNF1α promoter constructs. The luciferase value of the normal HNF1α promoter construct was arbitrarily set to 1.0. All samples were measured 24 h following transfection, and all experiments were repeated independently at least three times. ***, p < 0.001.
HNF4α-binding Site Is Only Partially Responsible for HNF1α Activation in Beta Cells
Next, we evaluated the role of HNF4α regulation of HNF1α in beta cells. Using site-directed mutagenesis, we constructed an HNF1α reporter with a mutant HNF4α-binding site (see Fig. 3A). As reported previously (21, 22, 37), HNF4α was required for transactivation in hepatic cells (Fig. 1C); however, mutation of the HNF4α-binding site only partially diminished transactivation in βTC3 cells (Fig. 1C). This demonstrates that beta cells are regulated by a separate response element that is different from hepatic cells.
FIGURE 3.

HNF1α promoter analysis. A, HNF1α promoter mutations. The locations of the NKX6.1- and HNF4α-binding sites are indicated in boxes on the promoter. Sequences below the shaded boxes show the induced mutations. B, HNF1α promoter species alignment. Mouse and rat (GenBankTM accession number X67649) HNF1α promoter sequences surrounding the NKX6.1-binding (upper panel) and HNF4α-binding (lower panel) sites are shown. Vertical lines indicate the close homology between species. The shaded boxes indicate core-binding sequences.
HNF1α Can Be Activated by NKX6.1
After finding an HNF1α regulatory response element (−2772 to −1820) that is beta cell-specific, we were interested in discovering which factors regulate transcription at this site. Several beta cell-specific transcription factors were screened (PDX1, NGN3, MafA, NeuroD1, PAX6, NKX6.1, HNF1α, HNF1β, HNF4α, and PBX1) using transient transfection assays in 3T3 cells with cotransfection of the HNF1α-luciferase reporter (Fig. 2A). Our results confirmed previous reports that HNF1α can be activated by HNF4α (21, 22, 37), and we found that NKX6.1 is also a strong activator. NKX6.1 activated HNF1α in a concentration-dependent manner (Fig. 2B) and is likely to be the regulator of the cis-regulatory element that is unique to beta cells.
FIGURE 2.
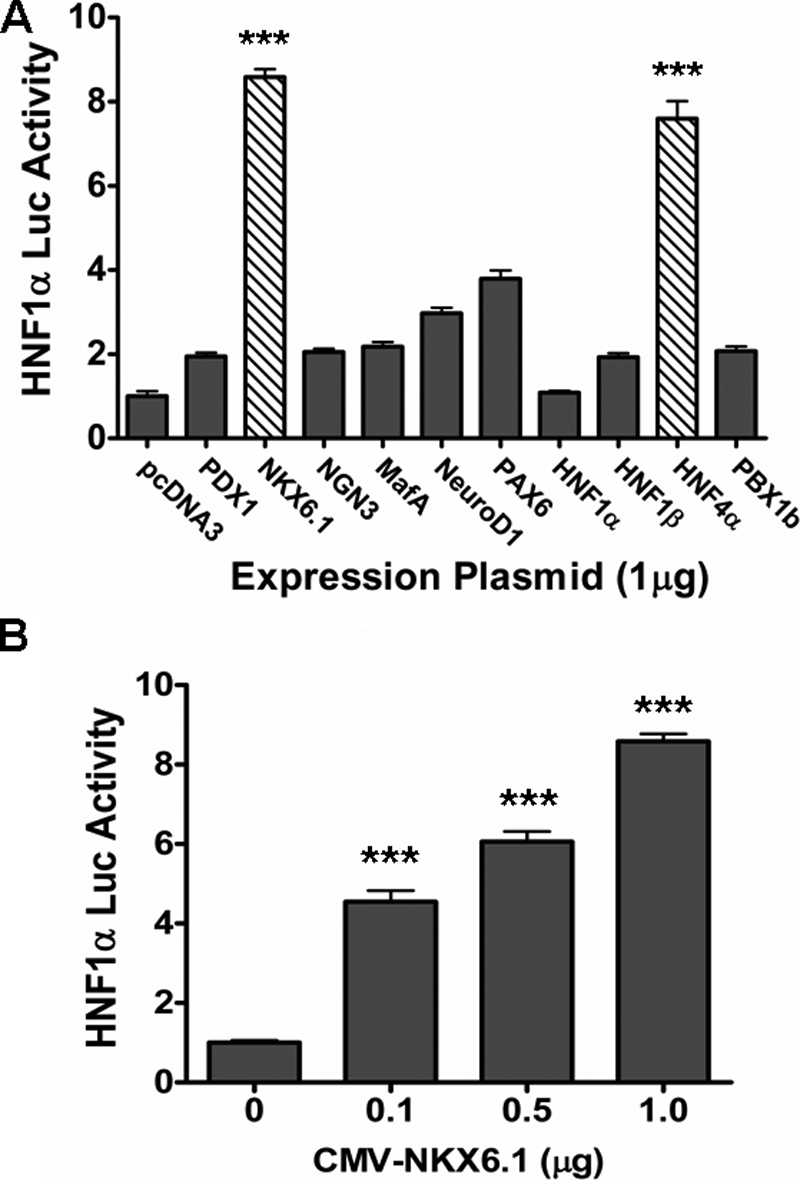
HNF1α regulation by NKX6.1. A, HNF1α promoter screening. The full-length (−2772) HNF1α promoter (1 μg) was cotransfected with various beta cell-specific transcription factor expression plasmids (1 μg) in 3T3 cells. B, NKX6.1 activation of the HNF1α promoter in a concentration-dependent manner. The full-length (−2772) HNF1α promoter (1 μg) was cotransfected in 3T3 cells with increasing amounts of NKX6.1 expression plasmid as indicated. pcDNA3 was used as a control to equalize DNA quantity used for each transfection. All samples were measured 24 h following transfection, and all experiments were repeated independently at least three times. Luc, luciferase; CMV, cytomegalovirus. ***, p < 0.001.
