

Abstract
Background
The inositol glycans (IGs) are glycolipid-derived carbohydrates produced by insulin-sensitive cells in response to insulin treatment. IGs exhibit an array of insulin-like activities including stimulation of lipogenesis, glucose transport and glycogen synthesis, suggesting that they may be involved in insulin signal transduction. However, because the natural IGs are structurally heterogeneous and difficult to purify to homogeneity, an understanding of the relationship between structure and biological activity has relied principally on synthetic IGs of defined structure.
Discussion
This article briefly describes what is known about the role of IGs in signal transduction and reviews the specific biological activities of the structurally defined IGs synthesized and tested to date.
Conclusion
A pharmacophore for IG activity begins to emerge from the reviewed data and the structural elements necessary for activity are summarized.
Historical perspective
The inositol glycans (IGs) are a class of naturally occurring, phosphorylated, inositol-containing pseudosaccharides first identified in 1986 from bovine liver treated with insulin [1]. The finding that isolated natural IGs, released in response to insulin, are competent in stimulating insulin-sensitive cells in the absence of insulin led to the hypothesis that IGs are the long-elusive second messengers for insulin action, as implied by earlier experiments [2]. However, it was quickly realized that the IG preparations obtained from natural sources were mixtures of closely related carbohydrates, and this microheterogeneity severely complicated the elucidation of the role of IGs in insulin signal transduction.
Using a combination of metabolic labeling and degradation studies, the basic structural features of the natural IGs were identified. All of the IGs contain a terminal inositol, but both chiro [3] and myo-inositol [1] isomers were found. Similarly, the second sugar is invariably an unacylated aminosugar, but both galactosamine [3] and glucosamine [1] were identified. The remaining structure of the glycan was less well established, but the presence of a number of mannose residues and one or more phosphate residues was reported. Given this information and the approximate molecular weight of 1400 Da [1], it became clear that the IGs are very closely related to the glycosylphosphatidylinositol (GPI) membrane anchors that many cells use to tether cell surface proteins to the plasma membrane and some of whose structures have been unequivocally identified. The close structural relationship between IGs and GPIs was confirmed by the ability of phosphatidylinositol-specific phospholipase C (PI-PLC) to release IGs from the cell surface [1] as well as by the observation that the trypanosome-derived variant surface glycoprotein GPI anchor, cleaved from the lipid with PI-PLC and from the protein with pronase, has significant insulin-like activity in rat hepatocytes and adipocytes [4,5].
The similarity between the partially known structures of the IGs and the known structures of the GPIs suggested a way of unraveling the relationship between IG structure and biological activity. The established GPI structures could be used as a template for the design and chemical synthesis of structurally defined IG analogues that could then be subjected to biological evaluation. The chemical synthesis of a bioactive IG analogue was first accomplished in 1992, when a simple pseudodisaccharide – comprising the glucosamine and myo-inositol terminus of the GPI anchor structure, but terminated by a cyclic phosphate as would be produced from PI-PLC hydrolysis of a glycolipid – was prepared and found to be able to mimic one of the actions of insulin: stimulation of lipogenesis in rat adipocytes [6]. This led to a burst of synthetic activity and over the next 16 years, various research groups synthesized a large number of IG analogues and evaluated them for biological activity. This has provided an extensive collection of data from which a pharmacophore for insulin-like activity is emerging.
This article reviews the published information on the relationship between IG structure and biological activity. The review covers only the compounds for which a fully defined structure and biological activity have been reported. Therefore, neither synthetic IGs that have not been tested in any bioassays nor natural IGs whose structures are not completely elucidated are included here. Earlier publications have reviewed the role of the natural IGs in signal transduction [7-13] and the chemical synthesis of the IGs [14], but this is the first comprehensive survey of the biological activity of structurally defined IGs.
Overview of insulin signaling
Type 2 diabetes mellitus is characterized by insulin resistance in the target tissues, effectively a failure in insulin signal transduction. Since the IGs are able to stimulate many insulin-like effects, but do not act via the insulin receptor (IR) they (or appropriate analogues) may be useful for the treatment of Type 2 diabetes. Any attempt to understand the role of IGs in producing insulin-like effects first requires a basic understanding of the molecular machinery of insulin signaling. A lot of information on the subject of insulin signal transduction has been published and several aspects have been reviewed elsewhere [15-18]. Insulin has two basic effects, mitogenesis and stimulation of anabolic metabolism. In this section we provide a brief and somewhat simplified overview of what is currently known about the stimulation of anabolism, including glucose transport, glycogen synthesis and lipogenesis. Each step in the process, described below, corresponds to a number in Figure 1:
Figure 1. Insulin signal transduction.

Inactive proteins are indicated by a paler color. Protein shape and color depend on the protein function: kinases (and insulin receptor substrates) are green and rectangular with rounded edges. Regulatory domains (e.g., the p85 domain of PI3 kinase) and adaptor proteins are light blue and oval. Phosphatases are dark blue and rectangular. Lyases and synthases are brown. Blue arrows denote a chemical transformation (e.g., PIP2→PIP3) as well as movement of proteins. Black arrows point to the protein target. A solid arrow indicates that the reaction has been established. Dashed arrows refer to pathways that are not yet well established.
