

Abstract
The transcription factor nuclear factor kappa B (NF-κB) is a key regulator of inflammatory processes in reactive glial cells. We utilized a transgenic mouse model (GFAP-IκBα-dn) where the classical NF-κB pathway is inactivated by overexpression of a dominant negative (dn) form of the inhibitor of kappa B (IκBα) in glial fibrillary acidic protein (GFAP) expressing cells, which include astrocytes, Schwann cells, and satellite cells of the dorsal root ganglion (DRG) and sought to determine whether glial NF-κB inhibition leads to a reduction in pain behavior and inflammation following chronic constriction injury (CCI) of the sciatic nerve. As expected, following CCI nuclear translocation, and hence activation, of NF-κB was detected only in the in the sciatic nerve of wild type (WT) mice, and not in GFAP-IκBα-dn mice, while upregulation of GFAP was observed in the in sciatic nerve and DRGs of both WT and GFAP-IκBα-dn mice, indicative of glial activation. Following CCI, mechanical and thermal hyperalgesia were reduced in GFAP-IκBα-dn mice compared to WT, as well as gene and protein expression of CCL2, CCR2 and CXCL10 in the sciatic nerve. Additionally, gene expression of TNF, CCL2, and CCR2 was reduced in the DRGs of transgenic mice compared to WT after CCI. We can therefore conclude that transgenic inhibition of NF-κB in GFAP expressing glial cells attenuated pain and inflammation after peripheral nerve injury. These findings suggest that targeting the inflammatory response in Schwann cells and satellite cells may be important in treating neuropathic pain.
Keywords: Pain, NF-kappa B, Chronic Constriction Injury, Peripheral Glia
Introduction
Nuclear factor kappa B (NF-κB) is a ubiquitous transcription factor which regulates the expression for many genes (e.g. cytokines, chemokines, iNOS) that are important in immunity, inflammation, central nervous system (CNS) injury, and neuropathic pain [18,20,26]. In non-stimulated cells, NF-κB dimers are maintained in the cytosol by binding to the inhibitors of κB (IκBs; e.g., IκBα, IκBβ, and IκBε). In response to cell stimulation (e.g., cytokines, glutamate, oxidative damage, growth factors, etc.), a multi-subunit kinase complex, the IκB kinase (IKK), is rapidly activated and phosphorylated in the N-terminal regulatory domain of the IκBs. This signals the ubiquitin proteasome pathway to degrade IκBα, releasing activated NF-κB which can translocate to the nucleus and initiate transcription of mediators that are involved in the pathogenesis of neuropathic pain.
While suppression of NF-κB appears to be an attractive concept in the approach to treating pain and inflammation, unselective and complete inhibition of NF-κB may lead to deleterious effects in terms of cell survival [18]. It has been postulated that strategies to selectively inhibit NF-κB activity would be of great benefit in terms of attenuating pain and inflammation while reducing unwanted side effects [18,26]. In a recent review by Niederberger et al. [18], a novel approach for pain therapy includes strategies which prevent IκB phosphorylation and selectively inactivate NF-κB transcription. For example, pharmacological agents that specifically inhibit IKK have been shown to reduce mechanical and thermal hyperalgesia after CCI and zymosan induced paw inflammation in rats [26].
To address the issue of selective inactivation of NF-κB as a modality to treat pain, we utilized a transgenic mouse model that overexpresses the dominant negative (dn) form of the inhibitor of kappa B alpha (IκBα) in glial cells, under the control of the glial fibrillary acidic protein (GFAP) promoter [3]. The transgene, GFAP-IκBα-dn, is present not only in astrocytes in the CNS, which constitutively express GFAP, but also in GFAP expressing non-myelinating Schwann cells, thereby making it a useful model to study pain in relation to the peripheral nervous system [3,12,26]. Previously, we found that transgenic inhibition of glial NF-κB reduced nociceptive behavior after formalin pain [11]. In the present study, we determined that transgenic inhibition of glial NF-κB attenuates the pain and inflammatory response following chronic constriction injury (CCI) of the sciatic nerve.
Methods
Animals
All experiments were carried out following the guidelines and protocols approved by the Animal Care and Use Committee of the University of Miami. GFAP-IκBα-dn mice were bred at the Transgenic Core Facility of the University of Miami. Details pertaining to the generation of GFAP-IκBα-dn mice were previously described [3]. All mice used in our experiments were 2-4 months old C57BL/6 males, weighing 25-35 grams. These mice were obtained by breeding heterozygous transgenic GFAP-IκBα-dn males with WT females. WT littermates from the same breeding group were used as controls. All animals were housed in a 12 hr light/dark cycle in a virus/antigen-free facility with controlled temperature.
RNA Isolation and RT-PCR of the GFAP-IκBα-dn transgene
Expression of the GFAP-IκBα-dn transgene was evaluated by RT-PCR in spinal cord tissue, sciatic nerve, and dorsal root ganglion (DRG). Extraction of total RNA was carried out with TRIzol (Invitrogen) according to manufacturer's instructions. Two μg of RNA were reverse transcribed with 200 U/sample SuperScript II (Invitrogen) and 250 ng/reaction of random primers (Promega). The GFAP-IκBα-dn transgene was amplified from 0.1 μg aliquots of cDNA in a standard PCR buffer (50 mM KCl, 1.5 mM MgCl2, and 10 mM Tris-HCl, pH 8.3) containing 10 pmol of forward GFAP (5-TTC ATA AAG CCC TCG CAT CC-3) and reverse IκBα (5-ACA GCC AGC TCC CAG AAG TG-3) primers and 0.5 U/sample of Ampli-Taq DNA polymerase (Applied Biosystems). Mouse β-actin was amplified as a control for the PCR reaction. Samples lacking reverse transcriptase were amplified as a control for genomic DNA contamination.
Chronic Constriction Injury (CCI)
Male mice were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) for surgical procedures. Chronic constriction of the sciatic nerve was performed according to the method described by Bennett and Xie [2]. Briefly, the left sciatic nerve was exposed at the level of the mid thigh. Three loose 6.0 silk ligatures were placed around the dissected nerve with a 1.0-1.5 mm interval between each ligature. The ligation was carefully manipulated so that the ligature was loosely tied around the nerve. The wound was closed with 6-0 Ethilon monofilament nylon in layers. The same procedure was used for sham surgeries except that the contralateral sciatic nerve was exposed but not ligated.