Mutational Analysis of the HNF1α Promoter
The literature pertaining to NKX6.1 binding suggests that the most common core DNA sequence utilized by NKX6.1 for binding is 5′-TAAT-3′ (33, 38–42) or its complement (5′-ATTA-3′), and it has also been shown to bind a similar sequence (5′-ATTT-3′) (42). We visually examined the HNF1α promoter sequence conferring strong transactivation in beta cells (−2772 to −1820) and found that there is a single 5′-TAAT-3′ DNA sequence in this entire region (Fig. 3A). An alignment of this promoter region between mouse and rat shows a very high level of homology similar to alignment of the promoter region near the known HNF4α-binding site (Fig. 3B). This prompted us to use site-directed mutagenesis to induce a mutation (Fig. 3A) at this potential NKX6.1-binding site to examine the effect on transactivation by NKX6.1. We also generated a double mutant HNF1α-luciferase reporter with both the HNF4α-binding site mutation and the potential NKX6.1-binding site mutation.
Using our mutant plasmids, we examined the ability of HNF4α or NKX6.1 to activate the HNF1α-luciferase reporter in 3T3 cells (Fig. 4A). Results were as expected and demonstrated that the normal HNF1α promoter can be activated by either HNF4α or NKX6.1. The mutant HNF4α-binding site failed to respond to HNF4α but could still be activated by NKX6.1. Similarly, the mutant NKX6.1-binding site failed to respond to NKX6.1 but could still be activated by HNF4α. The double mutant failed to respond to either HNF4α or NKX6.1. This result suggests that our mutant site (5′-TAAT-3′) is a real NKX6.1-binding site that is used for initiating HNF1α transcription.
FIGURE 4.

Analysis of HNF1α promoter mutations. A, activation of mutant HNF1α promoter constructs. Normal (upper left panel), mutant HNF4α-binding site (upper right panel), mutant NKX6.1-binding site (lower left panel), and double mutant (lower right panel) HNF1α promoter constructs (1 μg) were cotransfected into 3T3 cells with NKX6.1 (1 μg) or HNF4α (1 μg) expression plasmid as indicated. Closed boxes indicate mutation, and open boxes indicate normal sequence. pcDNA3 was used as a control, and the relative value obtained was arbitrarily set to 1.0. B, luciferase (Luc) activity of the mutant NKX6.1-binding site HNF1α promoter in beta cells. βTC3 cells were transfected with 1 μg of the indicated normal (open box) and mutant NKX6.1-binding site (closed box) HNF1α promoter constructs. The luciferase value of the normal HNF1α promoter construct was arbitrarily set to 1.0. All samples were measured 24 h following transfection, and all experiments were repeated independently at least three times. ***, p < 0.001.