(1) Insulin (Ins) delivers its signal by binding to the IR, a cell-surface receptor tyrosine kinase. A complex consisting of the IR and insulin is formed, leading to conformational changes of the IR. (2) The two intracellular domains of the IR, possessing kinase activity, phosphory-late each other. This leads to increased kinase activity as well as affinity toward other IR substrates. (3) Adaptor proteins (IRS-1, IRS-2) bind to the phosphorylated receptor and then (4) their tyrosine residues are phosphorylated by the IR kinase activity. IRS proteins are localized to the plasma membrane during this event, perhaps through the interaction with phosphatidylinositol-4,5-diphosphate [19]. (5) The p85 regulatory domain of phosphatidylinositol 3 kinase (PI3K) docks with the phosphorylated sites of IRS1/2. (6) This leads to phosphorylation of the lipid phosphatidylinositol-4,5-diphosphate and generation of phosphatidylinositol-3,4,5-triphosphate (PIP3). An elevated concentration of PIP3 leads to a cascade of protein phosphorylations. (7) First, binding of phosphoinositide-dependent kinase 1 (PDK1) to PIP3 occurs. (8) Then PDK1 phosphorylates and activates protein kinase B (PKB), also referred to as Akt. In fact, two kinases are required for Akt activation and the nature of the second one was unknown for a long time; however, the putative kinase was designated PDK2. Recently, the mTOR (mammalian target of rapamycin)–RICTOR (rapamycin-insensitive companion of mTOR) protein complex has been suggested as PDK2 [20]. Akt is believed to be responsible for the regulation of various metabolic pathways via phosphorylations. For example, (9) it catalyzes phosphorylation of glycogen synthase kinase (GSK)-3 that (10) leads to deactivation of GSK-3 and as a result (11) prevents the inactivation of glycogen synthase (GS) by phosphorylation, and hence increases glycogen synthesis [21]. Moreover, PIP3 activates protein phosphatase 1 (PP1) localized on glycogen particles by an unknown mechanism [18], and (13) PP1 dephosphorylates inactive GS leading to (12) active GS and, therefore, to a further increase in glycogen synthesis.
Likewise, (14) deactivation of GSK-3 leads to a decreased level of phosphorylation of ATP citrate lyase (ACL) at one site. (15) Phosphorylation at a different site on ACL by PKB (16) increases the activity of that enzyme [22]. ACL catalyzes conversion of citrate to acetyl-CoA in cytosol where it can be utilized in lipid biosynthesis. Citrate is actively transported from the mitochondria, where it is prepared from pyruvate (in two steps). Pyruvate in turn is a product of glycolysis. This path, therefore represents lipid biosynthesis from glucose.
In addition to activation of glucose and lipid metabolism, insulin greatly stimulates glucose uptake: the GLUT-4 glucose transporter is translocated from intracellular vesicles to the cell membrane. There are at least two signaling pathways that produce this result, but neither has been fully elucidated. In the first, PKB stimulates GLUT-4 exocytosis either (17) directly or (18) via activation of atypical protein kinases C: PKC-ζ and -γ (designated as aPKC in Figure 1). Several possible molecular regulators downstream of PKB and aPKC are under consideration [23]. The second pathway of GLUT-4 translocation starts right downstream of the IR [24]. Another IR substrate referred to as Cbl (existing in complex with Cbl-associated protein [CAP]) is (21) phosphorylated by IR kinase, however (19) binding to IR and (20) phosphorylation of adaptor protein APS is required before Cbl-CAP association with IR and APS and subsequent phosphorylation. Phosphorylated Cbl requires (22) adaptor protein CrkII together with guanine nucleotide exchange factor C3G. C3G (23) activates the TC10 protein, which belongs to the Rho family of proteins. TC10 (24) activates GLUT-4 vesicle translocation via an unknown mechanism, however it is hypothesized that this happens via actin cytoskeleton regulation – a function attributed to many of the Rho proteins. It is also believed that the two GLUT-4 translocation mechanisms described regulate different states of GLUT-4 transport and are not independent from each other.
Insulin mediates two more metabolic events through the inhibition of protein kinase A (PKA). The processes begin when (26) phosphodiesterase 3B (PDE3B) is activated at endoplasmic reticulum/Golgi [25]. The exact mechanism of the activation is not known, however (25) PI3K might be indirectly involved (perhaps through PKB) [26,27]. Activated PDE3B (27) hydrolyzes cAMP. Low cAMP level (28) reduces the activity of PKA since cAMP activates PKA by binding to its regulatory domain. As a result (29) PKA does not phosphorylate hormone sensitive lipase (HSL) and HSL is not converted into active form. Lipolysis is, therefore, decreased.
Similarly, (30) inactive PKA does not phosphorylate fructose 2,6-bisphosphatase (Fru-2,6P2ase), thus failing to convert it into active form. Inactive Fru-2,6P2ase (31) cannot catalyze conversion of fructose-2,6-biphosphate (Fru-2,6P2) into fructose-6-phosphate (Fru-6P). Hence, Fru-2,6P2 accumulates and (32) inhibits fructose 1,6-bisphosphatase (Fru-1,6P2ase) – an enzyme that is involved in gluconeogenesis [28].