Assessment of Pain Behavior after CCI
Mechanical hyperalgesia
Mice were habituated to the behavioral testing paradigms for 3–5 days before initiating data collection. Each mouse was placed individually beneath an inverted transparent plastic cage with a wire mesh bottom. Von Frey filaments (Touch-Test Sensory Evaluator, North Coast Medical, Morgan Hill, CA) with incremental stiffness (0.1-1.5g) were applied serially to the paw in ascending or descending order of stiffness depending on the foot withdrawal response of the mouse. The maximum and minimum cut-offs were at 1.5 and 0.1 g respectively. A single trial of stimuli consists of five applications of von Frey filaments to the plantar surface of the paw perpendicularly for about 4-5 s. Foot withdrawals (at least three times out of five applications) in response to the stimuli were considered positive. Depending on the positive or negative response, subsequent filaments were applied in order of descending and ascending intensity, respectively. The withdrawal threshold was measured 5 times and expressed as the tolerance level in grams.
Thermal hyperalgesia
Mice were habituated to the behavioral testing paradigms for 3–5 days before initiating data collection. Mice were placed in a transparent Perspex box on a thin glass platform. The hindpaws were in contact with a 1/4″ thick glass plate that is maintained at room temperature. A mobile infrared heat lamp device or plantar apparatus (UGO BASILE 7371 Plantar Test Apparatus, Italy) was then positioned underneath the targeted hind paw. The plantar apparatus directs a focused radiant light source from below the glass onto the plantar surface of one hindpaw. The paw withdrawal latency (PWL) was determined for the left and right hindpaw, with a 5-min inter-trial interval. The infrared intensity was set at 50 (corresponding to 196 mW/cm2), which produced baseline paw withdrawal latencies of 5-10 sec. The mean latency of withdrawal response of each hind paw was determined by 6 tests. A cut off of 20 sec was used.
Frequency of behavioral assessment
A blinded observer measured withdrawal threshold to mechanical stimuli and latency withdrawal to thermal stimuli. Baseline measurements were obtained two days prior to CCI surgery. At the following time points, including days 1, 3, 7, 10 and 14 after CCI, behavioral tests were performed to evaluate the effects of CCI on mechanical and thermal hyperalgesia.
Real Time RT-PCR
Total RNA was isolated from sciatic nerve, L4-L6 DRGs and L4-L6 spinal cord using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer's instructions. Total RNA (2 μg) was reverse-transcribed using Superscript II (Invitrogen) in a 20-μL reaction volume containing 4.5 μg/mL random hexamers (Promega, Madison, WI, USA), 0.5 mmol/L dNTPs, and 10 mmol/L dithiothreitol. Real time PCR was performed using the TAQurate green real-time PCR master mix (Epicentre Biotechnologies, Madison, WI, USA) following manufacturer's instructions and data were collected on the Rotor-Gene 2000 (Corbett Research, Mortlake, Australia).
Immunohistochemistry and immunofluorescence quantification
Animals were perfused with 4% paraformaldehyde in 0.1 M PBS, spinal cord, sciatic nerve and DRG removed and post-fixed overnight. Tissues were cryoprotected in 0.1 M PBS containing 20% sucrose and cryostat cut into 15 μm thick sections. Sections were blocked for 1 hr at 22°C in 0.1 M PBS containing 3% donkey serum and 0.4% Triton-X, prior to overnight incubation at 4° C with the primary antibodies anti-GFAP (Dako, USA), anti-NF-κB p65 phosphoSer276 (Millipore, USA) anti-CCL2 (Santa Cruz Biotechnology, Inc. USA), anti-CXCL10 (Biovision, USA), and anti-CCR2 (Epitomics, USA), followed by secondary species-specific fluorescent antibodies (Jackson ImmunoResearch Lab, Inc. USA) for 1 hr at 22°C. Images were collected on a Zeiss Axiovert 200M fluorescent microscope and on a Zeiss LSM 510 confocal microscope.
For quantification of the immunofluorescence signal, two sections of the spinal cord, sciatic nerve or DRG were randomly selected for each mouse and analyzed with the software NIH Image J (developed by the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/ij). Four animals per experimental condition were analyzed.
Statistical Analysis
Data are presented as mean ± SEM. For assessment of mechanical and thermal hyperalgesia, the thresholds or latencies to withdraw the paw on the injured side relative to that on the same side of sham-injured animals were compared by two-way analysis of variance with Bonferroni post-hoc analysis. Real time PCR measurements and fluorescence quantification were analyzed with one-way ANOVA followed by Tukey Multiple Comparison Test. P values of less than 0.05 were designated as statistically significant.
Results
Expression and function of the GFAP-IκBα-dn Transgene
Transgene expression, measured by RT-PCR, was detected in sciatic nerve, DRGs and spinal cord of GFAP-IκBα-dn mice (Fig. 1A). To assess whether the transgene was functional in inhibiting NF-κB activation, we performend immunostaining for activated phospho-p65 in WT and GFAP-IκBα-dn sciatic nerves 1 day after CCI. As expected, nuclear translocation of phospho-p65 was observed in WT but not in GFAP-IκBα-dn mice (Figure 1B), indicating the ability of the GFAP-IκBα-dn transgene to inactivate the NF-κB classical pathway. Reactive gliosis was observed following CCI in both WT and transgenic mice, as a marked upregulation of GFAP immunoreractivity was detected in sciatic nerve, DRGs, and spinal cord of both genotypes following CCI (Fig. 2). A quantification of the GFAP immunofluorescence was performed with the software NIH Image J and indicated no significant differences between the two mouse genotypes in all tissues tested (Table 1).
Figure 1.
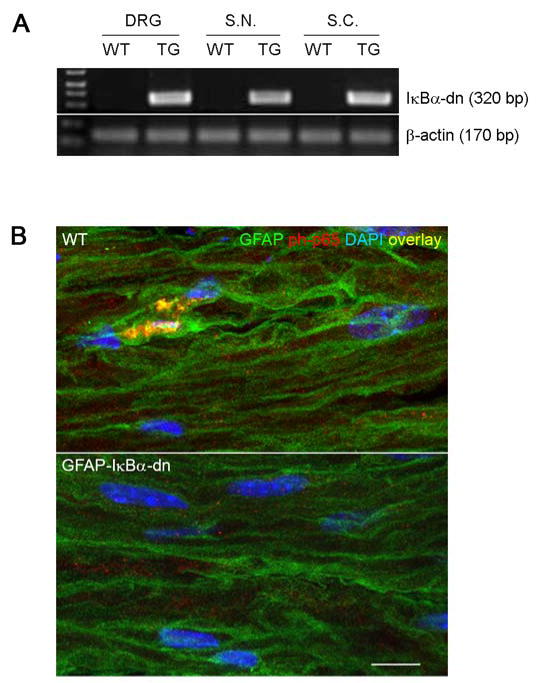
(A) RT-PCR analysis of GFAP-IκBα-dn transgene expression in WT and GFAP-IκBα-dn mice. Each gene was normalized to β-actin expression as a loading control for the PCR reaction. (B) Nuclear translocation of activated p65 in sciatic nerve of WT and GFAP-IκBα-dn mice 1 day after CCI. Green: GFAP; Red: phospho-p65; Blue: DAPI (scale bar = 10 μm). TG: transgenic mice; DRG: dorsal root ganglion; S.N.: sciatic nerve; S.C.: spinal cord.