NKX6.1-binding Site Mutation Reduces Activation of the HNF1α Promoter in Beta Cells
Because the NKX6.1-binding site mutation was sufficient to prevent NKX6.1-mediated activation of the HNF1α-luciferase reporter in 3T3 cells, we next assessed its function in beta cells. Transfection of the mutant NKX6.1-binding site construct showed ∼50% reduced activity compared with the normal HNF1α-luciferase reporter in βTC3 beta cells (Fig. 4B). Four other random 5′-TAAT-3′ mutant constructs were generated and had no change in activity compared with the normal promoter construct in similar assays (data not shown). Activity was not completely diminished in the mutant NKX6.1-binding site construct, suggesting that other beta cell-specific transcription factors, such as HNF4α and PAX6, may play a role in regulation of this gene. This result suggests that NKX6.1 is a major beta cell-specific activator of HNF1α in beta cells.
NKX6.1 Can Bind to the HNF1α Distal Regulatory Element
To further evaluate DNA-protein interaction between NKX6.1 and the HNF1α distal promoter element (−2772 to −1820), we designed biotin-labeled oligonucleotide probes spanning the potential NKX6.1-binding site (5′-TAAT-3′). Using EMSA, we demonstrated that NKX6.1 protein from beta cell lysate can bind to our probe (Fig. 5A, lane 2). The addition of anti-NKX6.1 polyclonal antibody caused a disruption and supershift of the DNA-protein band (lane 3), confirming that the protein is indeed NKX6.1. An unlabeled probe was able to compete for binding NKX6.1 (lanes 4 and 5), whereas a nonspecific probe was unable to compete (lane 6). This result confirms that NKX6.1 protein is capable of binding the HNF1α distal promoter element.
FIGURE 5.

NKX6.1 binding to the HNF1α promoter by EMSA and ChIP. A, EMSA. EMSAs were conducted with INS1 cell lysate and biotin-labeled HNF1α promoter oligonucleotide (Probe) as indicated. Arrows indicate binding and supershift. In competition assays, DNA binding reactions were preincubated with 10-fold (10×) or 100-fold (100×) unlabeled HNF1α oligonucleotide or nonspecific (NS) oligonucleotide as indicated. Ab, antibody. B, ChIP assays. NKX6.1 binding experiments are shown in the left panel with (+) or without (−) NKX6.1 overexpression. HNF4α binding experiments are shown in the right panel, with (+) or without (−) HNF4α overexpression. Representative images are shown following agarose gel electrophoresis of PCR products. Quantification of bands was done with the Microsoft Photoshop quantification tool.
NKX6.1 Occupies the HNF1α Distal Regulatory Element in Beta Cells
To confirm that NKX6.1 actually occupies the HNF1α promoter endogenously in beta cells, βTC3 cells were grown normally (−) or transfected with the NKX6.1 expression plasmid (+). Following ChIP with anti-NKX6.1 polyclonal antibody, we were able to amplify this DNA sequence from normal βTC3 cells (Fig. 5B, left panel). The βTC3 cells transfected with the NKX6.1 expression plasmid showed an increase in the intensity of the band, confirming NKX6.1 binding at this site. Our cumulative data demonstrate that NKX6.1 occupies the endogenous HNF1α promoter cis-regulatory element in βTC3 cells and that it is able to positively regulate its expression.
We also evaluated the occupancy of HNF4α at the proximal binding site of the HNF1α promoter as reported previously for hepatocytes. We performed a similar ChIP assay (Fig. 5B, right panel) with βTC3 cells that were grown normally (−) or transfected with the HNF4α expression plasmid (+). Following ChIP with anti-HNF4α polyclonal antibody, we were unable to amplify this DNA sequence from normal βTC3 cells, indicating that HNF4α may not be a major regulator of HNF1α in beta cells. However, the βTC3 cells transfected with the HNF4α expression plasmid (hepatocyte-specific isoform) showed a weak amplification band, indicating that the binding site may still have function to allow its detection. It may also be that HNF4α binding cannot be detected due to the relatively lower binding affinity of beta cell-specific HNF4α isoforms; therefore, we cannot exclude the possibility that HNF4α is involved in HNF1α regulation.