Another observation should be noted. At least some of the IRs are found in caveolae – small cave-like structures on the cell membrane. They represent a class of so-called lipid rafts or detergent/carbonate-insoluble glycolipid-enriched (DIG) microdomains. Like lipid rafts, the caveolae are rich in glycosphingolipids and cholesterol. Some proteins may contribute to DIG compositions; for example the IR is found to be associated with the protein caveolin (not shown in Figure 1), which is abundant in caveolae. If the caveolin-1 gene is disrupted, cells become insulin resistant.
Other molecular bases of insulin resistance have been found. It was shown that mutation of several proteins involved in insulin signal transduction, particularly in IRS-2 and the β isoform of PKB, cause the onset of diabetes or impair glucose metabolism and (or) insulin sensitivity [29]. An excess of adipose tissue may also cause diabetes and there is a correlation between Type 2 diabetes and obesity. So-called visceral adipocytes that are found in these patients are relatively resistant to antilipolytic insulin signaling and as a result they secrete a lot of nonesterified fatty acids (NEFA) into the bloodstream. NEFAs can block insulin signaling in other cells via several possible mechanisms or they can impair glucose metabolism in liver [16].
Mechanism of action of inositol glycans
How do IGs stimulate insulin-like effects in cells? Do IGs crosstalk with the insulin-signaling pathway? Are extracellularly-produced IGs transported into cells? The answer to this last question appears to be yes, at least in hepatocytes. The existence of an IG transporter is implied by the observation that a metabolically radiolabeled IG is transported into rat hepatocytes through an energy-requiring process [30], but whether this is due to a specific IG transporter or the normal detoxifying function of hepatocytes is unknown. However, both natural and synthetic IGs are able to modulate purified pyruvate dehydrogenase phosphatase [31,32] and pyruvate dehydrogenase kinase (PDK) [32] activities in cell free assays, suggesting not only an intracellular role for IGs, but also an intramitochondrial role.
Interestingly, IGs also appear to be able to stimulate cells by interacting with the exterior surface of the membranes. For example, a fluorescently-labeled synthetic IG analogue was found to stimulate lipogenesis in rat adipocytes despite the fact that it was not able to enter the cell [33]. Müller has suggested that the dynamics of plasma membrane microdomains may be a part of the insulin-signaling pathway and has collected considerable evidence suggesting that IGs act via this mechanism [34,35]. Müller’s theory is summarized in Figure 2 and the numbered description below.
Figure 2. Mechanism of inositol glycan (inositolphosphate glycan) action on cell surfaces.

GPI: Glycosylphosphatidylinositol; IPG: Inositolphosphate glycan.
There is evidence that DIG microdomains are heterogeneous: there are cholesterol-rich (hcDIGs) and cholesterol-depleted micro-domains (lcDIGs). Major fractions of certain GPI anchored proteins exist at hcDIG. Other lipid-modified signaling proteins (fatty acid-anchored or prenylated) may exist preferentially in different DIG areas depending on the structure of their lipid anchor. Another targeting signal for proteins that defines their location at plasma membrane microdomains is association with caveolin or other hcDIG proteins. Moreover, association of caveolin with a signaling protein (e.g., the kinase pp59Lyn) seems to be a way of regulating the latter’s activity. It was found that pp59Lyn in its inactive state resides in hcDIG bound to the caveolin scaffolding domain (CSD) of caveolin through caveolin binding domains (CBDs). One explanation for the loss of activity due to caveolin binding is that the interaction with caveolin may cause some conformation change in pp59Lyn, leading to its inactivation. On the other hand, this binding may keep the kinase away from its substrates. However, it was shown that CSDP directly interacts with CBD, blocking pp59Lyn activity, and therefore supporting the first hypothesis.
Receptors for the GPI epitope of natural GPI-anchored proteins (as well as probably for free GPIs) exist in hcDIG microdomains. It was shown that GPI-anchored proteins are freely distributed with some preference to lcDIG in membrane models. Accordingly, it is the binding to the receptor that favors accumulation at hcDIG. The nature of the receptor is not well established other than that it is a protein with a molecular weight of 115 kDa.
Müller’s hypothesis, illustrated in Figure 2, is that (1) insulin stimulates a glycosylphosphatidylinositol-specific phospholipase C (GPI-PLC) that (2) lipolytically cleaves GPI-anchored proteins (and, perhaps free GPIs) on the outer leaflet of the cell membrane. (3) The lipolytically cleaved protein–IG conjugate (and IGs) promotes GPI-anchored protein(s) to (4) dissociate from the receptor; GPI-anchored proteins are found to translocate to lcDIG microdomains after insulin treatment. (5) Redistribution of acylated non-receptor tyrosine kinases (NRTKs) such as pp59Lyn on the inner leaflet of the plasma membrane between hcDIGs and lcDIGs is also observed. (6) Kinase pp59Lyn activates another kinase pp125Fak that is able to (7) recruit IRS proteins for (8) phosphorylation by pp59Lyn at the sites that are recognized by PI(3)K. This event, which was discussed above, turns on the phosphorylation cascade and is a point of convergence between the IG mechanism of action and the insulin-signaling pathway described above.