Figure 2.
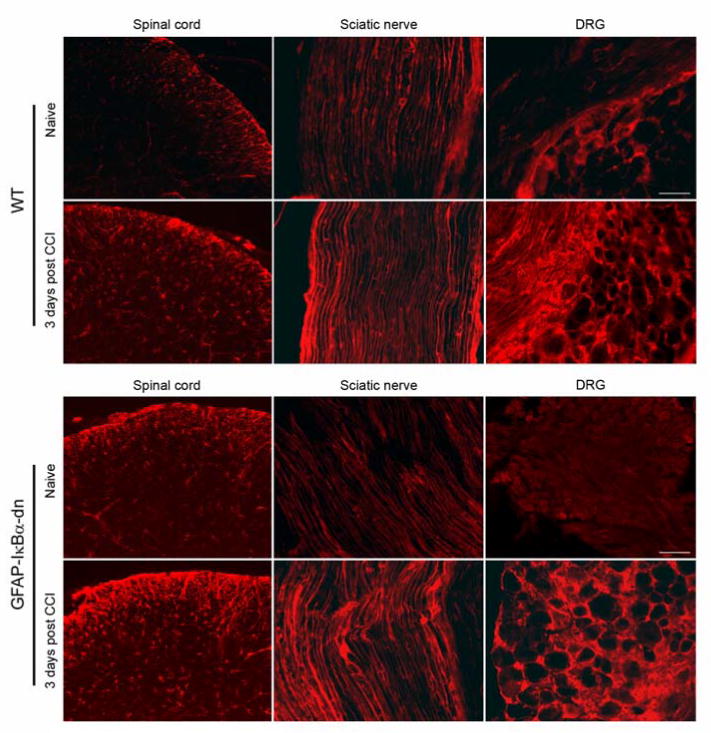
GFAP immunoreactivity in spinal cord, sciatic nerve and DRG of WT and GFAP-IκBα-dn mice under naïve conditions and 3 days after CCI (scale bar = 100 μm).
Table 1.
Quantification of GFAP immunoreactivity in spinal cord, sciatic nerve and dorsal root ganglion after CCI.
| Spinal cord | Sciatic nerve | Dorsal root ganglion | ||||
|---|---|---|---|---|---|---|
| WT | TG | WT | TG | WT | TG | |
| Naive | 31.5±2.9 | 35.9±2.7 | 9.5±1.1 | 9.4±.9 | 9.4±0.6 | 7.7±0.7 |
| 1 day CCI | 50.1±2.2* | 48.9±2.2* | 14.8±0.7* | 14.1±0.4* | 20.2±3.4* | 16.7±2.2* |
| 3 days CCI | 44.7±2.7* | 51.8±2.8* | 17.6±1.4* | 18.8±2.0* | 19.0±1.8* | 17.6±1.3* |
Comparison between wild type (WT) and transgenic (TG) GFAP-IκBα-dn mice under naïve conditions and at day 1 and 3 after CCI. Data are expressed in arbitrary units and presented as mean ± S.E.M. of 4 animals/condition.
p<0.05 vs corresponding naïve, one-way ANOVA, Tukey test.
Glial NF-κB Inhibition Reduces Mechanical and Thermal Hyperalgesia
To assess whether the blockade of NF-κB had any effect on pain behavior after peripheral nerve injury, we measured mechanical allodynia and thermal hypersensitivity on days 1, 3, 7, 10, and 14 after CCI. Fig. 3A shows the mechanical hyperalgesia response for the ipsilateral hindpaws in WT and GFAP-IκBα-dn mice after CCI. Compared to baseline (non-injured), decreases in withdrawal threshold occurred after CCI, as an indication of increasing mechanical allodynia. The decrease in threshold was greater in the ipsilateral hindpaw, which corresponded to the side of the sciatic lesion, versus the contralateral hindpaw. In the ipsilateral hindpaw, WT mice had greater mechanical hyperalgesia compared to GFAP-IκBα-dn mice (p<0.05, two-way ANOVA). Bonferroni post hoc analysis showed that the difference between groups was significant on days 1, 3, and 7 after CCI (Fig. 3A). In the contralateral hindpaw, WT mice had greater mechanical hyperalgesia than non-injured animals (p<0.05, two-way ANOVA), but no difference was found between WT and transgenic mice at any time point after CCI (Fig. 3B).
Figure 3.
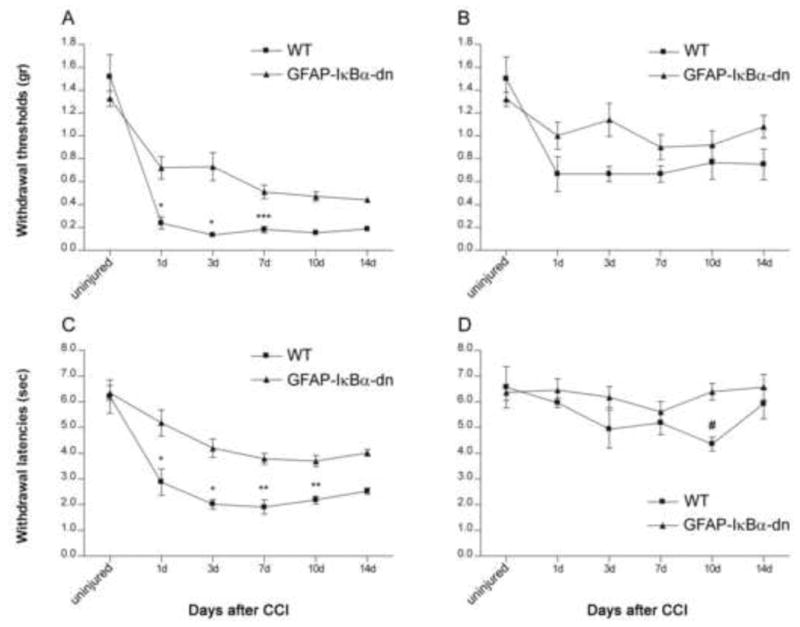
Nociceptive responses in the hindpaws in WT (n=6) and GFAP-IκBα-dn mice (n=10) after CCI. WT mice had greater mechanical hyperalgesia, as measured by the decrease in withdrawal thresholds in the ipsilateral (A) and contralateral (B) hindpaws. Curves are significantly different (p<0.05, two-way ANOVA). WT mice had greater thermal hyperalgesia, as measured by decreases in withdrawal latencies in the (C) ipsilateral hindpaw and (D) contralateral hindpaws. Curves are significantly different (p<0.05, two-way ANOVA). Bonferroni post hoc analysis: *p<0.001, **p<0.01, ***p<0.05, for WT ipsilateral hindpaw vs. GFAP-IκBα-dn ipsilateral hindpaw; #p<0.05 for WT contralateral hindpaw vs GFAP-IκBα-dn contralateral hindpaw.