NKX6.1 Regulates Gene Expression of HNF1α in Pancreatic Beta Cells
To determine a role of NKX6.1 in regulating HNF1α in beta cells, mouse beta cell lines (βTC3 and NIT1) were used for this study. First, we confirmed that NKX6.1 indeed can bind to the promoter probe of HNF1α by EMSA as shown in Fig. 6A. Next, we confirmed that both beta cell lines express mRNA for key pancreatic transcription factors (including insulin, Pdx1, Nkx6.1, and HNF1α) that are important for maintaining beta cell function (Fig. 6B). To determine whether NKX6.1 regulates HNF1α in beta cells, the effects of NKX6.1 overexpression or knockdown by its siRNA on HNF1α mRNA levels were investigated. As shown in Fig. 6C, overexpression of NKX6.1 in NIT1 beta cells markedly increased endogenous HNF1α expression by 3-fold over the control (upper panel), whereas Nkx6.1-specific siRNA knocked down endogenous expression of HNF1α by ∼80% of the original level (lower panel), providing further support that NKX6.1 is a regulator of HNF1α. Cyclin B expression was used as an internal control because it has been previously shown to be regulated by NKX6.1 through distal promoter binding (34). Our data confirm that NKX6.1 is a major activator of HNF1α in pancreatic beta cells.
FIGURE 6.

Effects of NKX6.1 overexpression or knockdown on HNF1α. A, EMSA. EMSAs were conducted with βTC3 and NIT1 cell lysates and biotin-labeled HNF1α promoter oligonucleotide (Probe) as indicated. Arrows indicate NKX6.1 binding and supershift. In competition assays, DNA binding reactions were preincubated with 10-fold (10×) or 100-fold (100×) unlabeled HNF1α oligonucleotide or nonspecific (NS) oligonucleotide as indicated. Ab, antibody. B, RT-PCR. Total RNA was isolated from the indicated cell lines, and expression of pancreatic genes was measured by RT-PCR. C, effect of NKX6.1 overexpression and knockdown on mRNA levels. NIT1 mouse beta cells were transfected with pCMV-NKX6.1 expression plasmid (upper panel) or Nkx6.1 siRNA (lower panel) for 48 h. Total RNA was isolated and subjected to reverse transcription. Target gene expression was quantified by real-time PCR and expressed as -fold over the control as indicated. Empty vector plasmid (pcDNA3) transfection was used as a negative control and arbitrarily set to 1.0 for all tested genes.
DISCUSSION
In this study, we have shown a distinct mechanism of regulation of HNF1α in beta cells. We have demonstrated that NKX6.1 is a key regulator for HNF1α by binding to the distal region of the HNF1α promoter in beta cells, which is different from the situation in hepatocytes. We confirmed a specific physical binding of the HNF1α promoter to NKX6.1 protein in beta cells by EMSA and ChIP assay. Furthermore, we also demonstrated that endogenous HNF1α gene transcription is indeed regulated by NKX6.1 in beta cells. To the best of our knowledge, this is the first time that NKX6.1 has been reported as a key regulator of HNF1α in pancreatic beta cells.
NKX6.1 is known to be involved in pancreatic differentiation and beta cell function (31–33). Embryonic expression of Nkx6.1 is dependent on NKX2.2 (43, 44), and in mature beta cells, it is regulated by PDX1 (44). NKX6.1 maintains beta cell phenotype in part by direct interaction with the glucagon promoter, suppressing its activity (33, 38). NKX6.1 inhibits glucagon expression by competing with PAX6 (glucagon activator) for occupancy of the G1 element on the glucagon promoter. NKX6.1 has also been linked to beta cell proliferation by up-regulating cyclins A, B, and E, as well as many regulatory kinases (34).
Studies in knock-out mice reveal that the Nkx6.1 gene is required for beta cell development, terminal differentiation, and biological function (43). Overexpression of NKX6.1 has been shown to increase GSIS in rat islets (34). To address what happens to gene expression in beta cells when NKX6.1 expression is altered, we performed functional studies in beta cell lines to show the effects of NKX6.1 overexpression and knockdown (Fig. 6). As expected, NKX6.1 overexpression led to an increase in HNF1α expression as measured by real-time PCR, whereas Nkx6.1 knockdown by siRNA led to decreased expression. In addition, Uchizono et al. (45) have shown that HNF1α deficiency severely constrains the extent of beta cell proliferation in mice, leading to significant changes in blood glucose levels. In the same study, by exploring gene expression profiles using immortalized beta cells generated from HNF1α-deficient homozygous mice, they also revealed decreased Nkx6.1 gene expression. The results also show changes in expression of genes involved in beta cell growth and proliferation, providing insight into the mechanism whereby HNF1α affects beta cell function. In terms of beta cell functional studies using islets, Newgard and co-workers (34) have shown that overexpression of NKX6.1 increases GSIS in rat and human islets by inducing beta cell replication. In contrast, knockdown of NKX6.1 in human islets leads to impaired GSIS (34).