There are several explanations for the fact that GPI-anchored proteins on the exterior leaflet and acylated proteins on the cytoplasmic side move at the same time, namely because there is communication between the leaflets within the microdomain [34]. The preference of GPI-anchored proteins for lcDIG could be considered a driving force. The lipid part of a migrating GPI may bring some other components of hcDIG (perhaps, cholesterol) with it, thus changing hcDIG composition on the cytoplasmic side. This in turn may somehow cause a protein–protein interaction-mediated signal to caveolin leading to its conformational change and to a decreased affinity toward pp59Lyn. Another possibility is that it is a change in the lipid composition of the inner leaflet that must occur after lipid rearrangement on the outer leaflet that drives pp59Lyn away.
Description of biological assays performed on structurally defined inositol glycans
Almost two dozen distinct assays or measurements have been used to probe how IG structure affects the molecule’s ability to interact with cells. To help the reader connect the mechanisms of action described above with Tables 1-7 correlating IG structure to specific activity in published assays, we have compiled a list of the assays with a brief description of each. The assays are listed in alphabetical order and, where appropriate, the step in Figure 1 or Figure 2 that is being probed is noted. Finally, the specific structurally defined IG(s) tested in the assay are listed.
cAMP-dependent protein kinase activity [36,37] (Figure 1, step 28, PKA): IG-41, 47. Rat adipocytes are prepared by collagenase digestion of epididymal fat pads. Approximately 20 μl of adipocyte suspension is mixed with 80 μl of solution containing 20 mM 3-(N-morpholino)propanesulfonic acid (pH 7.2) 4-mM DTT, 12.5-mM MgCl2, 0.2-mM PMSF, 1 mM isobutylmethylxanthine, 50-μM [32P]-ATP (2.5 μCi) with or without 1-μM cAMP. The resulting mixture is incubated at 30 °C for 10 min followed by the addition of 1 ml of ice-cold solution containing 20% trichloroacetic acid, 10 mM ATP, 5 mM NaPPi and 100 mM NaF. The resulting precipitates are separated by centrifugation and dissolved in 100 μl of 1 M NaOH. The precipitate is restored by the addition of 1 ml of a solution containing 20% trichloroacetic acid, 10-mM ATP and 5-mM NaPPi. The precipitate is filtered over a Whatman GF/C glass fiber filter, washed several times with washing solution (20% trichloroacetic acid, and 5-mM NaPPi) dried under vacuum and then counted for radioactivity. Activity of PKA is assayed by the incorporation of 32P from [32P]-ATP into histone H1.
cAMP phosphodiesterase activity (Figure 1, step 27, PDE3B) [1]: IG-2, -4, -9 and -10. The activity of cAMP phosphodiesterase, obtained from rat adipocytes, is measured at 30 °C by the method of Thompson and Appleman [38]. Membranes are incubated with Tris-HCl at pH 7.4, MgCl2, dithiothreitol and [2,8-32 H]-cAMP. The activity of the enzyme is measured by the conversion of [3H]-cAMP to [3H]-AMP and normalized to the protein concentration.
Cell proliferation and Fos oncoprotein expression in chicken embryo otic vesicles [39]: IG-2. fresh otic vesicles obtained from stage 18 chicken embryos [40] and the desired analyte: IGF-I, synthetic IG or no addition (control) are incubated at 37 °C in a water-saturated atmosphere containing 5% CO2 for 24 h. The vesicles are incubated in the absence of serum for 24 h. This makes them inactive or dormant. Cell proliferation is then estimated by scintillation counting of the incorporated [3H]-thymidine into trichloroacetic acid precipitated explants [40]. The extent of incorporation of Fos protein in these explants is measured using Western blotting analysis.
Fructose-1,6-bisphosphatase activity (Figure 1, step 32, Fru-1,6P2ase) [4]: IG-42. Fructose-1,6-bisphosphatase catalyzes the hydrolysis of fructose-1,6-bisphosphate to Fru-6P. IGs inhibit the activity of fructose-1,6-bisphosphatase. The assay for the activity of this enzyme is carried out with the cytosolic fractions of rat livers [41]. These fractions are incubated in 50 mM TRIS-HCl (pH 7.4) containing 5 mM MgCl2, 0.1 mM EDTA, 150 mM KCI, 0.1 mM NADP+, 0.8 unit of phosphoglucose isomerase, and 0.8 unit of glucose-6-phosphate dehydrogenase, at 25 °C for 10 min. To assay the activity of the enzyme, 1 mM fructose-1,6-bisphosphate is added to the sample of incubated cytosol and the resulting solution is analyzed spectrophotometrically for approximately 12 min. Increase in the absorbance (340 nm) indicates the rate of conversion of NADP+ to NADPH, which in turn indicates the activity of fructose-1,6-bisphosphatase.
Gluconeogenesis (Figure 1) [4]: IG-2, -4, -9, -10 and -42. Gluconeogenesis is determined by the conversion of [14C]-pyruvate into [14C]-glucose. The assay is carried out on male Sprague–Dawley rat hepatocytes isolated by collagenase digestion [42]. The hepatocytes are suspended in Hanks’ buffer containing 2% bovine serum albumin with the desired IG. The solution is incubated in a shaking water bath at 37 °C for 1h. [14C]-pyruvate (4 μCi/ml) is added and the incubation is continued for 40 min. The solution is centrifuged (12,000βg for 5 min) to remove the hepatocytes and the supernatant is deproteinized by the addition of ZnSO4 and Ba(OH)2. The solution is centrifuged again for 5 min and the precipitate is removed. The supernatant is eluted through a trilayered column consisting of 1 ml of Dowex-1 anion exchange resin (100 mesh) 1 ml of Amberlite XAD-2, and 1 ml of Dowex-1 anion exchange resin (100 mesh). [14C]-glucose production is quantified by scintillation counting.