Next we measured the thermal sensitivity response for both right and left hindpaws in WT and GFAP-IκBα-dn mice after CCI. Compared to baseline (non-injured), both genotypes showed a decrease in latency withdrawal after CCI, as an indication of increasing thermal sensitivity (Fig. 3C and D). As expected, such decrease in latency was greater in the ipsilateral hindpaw (Fig. 3C), which corresponded to the side of the sciatic nerve injury, versus the contralateral hindpaw (Fig. 3D). Interestingly, WT mice displayed a significantly higher thermal hyperalgesia compared to GFAP-IκBα-dn mice in the ipsilateral hindpaw at all time points tested (Fig. 3C, p<0.05, two-way ANOVA). WT mice showed increased thermal hyperlagesia compared to transgenic mice also in the contralateral hindpaw, but this was statistically significant only at the 10 days time point (Fig. 3D).
Glial NF-κB Inhibition Reduces Expression of Inflammatory Mediators in Sciatic Nerve and DRGs
To investigate the molecular mechanisms relevant to the decreased pain response in GFAP-IκBα-dn mice, we evaluated the expression of NF-κB-dependent inflammatory mediators, such as cytokines (IL-1β, IL-6, TNF), chemokines (CCL2, CXCL9, CXCL10) and chemokine receptors (CCR2, CXCR3). In the sciatic nerve, no increase in the gene expression of IL-6, CXCL9, and CXCR3 were detected in either genotypes after CCI (data not shown). Gene expression for both IL-1β, and TNF was significantly upregulated 1 day after CCI as compared to the corresponding naïve mice, however no difference was measured between WT and GFAP-IκBα-dn mice (Fig. 4). Even thought CCL2 gene expression was upregulated in the sciatic nerve (ipsilateral side) of both WT and GFAP-IκBα-dn mice after CCI (Fig. 5), this increase was significantly lower in GFAP-IκBα-dn mice compared to WT, particularly at the 1 day time point. A very similar pattern of expression was detected for CCR2, the receptor for CCL2, where the CCI-induced upregulation of gene expression was lower in the GFAP-IκBα-dn mice compared to WT at 1 day after injury (Fig. 5). As for CXCL10, another powerful chemoattractant for peripheral leukocytes, increased expression was measured following CCI in both genotypes, but this was significantly diminished in transgenic mice compared to WT (Fig. 5).
Figure 4.
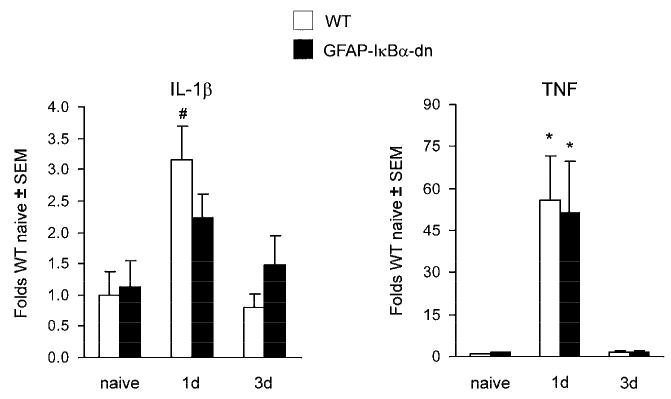
Gene expression of IL-1β and TNF in the ipsilateral sciatic nerve of WT mice and GFAP-IκBα-dn mice following CCI. For each gene, results are expressed as folds of WT naïve mice ± S.E.M., after normalization to β-actin. Four animals/group/time point were analyzed. *p<0.001, #p<0.05 compared to corresponding naïve mice, one-way ANOVA, Tukey test.
Figure 5.
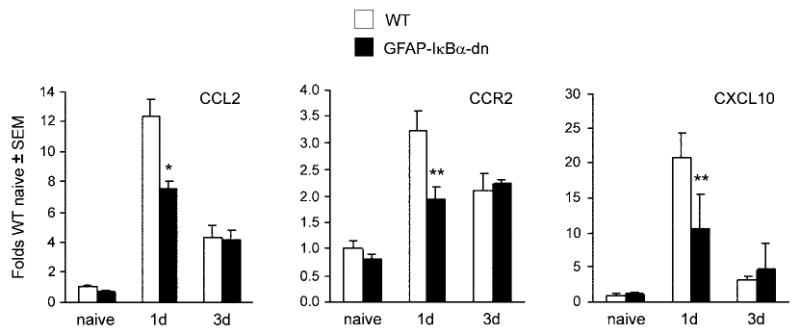
Gene expression of CCL2, CCR2, and CXCL10 in the ipsilateral sciatic nerve of WT and GFAP-IκBα-dn mice following CCI. For each gene, results are expressed as folds of WT naïve ± S.E.M., after normalization to β-actin. Four animals/group/time points were analyzed. *p<0.01, **p<0.05 compared to WT 1 day, one-way ANOVA, Tukey test.