A recent report demonstrated that mutations affecting the expression of the B-lymphocyte kinase gene (BLK) were found to be responsible for some patients with MODY symptoms but without mutations in known MODY genes (46). BLK is a previously unidentified modulator of insulin synthesis and GSIS in beta cells by the mechanism of enhancing the expression of NKX6.1, providing further evidence that NKX6.1 may be a new candidate gene involved in MODY.
NKX6.1 homeodomain constructs have been shown to bind sequences containing the homeodomain core-binding site (5′-TAAT-3′ or 5′-ATTA-3′) and direct both gene repression and gene activation (39, 40). In its own promoter, NKX6.1 has been shown to bind a similar sequence (5′-ATTT-3′) to positively regulate its own expression (42). NKX6.1 has the ability to function as both a transcriptional activator and repressor, which may be sequence-dependent (42). The transcriptional repression domain has been isolated to the N terminus (40), whereas the transcriptional activation domain has been shown to be dependent on the C terminus (42). The C terminus has also been observed to interfere with DNA binding but greatly enhance specificity for homeodomain core-containing sequences (41).
Previous work on HNF1α expression has established that alternative isoforms of HNF1α are generated by post-translational modification (14, 23). Three separate isoforms have been identified: HNF1αA, HNF1αB, and HNF1αC. These isoforms have been confirmed by real-time PCR, and no evidence for additional forms was found (23). The different isoforms are generated through differential selection of polyadenylation and alternative splicing, generating distinct 3′-ends (23). Furthermore, HNF1α transcription does not use an alternative promoter as demonstrated by 5′-rapid amplification of cDNA ends analysis (23). The different HNF1α isoforms are distributed differently between tissues and during different time points in development, suggesting that they may control gene expression in a temporal and tissue-specific manner. The alternative isoform ratios may also contribute to differential gene regulation between cell types. Alternative splicing of transcripts increases versatility of function of products, and several documented proteins act as activators and repressors from the same gene (47).
Several studies in mice have demonstrated the importance of HNF1α. HNF1α knock-out mice develop many symptoms of diabetes (7). Beta cell dysfunction occurs in HNF1α knock-out mice, marked by hyperglycemia and impaired glucose tolerance without loss of beta cell mass (5). Genes involved in beta cell regulation and metabolism are expressed abnormally in HNF1α null mice, such as Glut2, pyruvate kinase, and insulin, as well as the islet-enriched transcription factors Pdx1, HNF4α, and NeuroD1 (6). HNF1α cellular concentration is critical for beta cell homeostasis. HNF1α knock-out mice have abnormal beta cell gene expression, impaired glucose tolerance, and fasting hyperglycemia, leading to the development of diabetes (4–7). Mice engineered to overexpress HNF1α also develop a diabetic phenotype (8), marked by compromised islet morphology and reduced beta cell mass. HNF1α cellular concentration is also critical for epigenetic maintenance. Studies of HNF1α null murine tissues demonstrate that loss of HNF1α leads to modification of chromatin structure and alters gene expression (3).
MODY is a form of monogenetic diabetes caused by an autosomal dominant mutation in one of several genes. The most prominent form of this disease (>70%) is MODY3, caused by mutations affecting expression of HNF1α (48–51). MODY3 is characterized by adolescent onset of hyperglycemia that progressively worsens with age and most often requires pharmacological treatment. Patients with MODY3 often suffer from diabetic complications.