Glycogen synthesis in insulin-sensitive hepatoma H4IIE cells (Figure 1) [43]: IG-3. To assay the incorporation of [14C]-glucose into [14C]-glycogen, H4IIE hepatoma cells are grown in HAM’s F-12/DMEM (1:1) with 10% fetal bovine serum containing 5-mM glucose. Before the assay, the cells are washed twice with PBS and incubated with [14C] glucose (0.2 μCi) along with unlabeled glucose for 60 min at 37 °C. After the incubation the cells are washed twice with PBS and incubated with 30% KOH for 60 min at 37 °C. The solution is heated to 100 °C and glycogen is precipitated using ethanol. Radioactivity of the resulting glycogen is measured using a scintillation counter.
Glucose concentration; decrease in hyperglycemia in STZ diabetic rats [43]: IG-3. Type 2 diabetes is induced in male Sprague–Dawley rats by injecting low-dose streptozotocin (50 mg/kg) intravenously into the tail vein. As diabetes is induced, the blood glucose level increases to approximately 250−400 mg/100 ml. These diabetic rats are anesthetized with ketamine (2-[2-chlorophenyl]-2-[methylamino] cyclohexanone). Their tails are clipped off and the blood glucose level is determined. This corresponds to the zero time blood glucose level reading. Next the synthetic IG diluted with the saline, or the saline alone (control) is injected intravenously into the tail veins and the corresponding blood glucose level is recorded using a glucometer at different time intervals. A decrease in the blood glucose level with time indicates decrease in hyperglycemia.
Glucose-6-phosphatase activity [4]: IG-42. Glucose-6-phosphatase catalyses the hydrolysis of glucose-6-phosphate to glucose and inorganic phosphate. Microsomal fractions of rat liver are isolated [41] and suspended in 50 mM TRIS-HCl (pH 7.4) that contains Triton X-100 0.1% with some additive. The solution is incubated at 25 °C for 10 min followed by the addition of 50 mM glucose-6-phosphate solution in 0.2 M imidazole (pH 6.5). The resulting solution is further incubated at 37 °C for 20 min. The reaction is stopped by the addition of trichloroacetic acid 10%. Any precipitates are removed by centrifugation and the supernatant is assayed for the presence of inorganic phosphate [44].
Glucose transport (Figure 1) [37,45,46]: IG-32, -47, -74 and -78. Glucose transport in adipocytes is measured as the uptake of 2-deoxy-[3H]-glucose (0.5-μCi, 100-μM final concentration). Adipocytes in KRH (1 ml, 3 × 105 cells) are incubated with different concentrations of insulin or IG in 15-ml polystyrene tubes for 20 min at 37 °C. KRH (50 μl) containing 0.5-μCi 2-deoxy-[3H]-glucose is added and the solution is incubated for another 5 min at room temperature. The reaction is stopped by the addition of cytochalasin B followed by immediate separation of 100-μl aliquots of the cells by centrifugation through dinonylphthalate oil. Radioactivity is measured to give the initial rates of glucose transport. Glucose uptake by diffusion and trapping of glucose in the intercellular spaces of the packed adipocytes is corrected by preincubating the cells with cytochalasin B before addition of the labeled glucose.
Glucose transporter isoform 4 translocation (Figure 1, step 24, GLUT-4) [37,45]: IG-32, -47, -74 and -78. Approximately 7 × 106 adipocytes are incubated with KRH buffer containing glucose and synthetic IG. The solution is shaken for 20 min at 37 °C. The cells are washed with KRH containing 2-mM KCN and are suspended in 20-mM Tris/HCl buffer (pH 7.4) containing 250-mM sucrose, 1-mM EDTA, 0.2 mM PMSF, 100-μM benzamidine and 20 μg/ml each of leupeptin, pepstatin, aprotinin and antipain. This solution is homogenized ten times in a Teflon-in-glass homogenizer. Plasma membranes and low-density microsomes are isolated and the GLUT4 is quantitatively immunoprecipitated using rabbit anti-GLUT4 antibodies raised against the carboxyl terminal 16 amino acids of rat GLUT4 and protein A Sepharose. The immunoprecipitated GLUT4 is separated by SDS-PAGE and its amount evaluated by immunoblotting with the same antibodies and [125I] protein A followed by autoradiography of the dried gel (Kodak X–Omat AR). Quantitation is performed by phosphorimaging of the dried gel.
Glycerol-3-phosphate acyltransferase (GPAT) activity [37,45]: IG-32, -47, -74 and -78. GPAT activity is assayed as incorporation of [3H]-glycerol-3-phosphate into butanol-extractable lipids [37,47]. The crude microsomal cell extracts can be prepared as described [47]. The resulting pellet is suspended in 25-mM Tris/HCl (pH 7.4) 250-mM sucrose, 0.5-mM EDTA, 1-mM DTT, 0.1-mM PMSF at 2-mg protein/ml and stored in liquid nitrogen until determination of GPAT activity. The assay mixture containing 0.2 mM [3H]-glycerol-3-phosphate (0.5-μCi), 150-μM palmitoyl-CoA and 100-mg microsomal protein in a total volume of 0.5 ml is incubated for 3 min at 37 °C. The incubation is stopped by addition of water-saturated butanol, followed by butanol-saturated water. The butanol phase is separated and measured for radioactivity.