We determined by immunohistochemistry that CCI induced increases also in the protein expression of chemokines and chemokine receptors in the sciatic nerve (Fig. 6-8), and such changes were quantified with the software NIH Image J (Table 2). CCL2 upregulation peaked at day 1, but was significantly reduced in GFAP-IκBα-dn mice compared to WT. CCR2, the receptor for CCL2, was also upregulated after CCI, however both at day 1 and 3 post-injury its expression was significantly reduced in the transgenic mice compared to the WT. As for CXCL10, expression was significantly reduced in GFAP-IκBα-dn mice compared to WT at day 3 post-CCI (Table 2). Although in most animals tested CXCL10 expression appeared reduced also at day 1 in transgenic mice compared to WT, the quantification of the immunofluorescence staining did not show a statistically significant difference between the two groups (Table 2). These data correlate well with our gene expression data and are consistent with findings reported in the literature [13]. We also quantified the expression of these molecules in the spinal cord and only found a mild upregulation after CCI, with no significant differences between the two genotypes (Table 2).
Figure 6.
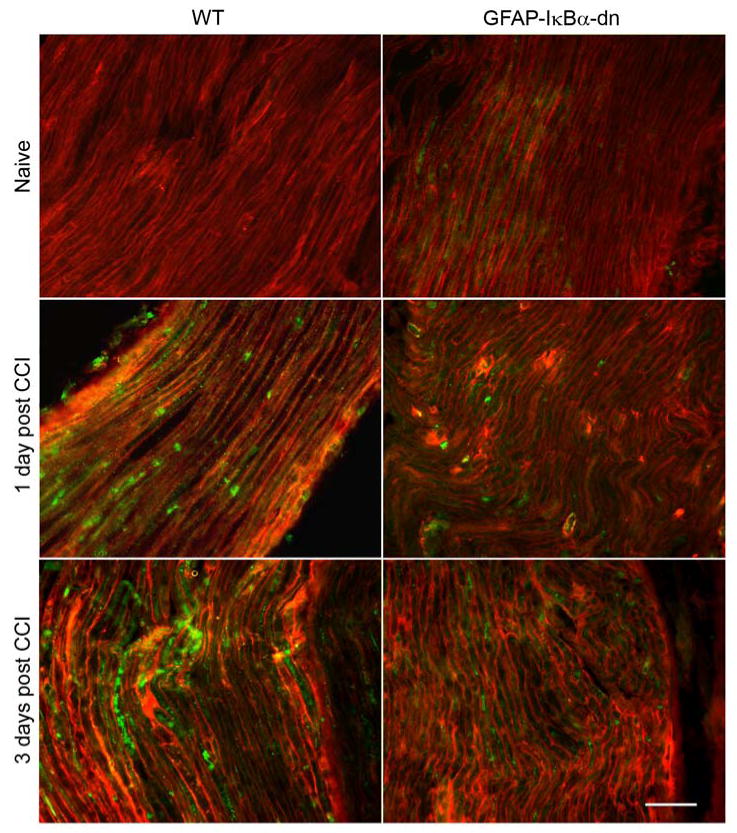
Immunostaining for CCL2 (green) and GFAP (red) in the ipsilateral sciatic nerve of WT and GFAP-IκBα-dn mice. CCI induced increased immunoreactivity for both GFAP and CCL2. On day 1, greater CCL2 immunoreactivity was present in WT mice compared to GFAP-IκBα-dn (scale bar = 50 μm).
Figure 8.
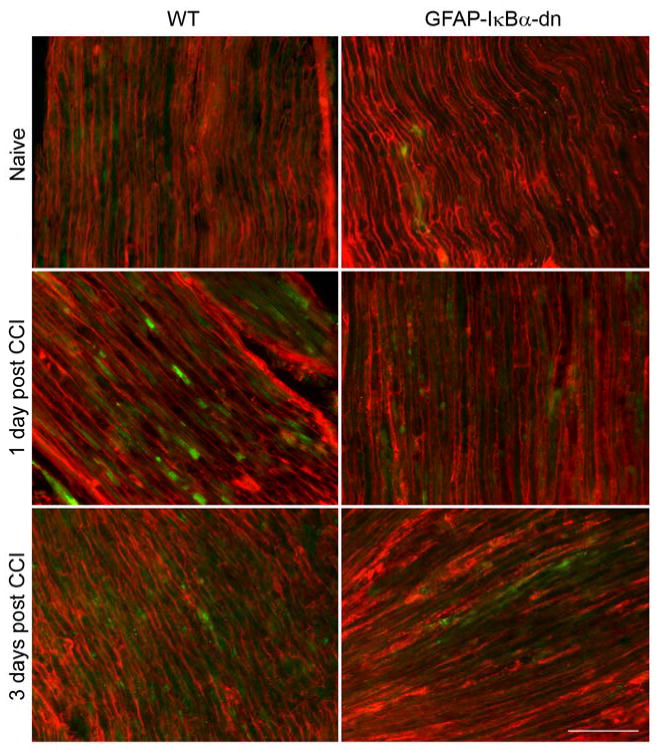
Immunostaining for CXCL10 (green) and GFAP (red) in the ipsilateral sciatic nerve of WT and GFAP-IκBα-dn mice. CCI induced increased immunoreactivity for both GFAP and CXCL10. On day 3, greater CXCL10 immunoreactivity was present in WT mice compared to GFAP-IκBα-dn (scale bar = 50 μm).
Table 2.
Quantification CCL2, CCR2 and CXCL10 immunoreactivity in spinal cord and sciatic nerve after CCI.
| Spinal cord | Sciatic nerve | ||||
|---|---|---|---|---|---|
| WT | TG | WT | TG | ||
| CCL2 | Naive | 9.5±1.7 | 10.4±1.8 | 9.0±1.2 | 9.0±1.6 |
| 1 day CCI | 14.5±1.1* | 14.1±0.5* | 54.8±7.0* | 38.2±5.7*# | |
| 3 days CCI | 14.8±1.2* | 12.7±1.6* | 23.3±2.6* | 22.7±1.8* | |
| CCR2 | Naive | 10.1±1.8 | 11.0±1.5 | 27.1±2.5 | 23.7±2.5 |
| 1 day CCI | 15.5±1.4* | 15.0±2.2* | 43.6±3.2* | 31.2±2.5*# | |
| 3 days CCI | 15.0±1.5* | 14.4±1.2* | 41.5±3.3* | 25.6±3.4# | |
| CXCL10 | Naive | 10.1±1.5 | 11.7±1.5 | 13.4±1.8 | 12.4±2.0 |
| 1 day CCI | 13.2±1.2 | 11.1±1.0 | 42.0±3.3* | 31.0±3.3* | |
| 3 days CCI | 17.2±2.1* | 16.3±1.5* | 16.5±1.6 | 10.0±1.1# | |
Comparison between wild type (WT) and transgenic (TG) GFAP-IκBα-dn mice under naïve conditions and at day 1 and 3 after CCI. Data are expressed in arbitrary units and presented as mean ± S.E.M. of 4 animals/condition.
p<0.05 vs corresponding naïve;
p<0.05 vs corresponding WT; one-way ANOVA, Tukey test.