Defining the etiology of MODY is essential for proper pharmacogenetic treatment of the disease (49, 52–54). HNF1α regulates the expression of a plethora of genes involved in GSIS (51), and mutations result mainly in defective glucose metabolism and insulin secretion in beta cells (51). For this reason, it is not surprising that MODY3 patients respond well to treatment with oral sulfonylureas, given that these drugs bind the KATP channel, which is downstream of glucose metabolism in the insulin secretion pathway (53). Sulfonylurea therapy can close the KATP channel, which increases intracellular Ca2+ and stimulates insulin secretion by an ATP-independent pathway. Patients who are misdiagnosed with type 1 or 2 diabetes have successfully transferred to sulfonylurea treatment without deterioration in glycemic control (48, 55, 56). Following etiologic identification of MODY3, patients have been transferred from insulin to oral sulfonylurea treatment and have shown improved glycemic control, reduced risks of hypoglycemia, and delay or prevention of diabetic complications (48, 55, 56). However, etiologic identification is essential to alter treatment in MODY patients because sulfonylureas are not effective for all other forms of this disease (49, 52–54). Current screening for HNF1α mutations is restricted to the coding sequence and proximal promoter (50, 52).
In up to 20% of MODY patients, the etiology of the disease is unknown, making it very difficult to predict the clinical course of the disease or provide the most effective treatment. We have demonstrated that NKX6.1 is a major regulator of HNF1α and that this regulatory system is unique to beta cells. Our work provides potential novel regulatory elements that should be included when screening for mutations that affect HNF1α expression. NKX6.1 is uniquely expressed in mature beta cells of the pancreas (32) but is not expressed in the liver (57). This novel regulatory network may provide potential new targets for diagnosis and MODY and possibly implicate a new gene (NKX6.1) involved in this disease. Identification of novel molecular targets that cause MODY has the potential to greatly improve treatment and the quality of life for a great deal of diabetic patients.
This work was supported in part by National Institutes of Health Grants DK071831 and DK64054 (to L.-J. Y.) from NIDDK.
All sequences of primer pairs are available upon request.
- HNF1α
- hepatic nuclear factor 1α
- GSIS
- glucose-stimulated insulin secretion
- MODY
- maturity-onset diabetes of the young
- EMSA
- electrophoretic mobility shift assay
- ChIP
- chromatin immunoprecipitation
- RT
- reverse transcription
- siRNA
- small interfering RNA.
REFERENCES
- 1.Ryffel G. U. (2001) J. Mol. Endocrinol. 27, 11–29 [DOI] [PubMed] [Google Scholar]
- 2.Fajans S. S., Bell G. I., Polonsky K. S. (2001) N. Engl. J. Med. 345, 971–980 [DOI] [PubMed] [Google Scholar]
- 3.Pontoglio M., Faust D. M., Doyen A., Yaniv M., Weiss M. C. (1997) Mol. Cell. Biol. 17, 4948–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pontoglio M., Barra J., Hadchouel M., Doyen A., Kress C., Bach J. P., Babinet C., Yaniv M. (1996) Cell 84, 575–585 [DOI] [PubMed] [Google Scholar]
- 5.Pontoglio M., Sreenan S., Roe M., Pugh W., Ostrega D., Doyen A., Pick A. J., Baldwin A., Velho G., Froguel P., Levisetti M., Bonner-Weir S., Bell G. I., Yaniv M., Polonsky K. S. (1998) J. Clin. Invest. 101, 2215–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shih D. Q., Screenan S., Munoz K. N., Philipson L., Pontoglio M., Yaniv M., Polonsky K. S., Stoffel M. (2001) Diabetes 50, 2472–2480 [DOI] [PubMed] [Google Scholar]