Glycogen synthesis in hemidiaphragms (Figure 1) [37]: IG-32, -47, -74 and -78. Hemi-diaphragms from male Wistar rats are incubated in DMEM (15 ml/hemidiaphragm) containing 10% FCS, 5.5-mM glucose, 1% BSA, 50 U penicillin/ml, 50-μg streptomycin sulfate/ml under constant bubbling of O2/CO2 (95:5) for 60 min at 37 °C. For the assay, these diaphragms are incubated with insulin or the synthetic IG in DMEM (15 ml/hemidiaphragm) for 60 min at 37 °C and then washed with the buffer. Washed hemidiaphragms are incubated with [U-14C]-glucose in a solution of KRH containing 2 mM glucose for 20 min at 37 °C. After washing and homogenization of the frozen hemidiaphragms, the resulting supernatant is precipitated with ethanol. The extent of glycogen synthesis is assayed by measuring the incorporation of [U-14C]-glucose into ethanol-precipitable glycogen.
Glycogen synthase (Figure 1, step 12, GS) [37,48]: IG-47. Rat adipocytes are incubated with insulin or IG under 5% CO2 for 30 min at 37 °C. These cells are washed with a buffer and are frozen in liquid N2 followed by homogenization and centrifugation. The resulting infranatant solution is used as a source of GS. This homogenate is incubated with a solution containing 33-mM Tris/HCl (pH 7.8), 0.2-mM [14C]-UDP-glucose (4 μCi) glycogen, KF and glucose-6-phosphate for 20 min at 30 °C. The reaction is terminated by adding 66% ethanol and 10 mM LiBr followed by filtration over pre-wetted Whatman GF/C glass-fiber discs. These filters are washed with 66% ethanol at 25 °C, dried and are measured for radioactivity. The GS activity is assayed by measuring the incorporation of radioactivity from [14C ]-UDP-glucose into glycogen.
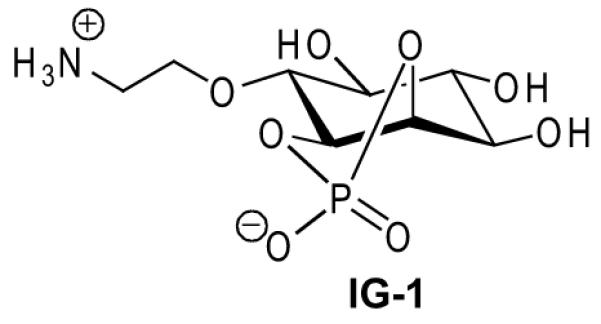
Lipogenesis (Figure 1, Lipid Synthesis) [6,33,36,45,49-51]: IG-1, -2, -4, -5, -7–10, -29–33, -35–41 and -43–83. Rat adipocytes are prepared by collagenase digestion of epididymal fat pads and incubated with radioactive [3-3H/6-3H/U-14C]-glucose along with the analyte for 1.5 h at 37 °C according to the method of Rodbell [52]. The assay is stopped by addition of water and a toluene-based scintillation cocktail. The solution is vortexed and centrifuged followed by scintillation counting to measure the incorporation of radioactive glucose into the toluene-extractable lipids (triglycerides). Incorporation of 3H or 14C into lipids is expressed as a percent of the maximal insulin response (%MIR).
Lipolysis (Figure 1) [4,37]: IG-32, -42, -47, -74 and -78. A suspension of isolated adipocytes, prepared by collagenase digestion of epididymal fat pads of male Sprague–Dawley rats, in Krebs-Ringer HEPES buffer (pH 7.4) is pre-incubated with insulin or inositol phosphate-glycan analogue at 37 °C for 30 min under an atmosphere of 5% CO2 followed by the addition of 50 nM–1-μM isoproterenol and 1 unit of adenosine deaminase. The incubation is continued for 30–60 min and the assay is finally terminated by the addition of 50 μl of 4.6 M perchloric acid. Samples are vortexed, diluted with chloroform and centrifuged at 12,000βg for 5–15 min. The aqueous layer is separated and neutralized with 5 N KOH and then assayed for glycerol content [53].
Other biophysical/chemical studies. Conformational studies [39]: IG-2. To understand the basic structural features and dynamic properties of these compounds, several conformational studies were carried out using NMR spectroscopy, molecular mechanics and molecular dynamics calculations; optical studies [33]: IG-5. Fluorescence microscopy and fluorescence-assisted cell sorting are utilized to determine whether fluorescent IG-5 is transported to the interior of living adipocytes.
PI3K activity (Figure 1, step 6) [54]: IG-47. IRS-1 immunoprecipitates to which phosphatidylinositol (0.2-mg/ml) is added are incubated in kinase buffer for 5 min at room temperature. [32P]-ATP (40 μM, 0.1 μCi/μl) is added, the reaction is run for 20 min, and then terminated by the addition of 30 μl of 4 M HCl and 130 μl of chloroform/methanol (1:1, v/v). The organic phase is spotted on a silica gel TLC plate, visualized and analyzed using a bioimaging analyzer.