In DRGs, no increases in the gene expression of IL-6, CXCL9, and CXCR3 were present in WT and GFAP-IκBα-dn mice after CCI (data not shown). Interestingly, CCI induced a significant upregulation of CCL2, CCR2, and TNF mRNA in the DRGs at day 1 post CCI (Fig. 9) and this was completely prevented in the GFAP-IκBα-dn mice.
Figure 9.
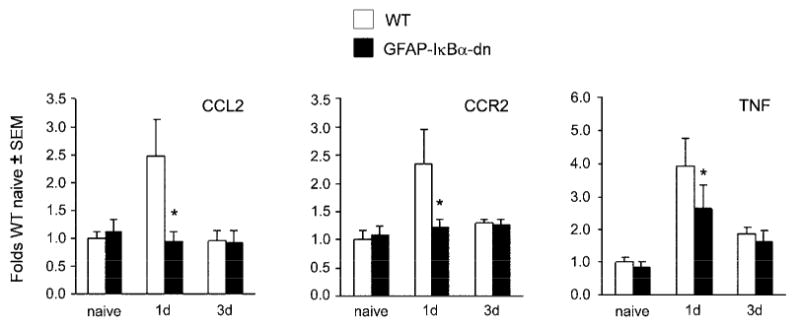
Gene expression of CCL2, CCR2, and TNF in the ipsilateral DRGs of WT mice and GFAP-IκBα-dn mice. For each gene, results are expressed as fold of corresponding WT naïve ± S.E.M., after normalization to β-actin. Four animals/group/time point were analyzed. *p<0.05 vs WT 1d; one-way ANOVA, Tukey test.
Discussion
In chronic pain states following peripheral nerve damage, peripheral glia, which includes Schwann cells and DRG satellite cells, become activated. Schwann cells regulate a microenvironment rich in extracellular mediators such as cytokines, chemokines, growth factors, and matrix metalloproteases [5-6]. While satellite cells are known to express cytokines, as an indication of an activated state, further work is needed to define the role of satellite cells as a site of action in the DRGs for mediating pain after peripheral nerve injury [16,19]. Non-myelinating Schwann cells [12,26] and DRG satellite cells [16,19,34] are peripheral glial cells that express GFAP. GFAP upregulation in DRG satellite cells [16,19] has been reported following neuropathic injury. In the present work, we demonstrated that GFAP upregulation occurs in both sciatic nerve and DRG after CCI, indicative of peripheral activation of GFAP expressing cells.
The GFAP-IκBα-dn transgenic mice used in this study have a functional inactivation of the NF-κB classical pathway in GFAP expressing cells in the central and peripheral nervous system. These mice were initially characterized by Brambilla et al. [3] in a model of spinal cord contusion, in which GFAP-IκBα-dn mice had improved motor function and decreased inflammation compared to WT mice. Brambilla et al. [3] found that the GFAP-IκBα-dn transgene was expressed in brain, spinal cord and sciatic nerve with no ectopic expression in heart, lung, kidney, liver or spleen. In addition to spinal cord and sciatic nerve tissue, we detected the presence of the GFAP-IκBα-dn transgene in the DRG. This finding can be explained by evidence that GFAP is expressed in DRG satellite cells [16,19]. These cells appear to play a role in chronic pain states, as they have been recently shown to interact with DRG neurons via chemical signaling following peripheral nerve inflammation [9].
In GFAP-IκBα-dn mice, we did not find evidence of nuclear translocation of activated p65 in the sciatic nerve after CCI, confirming the effectiveness of the transgene in suppressing NF-κB activation. GFAP-IκBα-dn mice had less mechanical and thermal hyperalgesia, which correlated with reduced expression of NF-κB-dependent inflammatory mediators in sciatic nerve and DRGs. Our behavioral and biochemical data are consistent with previous studies which have shown that inflammatory mediators in the periphery are involved in neuropathic pain and that anti-inflammatory strategies reduce nociception [13,14,21].
Kleinschnitz et al. [13,14] found that CCI induced the production of cytokines and chemokines not only in the injured sciatic nerve but also in the uninjured contralateral sciatic nerve, which may explain the behavioral changes we found in the contralateral hindlimb between WT and GFAP-IκBα-dn mice. In a study involving CCI in rats, Schäfers et al. [21] found that single cytokine antibody treatment (e,g, TNF antibody or IL-1 receptor antibody) reduced thermal hyperalgesia at all time points, while mechanical hyperalgesia was reduced on days 5 and 7 but not on day 10. Schäfers et al. [21] also demonstrated that the combined treatment with both antibodies reduced mechanical hyperalgesia at all time points, suggesting additive or synergistic suppression of the cytokine response. We had similar results in which GFAP-IκBα-dn mice had reduced thermal hyperalgesia at all time points but mechanical hyperalgesia was not reduced significantly after day 7. It is important to note that the GFAP-IκBα-dn transgene is not present in neurons and other non-GFAP-expressing glial cells, which also regulate the production of cytokines and chemokines [1,10,31,35]. Therefore, incomplete suppression of inflammation in GFAP-IκBα-dn mice may explain why mechanical hyperalgesia was not attenuated at all time points after CCI in GFAP-IκBα-dn mice.
In this study, CCI induced gene upregulation of pro-inflammatory cytokines TNF, IL-1β, and chemokine ligands/receptors CCL2, CXCL10, and CCR2 in the ipsilateral sciatic nerve. Both myelinating and non-myelinating Schwann cells function as immune cells, which are involved in the sensory processing of neuropathic pain [5, 7-8, 31]. Schwann cells are also known to produce and regulate TNF, IL-1β, and CCL2 [5-6,17,22,27,28,31]. In addition, the upregulation of the chemokine ligand CCL2 and its receptor CCR2 has been shown to occur after peripheral nerve injury and transection [1,23,25,29]. We found that upregulation of CCL2, CCR2 and CXCL10 after CCI were reduced in GFAP-IκBα-dn mice. We also observed that CCI induced protein expression of CCL2, CXCL10, and CCR2, with greater immunoreactivity in WT mice compared to GFAP-IκBα-dn. These RT-PCR and immunohistochemistry findings in the sciatic nerve indicate that NF-κB inhibition in GFAP expressing glial cells attenuates production of these inflammatory mediators.