- 7.Lee Y. H., Sauer B., Gonzalez F. J. (1998) Mol. Cell. Biol. 18, 3059–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luco R. F., Maestro M. A., del Pozo N., Philbrick W. M., de la Ossa P. P., Ferrer J. (2006) Diabetes 55, 2202–2211 [DOI] [PubMed] [Google Scholar]
- 9.Wobser H., Düssmann H., Kögel D., Wang H., Reimertz C., Wollheim C. B., Byrne M. M., Prehn J. H. (2002) J. Biol. Chem. 277, 6413–6421 [DOI] [PubMed] [Google Scholar]
- 10.Baumhueter S., Mendel D. B., Conley P. B., Kuo C. J., Turk C., Graves M. K., Edwards C. A., Courtois G., Crabtree G. R. (1990) Genes Dev. 4, 372–379 [DOI] [PubMed] [Google Scholar]
- 11.Blumenfeld M., Maury M., Chouard T., Yaniv M., Condamine H. (1991) Development 113, 589–599 [DOI] [PubMed] [Google Scholar]
- 12.Kuo C. J., Conley P. B., Hsieh C. L., Francke U., Crabtree G. R. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 9838–9842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendel D. B., Crabtree G. R. (1991) J. Biol. Chem. 266, 677–680 [PubMed] [Google Scholar]
- 14.Harries L. W., Ellard S., Stride A., Morgan N. G., Hattersley A. T. (2006) Hum. Mol. Genet. 15, 2216–2224 [DOI] [PubMed] [Google Scholar]
- 15.Emens L. A., Landers D. W., Moss L. G. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 7300–7304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ban N., Yamada Y., Someya Y., Miyawaki K., Ihara Y., Hosokawa M., Toyokuni S., Tsuda K., Seino Y. (2002) Diabetes 51, 1409–1418 [DOI] [PubMed] [Google Scholar]
- 17.Satoh S., Noaki T., Ishigure T., Osada S., Imagawa M., Miura N., Yamada K., Noguchi T. (2005) J. Biol. Chem. 280, 39827–39834 [DOI] [PubMed] [Google Scholar]
- 18.Gregori C., Kahn A., Pichard A. L. (1993) Nucleic Acids Res. 21, 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boj S. F., Parrizas M., Maestro M. A., Ferrer J. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14481–14486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulkarni R. N., Kahn C. R. (2004) Science 303, 1311–1312 [DOI] [PubMed] [Google Scholar]
- 21.Miura N., Tanaka K. (1993) Nucleic Acids Res. 21, 3731–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian J. M., Schibler U. (1991) Genes Dev. 5, 2225–2234 [DOI] [PubMed] [Google Scholar]
- 23.Bach I., Yaniv M. (1993) EMBO J. 12, 4229–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harries L. W., Locke J. M., Shields B., Hanley N. A., Hanley K. P., Steele A., Njølstad P. R., Ellard S., Hattersley A. T. (2008) Diabetes 57, 1745–1752 [DOI] [PubMed] [Google Scholar]
- 25.Hansen S. K., Párrizas M., Jensen M. L., Pruhova S., Ek J., Boj S. F., Johansen A., Maestro M. A., Rivera F., Eiberg H., Andel M., Lebl J., Pedersen O., Ferrer J., Hansen T. (2002) J. Clin. Invest. 110, 827–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J., Levitsky L. L., Rhoads D. B. (2009) Exp. Cell Res. 315, 1200–1211 [DOI] [PubMed] [Google Scholar]
- 27.Eeckhoute J., Moerman E., Bouckenooghe T., Lukoviak B., Pattou F., Formstecher P., Kerr-Conte J., Vandewalle B., Laine B. (2003) Endocrinology 144, 1686–1694 [DOI] [PubMed] [Google Scholar]
- 28.Ihara A., Yamagata K., Nammo T., Miura A., Yuan M., Tanaka T., Sladek F. M., Matsuzawa Y., Miyagawa J., Shimomura I. (2005) Biochem. Biophys. Res. Commun. 329, 984–990 [DOI] [PubMed] [Google Scholar]
- 29.Nakhei H., Lingott A., Lemm I., Ryffel G. U. (1998) Nucleic Acids Res. 26, 497–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang J., Karakucuk V., Levitsky L. L., Rhoads D. B. (2008) Diabetes Metab. Res. Rev. 24, 533–543 [DOI] [PubMed] [Google Scholar]
- 31.Oster A., Jensen J., Serup P., Galante P., Madsen O. D., Larsson L. I. (1998) J. Histochem. Cytochem. 46, 707–715 [DOI] [PubMed] [Google Scholar]
- 32.Lyttle B. M., Li J., Krishnamurthy M., Fellows F., Wheeler M. B., Goodyer C. G., Wang R. (2008) Diabetologia 51, 1169–1180 [DOI] [PubMed] [Google Scholar]