Protein phosphorylation/dephosphorylation (Figure 1) [5]: IG-42. Rat adipocytes are prepared by collagenase digestion of epididymal fat pads and incubated with [32P]-orthophosphate at 37 °C for 2 h with constant shaking. These adipocytes are further incubated with 50-nM isoproterenol (IPT) insulin, or the desired IG at 37 °C for 20 min. The assay is stopped by using poly(dimethylsiloxane) and the cells are lysed using a solution of ethanol containing sodium dodecyl sulfate 4% (SDS) 1.5-mM EDTA, 10-μg/ml leupeptin, 10-μg/ml pepstatin, and 100-μM phenylmethylsulfonylfluoride. The resulting solution is boiled in sample buffer, vortexed and centrifuged at 12,000βg for 5 min. Electrophoresis (with 8% SDS-polyacrylamide gels) on the supernatant solution gives the extent of phosphorylated protein.
In vitro phosphorylation/dephosphorylation is measured by incubation of frozen adipocyte cytosols with 1-μM (γ-32P)-ATP for 10 min, followed by 100-μM unlabeled ATP and analyte for various times and SDS-PAGE, as above. Under these conditions changes in the observed phosphorylation state is caused by the direct dephosphorylation reaction.
Pyruvate dehydrogenase kinase inhibition [32]: IG-2, -12–24, -26, -27 and -34. PDK is found to be tightly associated with the PDC complex. It is not isolated as free PDK. To determine the activity of PDK, the ATP-dependent reduction in activity of PDC is measured. First, the residual activity of nonphosphorylated (active) PDC is measured spectrophotometrically [55]. A 10-μl sample of activated pyruvate dehydrogenase complex (PDC) (10 units/ml) is incubated in 10-μl buffer solution pH 7.0 and 80 μl of inactivation mixture (containing 1.25-mM ATP) at 30 °C. 50 μl of this incubated mixture is transferred to pre-prepared Eppendorf tubes containing 10 μl of the desired IG analogue, followed by further incubation for 10 min at 30 °C. PDK activity is inferred from the rate of inactivation of PDC, measured spectrophotometrically.
Pyruvate dehydrogenase phosphatase (PDH phosphatase) activity [36,43]: IG-3 and -41. IGs stimulate PDH phosphatase by enhancing its sensitivity to Mg2+ [56]. PDH phosphatase activity can be assayed in several ways: by monitoring the release of phosphate from 32P-labeled PDC [57], by measuring the conversion of β-NAD+ spectrophotometrically [58], or by measuring the release of [14C]-CO2 from pyruvic acid [56]. The assay for PDH phosphatase is based upon the initial rates of activation of inactivated, phosphorylated PDC complex. This assay is done in two phases; the first stage consists of dephosphorylation of PDC by the IG-activated phosphatase and the second stage involves the quantification of the activated PDC. The inactivated PDC complex prepared from bovine heart [59] is preincubated at 30 °C for 2–3 min with a solution containing 1-mg/ml fat-free bovine serum albumin, 10-mM MgCl2, 0.1-mM CaCl2, 1-mM dithiothreitol in 20-mM potassium phosphate buffer, pH 7. After 3 min 5 μl of metal, 10 μl of IG and 10 μl of PDH phosphatase are added and the solution is further incubated for 3 min. At this stage, the addition of NaF and [1–14C]-pyruvic acid inhibits the phosphatase and initiates the second stage of the assay, respectively. The second stage involves the quantitation of the PDC complex by measuring the rate of production of the reduced form by using absorbance data at 340 nm for approximately 5 min or by measuring the radioactivity of the released [14C]-CO2 with a liquid scintillation counter.
Testosterone biosynthesis by polycystic ovary syndrome (PCOS) cells [60]: IG-3. Thecal cells (106 cells/ml) isolated from the ovaries of women with PCOS and non-PCOS, are incubated with serum-free culture medium and the corresponding analyte (no addition: negative control; hCG: positive control and insulin or synthetic IG) in a water-saturated atmosphere containing 5% CO2 at 37 °C for approximately 144 h. Anti-inositolglycan antibodies corresponding to the respective IG receptors are also incubated with this solution for approximately 15 min before the addition of the analytes. Rapid freezing in an acetone-dry ice bath terminates the incubation. Testosterone content of cells and media are analyzed in duplicate by RIA (Diagnostic Systems Laboratories, Webster, TX).
Tyrosine phosphorylation of caveolin or IRS-1 [45,48,51,54]: IG-32 and -47. After incubation with insulin or the synthetic IG, detergent insoluble complexes (caveolin) [51] or hemidiaphragms (IRS-1) [45] are collected and solubilized in 1 ml of TEST buffer followed by centrifugation (15,000 g, 10 min, 4 °C). The supernatant is precleared with protein A/G Sepharose and incubated with rabbit anticaveolin antibodies or with rabbit anti-carboxyterminal IRS-1 antibodies for 4 h at 4 °C, and then with protein A/G Sepharose for 16 h at 4 °C. The precipitates are collected by centrifugation (15,000 g, 2 min, 4 °C) and washed successively with a series of buffers. The immunoprecipitants are suspended in Laemmli sample buffer for 5 min at 95 °C followed by centrifugation (15,000 g, 2 min). The supernatant is subjected to SDS-PAGE ( 8 % resolving gel) followed by immunoblotting with either monoclonal antiphosphotyrosine antibodies or anti-IRS-1 antibodies using radioactive [125I] protein A. The radiolabeled blots are analyzed by phosphorimaging.