Interestingly, no difference in the upregulaton of TNF and IL-1β in sciatic nerve was present in WT and GFAP-IκBα-dn mice. Perhaps, myelinating Schwann cells are the predominant source for TNF and IL-1β in sciatic nerve after peripheral nerve injury. Our results were consistent with the findings by Brambilla et al. [3] who found no difference in spinal cord TNF production between WT and GFAP-IκBα-dn mice after contusion injury, but decreased production of CCL2 and CXCL10 in TG mice. Brambilla et al. [3] postulated that non-astrocytic cells may be the predominant source on TNF following spinal cord injury.
We found that in DRGs, CCI-induced upregulation of TNF, CCL2, and CCR2. Increased production of TNF and TNF receptor occurs in both neuronal and GFAP immunoreactive non-neuronal cells in DRGs [10,16,19,34]. Moreover, upregulation of CCL2 and CCR2 occurs after peripheral nerve injury in DRGs [1,24,35]. In this study, Iower expression of TNF, CCL2, and CCR2 in DRGs was present in transgenic mice, which may be attributed to selective glial NF-κB inhibition. Our TNF results in the DRGs were similar to the findings from Wei et al. [32], who demonstrated that non selective NF-κB inhibition prevented mechanical hyperalgesia and TNF upregulation in DRGs after peripheral nerve injury. While most studies suggest that production of CCL2 and CCR2 in DRG is primarily neuronal [1,29,35]. White et al. [33] did observe strong expression of CCL2 mRNA in non neuronal cells in DRGs following compression injury of the DRG. Our results of decreased gene expression of CCL2 and CCR2 in the DRGs of GFAP-IκBα-dn mice suggest that GFAP expressing cells in DRGs have a role in regulating CCL2 and CCR2.
In summary, the behavioral and biochemical findings presented in this study found the following: 1) presence of the GFAP-IκBα-dn transgene in sciatic nerve and DRGs; 2) inhibition of nuclear translocation of activated phospho-p65 in GFAP-IκBα-dn mice; 3) upregulation of GFAP in sciatic nerve and DRGs after peripheral nerve injury; 4) reduced mechanical and thermal hyperalgesia in GFAP-IκBα-dn mice after CCI; 5) increased expression of proinflammatory cytokines, chemokines, and chemokine receptors in sciatic nerve and DRGs during the first several days after CCI, and 6) decreased expression of inflammatory mediators in GFAP-IκBα-dn mice after CCI. Transgenic NF-κB inhibition in glial cells attenuates neuropathic pain by blunting the production of NF-κB-dependent mediators in the periphery. Therefore, targeting the inflammatory response in GFAP expressing non-myelinating Schwann cells and DRG satellite cells appears to be an effective and promising strategy in the treatment of neuropathic pain.
Figure 7.
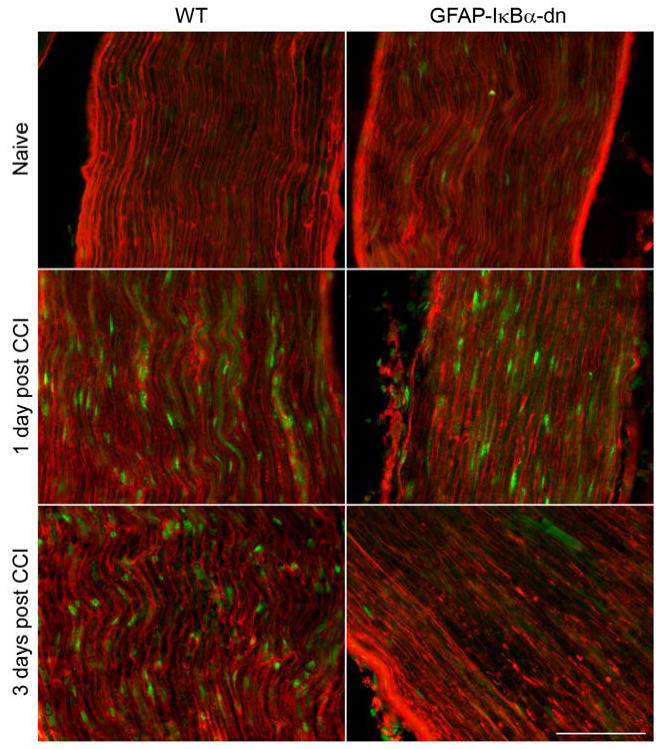
Immunostaining for CCR2 (green) and GFAP (red) in the ipsilateral sciatic nerve of WT and GFAP-IκBα-dn mice. CCI induced increased immunoreactivity for both GFAP and CCL2. On day 1 and day 3, greater CCR2 immunoreactivity was present in WT compared to GFAP-IκBα-dn mice (scale bar = 50 μm).