- 33.Schisler J. C., Jensen P. B., Taylor D. G., Becker T. C., Knop F. K., Takekawa S., German M., Weir G. C., Lu D., Mirmira R. G., Newgard C. B. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 7297–7302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schisler J. C., Fueger P. T., Babu D. A., Hohmeier H. E., Tessem J. S., Lu D., Becker T. C., Naziruddin B., Levy M., Mirmira R. G., Newgard C. B. (2008) Mol. Cell. Biol. 28, 3465–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lupi R., Bugliani M., Del Guerra S., Del Prato S., Marchetti P., Boggi U., Filipponi F., Mosca F. (2006) Nutr. Metab. Cardiovasc. Dis. 16, e7–e8 [DOI] [PubMed] [Google Scholar]
- 36.Koya V., Lu S., Sun Y. P., Purich D. L., Atkinson M. A., Li S. W., Yang L. J. (2008) Diabetes 57, 757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber H., Strandmann E. P., Holewa B., Bartkowski S., Zapp D., Zoidl C., Ryffel G. U. (1996) Int. J. Dev. Biol. 40, 297–304 [PubMed] [Google Scholar]
- 38.Gauthier B. R., Gosmain Y., Mamin A., Philippe J. (2007) Biochem. J. 403, 593–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jørgensen M. C., Vestergård Petersen H., Ericson J., Madsen O. D., Serup P. (1999) FEBS Lett. 461, 287–294 [DOI] [PubMed] [Google Scholar]
- 40.Mirmira R. G., Watada H., German M. S. (2000) J. Biol. Chem. 275, 14743–14751 [DOI] [PubMed] [Google Scholar]
- 41.Taylor D. G., Babu D., Mirmira R. G. (2005) Biochemistry 44, 11269–11278 [DOI] [PubMed] [Google Scholar]
- 42.Iype T., Taylor D. G., Ziesmann S. M., Garmey J. C., Watada H., Mirmira R. G. (2004) Mol. Endocrinol. 18, 1363–1375 [DOI] [PubMed] [Google Scholar]
- 43.Sander M., Sussel L., Conners J., Scheel D., Kalamaras J., Dela Cruz F., Schwitzgebel V., Hayes-Jordan A., German M. (2000) Development 127, 5533–5540 [DOI] [PubMed] [Google Scholar]
- 44.Watada H., Mirmira R. G., Leung J., German M. S. (2000) J. Biol. Chem. 275, 34224–34230 [DOI] [PubMed] [Google Scholar]
- 45.Uchizono Y., Baldwin A. C., Sakuma H., Pugh W., Polonsky K. S., Hara M. (2009) Diabetes Res. Clin. Pract. 84, 19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borowiec M., Liew C. W., Thompson R., Boonyasrisawat W., Hu J., Mlynarski W. M., El Khattabi I., Kim S. H., Marselli L., Rich S. S., Krolewski A. S., Bonner-Weir S., Sharma A., Sale M., Mychaleckyj J. C., Kulkarni R. N., Doria A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 14460–14465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foulkes N. S., Sassone-Corsi P. (1992) Cell 68, 411–414 [DOI] [PubMed] [Google Scholar]
- 48.Della M. T. (2007) J. Pediatr. 83, S178–S183 [Google Scholar]
- 49.Ellard S., Bellanné-Chantelot C., Hattersley A. T. (2008) Diabetologia 51, 546–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCarthy M. I., Hattersley A. T. (2008) Diabetes 57, 2889–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitchell S. M., Frayling T. M. (2002) Mol. Genet. Metab. 77, 35–43 [DOI] [PubMed] [Google Scholar]
- 52.Hattersley A., Bruining J., Shield J., Njolstad P., Donaghue K. (2006) Pediatr. Diabetes 7, 352–360 [DOI] [PubMed] [Google Scholar]
- 53.Hattersley A. T. (2005) Clin. Med. 5, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphy R., Ellard S., Hattersley A. T. (2008) Nat. Clin. Pract. Endocrinol. Metab. 4, 200–213 [DOI] [PubMed] [Google Scholar]
- 55.Pearson E. R., Liddell W. G., Shepherd M., Corrall R. J., Hattersley A. T. (2000) Diabet. Med. 17, 543–545 [DOI] [PubMed] [Google Scholar]
- 56.Shepherd M., Pearson E. R., Houghton J., Salt G., Ellard S., Hattersley A. T. (2003) Diabetes Care 26, 3191–3192 [DOI] [PubMed] [Google Scholar]
- 57.Fodor A., Harel C., Fodor L., Armoni M., Salmon P., Trono D., Karnieli E. (2007) Diabetologia 50, 121–130 [DOI] [PubMed] [Google Scholar]