Acknowledgments
This work was supported by the National Institutes of Health Grants NS37130 and NS051709 to JRB
Footnotes
Conflict of interests statement: The authors state that no conflicting financial interests are in place.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abbadie C, Lindia JA, Cumiskey AM, Peterson JB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Nat Acad Sci. 2003;100:7947–52. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 3.Brambilla R, Bracchi-Richard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–56. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campana WM, Li X, Shubayev VI, Angert M, Cai K, Myers RR. Erythropoietin reduces Schwann cell TNF-α, Wallerian degeneration and pain-related behaviors after peripheral nerve injury. Eur J Neurosci. 2006;23:617–26. doi: 10.1111/j.1460-9568.2006.04606.x. [DOI] [PubMed] [Google Scholar]
- 5.Campana WM. Schwann cells: activated peripheral glia and their role in neuropathic pain. Brain Behav Immun. 2007;21:522–27. doi: 10.1016/j.bbi.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chattopadhyay S, Myers RR, Janes J, Shubayev V. Cytokine regulation of MMP-9 in peripheral glia: implications for pathological processes and pain in injured nerves. Brain Behav Immun. 2007;21:561–8. doi: 10.1016/j.bbi.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G. Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat Neurosci. 2003;11:1186–93. doi: 10.1038/nn1139. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Velardez MO, Warot X, Yu Zx, Miller SJ, Cros D, Corfas G. Neuroregulin 1-erbB signaling is necessary for normal myeline and senory function. J Neurosci. 2006;26:3079–86. doi: 10.1523/JNEUROSCI.3785-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dublin P, Hanani M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav Immun. 2007;27:592–8. doi: 10.1016/j.bbi.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 10.Dubovy P, Jancalek R, Klusakova I, Svizenska I, Pejcalova K. Intra- and extraneuronal changes of immunofluorescence staining for TNF-α and TNFR1 in the dorsal root ganglia of rat peripheral neuropathic pain models. Cell Mol Neurobiol. 2006;26:1205–17. doi: 10.1007/s10571-006-9006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu ES, Zhang YP, Sagen J, Yang ZQ, Bethea JR. Transgenic glial nuclear factor kappa B inhibition decreased formalin pain in mice. Neuroreport. 2007;18:713–7. doi: 10.1097/WNR.0b013e3280d9e869. [DOI] [PubMed] [Google Scholar]
- 12.Jessen KR, Morgan L, Stewart HJS, Mirsky R. Three markers of adult non-myelin-forming Schwann cells, 217c(Ran-), A5E3 and GFAP: development and regulation by neuron-Schwann cell interactions. Development. 1990;109:91–103. doi: 10.1242/dev.109.1.91. [DOI] [PubMed] [Google Scholar]
- 13.Kleinschnitz C, Brinkhoff J, Zelenka M, Sommer C, Stoll G. The extent of cytokine induction in peripheral nerve lesions depends on the mode of injury and NMDA receptor signaling. J Neuroimmunol. 2004;149:77–83. doi: 10.1016/j.jneuroim.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 14.Kleinschnitz C, Brinkhoff J, Sommer C, Stoll G. Contralateral cytokine gene induction after peripheral nerve lesions: Dependence on the mode of injury and NMDA receptor signaling. Mol Brain Res. 2005;136:23–8. doi: 10.1016/j.molbrainres.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 15.Ma W, Bisby MA. Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Br Res. 1998;797:243–54. doi: 10.1016/s0006-8993(98)00380-1. [DOI] [PubMed] [Google Scholar]
- 16.Miyagi M, Ohtori S, Ishikawa T, Aoki Y, Ozawa T, Doya H, Saito T, Moriya H, Takahashi K. Up-regulation of TNFα in DRG satellite cells following lumbar facet injury in rats. Eur Spine J. 2006;15:953–8. doi: 10.1007/s00586-005-1031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers RR, Campana WM, Shubayev VI. The role of neuroinflammation in neuropathic pain: mechanisms and therapeutic targets. Drug Discov Today. 2006;11:8–20. doi: 10.1016/S1359-6446(05)03637-8. [DOI] [PubMed] [Google Scholar]
- 18.Niederberger E, Geisslinger G. The IKK-NF-κB pathway: a source for novel molecular drug targets in pain therapy. FASEB. 2008;22:3433–42. doi: 10.1096/fj.08-109355. [DOI] [PubMed] [Google Scholar]
- 19.Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-α and TNF-α receptor type 1 upregulation in glia and neurons after peripheral nerve injury. Spine. 2004;29:1082–8. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- 20.O'Neill LAJ, Kaltschmidt C. NFκB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–8. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 21.Schäfers M, Brinkhoff J, Neukirchen S, Marziniak M, Sommer C. Combined epineural therapy with neutralizing antibodies to tumor necrosis factor-alpha and interleukin-1 receptor has an additive effect in reducing neuropathic pain in mice. Neurosci Lett. 2001;310:113–6. doi: 10.1016/s0304-3940(01)02077-8. [DOI] [PubMed] [Google Scholar]
- 22.Shamash S, Reichert F, Rotshenker S. The cytokine network of Wallerian degeneration: tumor necrosis factor-α, interleukin-1α, and interleukin-1β. J Neurosci. 2002;22:3052–60. doi: 10.1523/JNEUROSCI.22-08-03052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subang MC, Richardson PM. Influence of injury and cytokines on synthesis of monocyte chemoattractant protein-1 mRNA in peripheral nervous tissue. Eur J Neurosci. 2001;13:521–8. doi: 10.1046/j.1460-9568.2001.01425.x. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–9. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Taskinen HS, Röyttä M. Increased expression of chemokines (MCP-1, MIP-1α, RANTES) after peripheral nerve transection. J Peripher Nerv Syst. 2000;5:75–81. doi: 10.1046/j.1529-8027.2000.00009.x. [DOI] [PubMed] [Google Scholar]
- 26.Tegeder I, Niederberger E, Schmidt R, Kunz S, Gühring H, Ritzeler O, Michaelis M, Geisslinger G. Specific inhibition of IκB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J Neurosci. 2004;24:1637–45. doi: 10.1523/JNEUROSCI.3118-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thacker MA, Clark AK, Marchand F, McMahon SB. Pathophysiology of peripheral neuropathic pain; immune cells and molecules. Anesth Analg. 2007;105:838–847. doi: 10.1213/01.ane.0000275190.42912.37. [DOI] [PubMed] [Google Scholar]
- 28.Tofaris GK, Patterson PH, Jessen KR, Mirsky R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J Neurosci. 2002;22:6696–6703. doi: 10.1523/JNEUROSCI.22-15-06696.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toews AD, Barrett C, Morell P. Monocyte chemoattractant protein 1 is responsible for macrophage recruitment following injury to sciatic nerve. J Neurosci Res. 1998;53:260–267. doi: 10.1002/(SICI)1097-4547(19980715)53:2<260::AID-JNR15>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 30.Triolo D, Dina G, Lorenzetti I, Malaguti M, Morana P, Del Carro U, Comi G, Messing A, Quattrini A, Previtali SC. Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration. J Cell Sci. 2006;119:3981–93. doi: 10.1242/jcs.03168. [DOI] [PubMed] [Google Scholar]
- 31.Wagner R, Myers RR. Schwann cells produce tumor necrosis factor alpha: expression in injured and non-injured nerves. Neurosci. 1996;73:625–9. doi: 10.1016/0306-4522(96)00127-3. [DOI] [PubMed] [Google Scholar]
- 32.Wei XH, Zang Y, Wu CY, Xu JT, Xin WJ, Liu XG. Peri-sciatic administration of recombinant rat TNF-alpha induces mechanical allodynia vai upregulation of TNF-alpha in dorsal root ganglion and in spinal dorsal horn: the role of NF-kappa B pathway. Exp Neurol. 2007;205:471–84. doi: 10.1016/j.expneurol.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 33.White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Stefik J, Cortright DN, LaMotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is upregulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Nat Acad Sci. 2005;102:14092–7. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woodham P, Anderson PN, Nadim W, Turmaine M. Satellite cells surrounding axotomised rat dorsal horn ganglion cells increase expression of a GFAP-like protein. Neurosci Lett. 1989;98:8–12. doi: 10.1016/0304-3940(89)90364-9. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–83. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]