Table 1.
Monosaccharides.
Details of assays (in alphabetical order) are given in the text.
The number assigned to this compound in the cited publication.
Lit. no.: Literacy number.
Table 7.
Heptasaccharides (cont.).
 | |||||
|---|---|---|---|---|---|
| Study | Compound | Assays* | Activity | Lit. no.‡ |
Ref. |
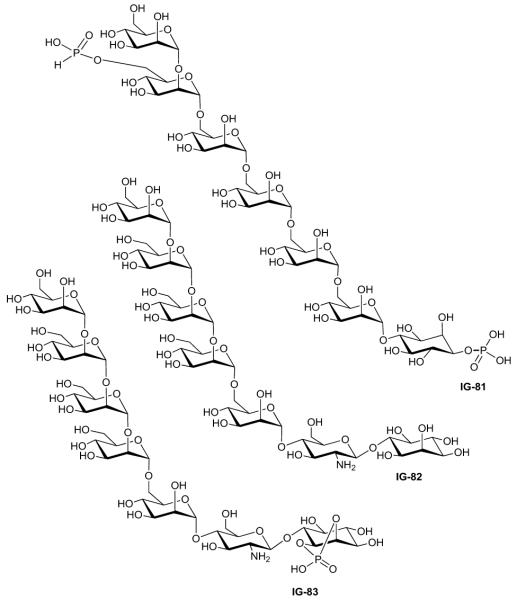
| Frick (1998) | IG-81 | Lipogenesis | Activity ‘B’§ | 12 | [45] |
| Frick (1998) | IG-82 | Lipogenesis | Activity ‘B’§ | 24 | [45] |
| Frick (1998) | IG-83 | Lipogenesis | Activity ‘B’§ | 28 | [45] |
Details of assays (in alphabetical order) are given in the text.
The number assigned to this compound in the cited publication.
Note: for Muller’s SAR study [45] aggregate activities were reported as follows: “A”, MIR < 20%; “B”, MIR 20–49%, EC20 25–200 μM; “C”, MIR 50–80%, EC50 10–100 μM,; “D”, MIR > 80%, EC50 3–30 μM.
Lit. no.: Literacy number; MIR: Maximal insulin response.
Biological activity of structurally defined inositol glycans
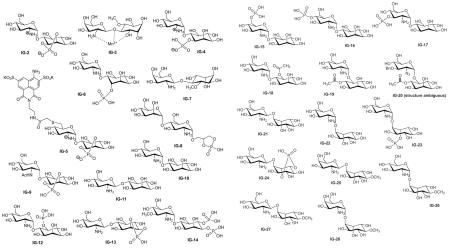
The structures and the biological activities of all of the published IGs that have been tested to date are compiled in Figures 1 & 2 and Tables 1-7. They are organized by the number of sugar (or pseudosugar) units and span the range of monosaccharides to heptasaccharides. The tables list our compound number (designated with an IG-prefix) to allow easy correlation with the structures, the assay performed (see the preceding section for a brief description of each assay), the results of that assay, the compound number in the literature (for easy comparison when reading the primary source) and the reference.
Conclusions
The large collection of structures listed above (largely due to the prolific work of Dr Müller’s team) suggests a preliminary IG pharmacophore for insulin-like action. Structural elements that appear to most often contribute to high activity are a cyclic phosphate on the inositol, though the inositol isomer is less important, a free (unacylated) amino group on the second sugar, and a distal anionic group on one or more of the mannose residues (especially the fourth residue from the inositol terminus) though the nature of the ionic group (phosphonate, phosphate, sulphate) is less important. Curiously, the anomeric configuration between the aminosugar and the inositol is relatively unimportant. The observation that a hybrid compound, IG-5, bearing a distal anionic group pendant on a noncarbohydrate scaffold attached to a disaccharide is more active than the disaccharide, IG-2, from which it was derived, suggests the possibility of noncarbohydrate or partially carbohydrate-based structures with IG receptor agonist properties. This may lead to the development of compounds with useful pharmacological properties that are also considerably simpler to synthesize than the currently most active IGs such as IG-78 or IG-39.
Future perspective
The role of the inositol glycans in insulin signal transduction has been controversial since the IGs were first identified. While there is no doubt that IGs are produced in response to insulin (and indeed to other biological signals [61-70]) and that the IGs are able to mimic the metabolic effects of insulin on insulin-sensitive cells, questions remain regarding the extent to which IGs are necessary and/or sufficient for insulin’s metabolic action.
Regardless of their role in insulin signaling, the fact that the IGs elicit insulin-like effects downstream of the IR suggests their use in the treatment of diabetes and possibly other metabolic diseases. Several pharmaceutical companies with interest in developing therapies for Type 2 diabetes have dabbled in the IG area, but most have abandoned this avenue of research, presumably daunted by the lack of ’drug-like’ structures among the IGs and the extremely lengthy syntheses required to prepare the most active substances. The field is still in need of considerable basic research to elucidate better the nature of the IG receptor on the cell surface, develop more efficient syntheses, establish structure–activity relationships differentiating mitogenic and metabolic activity, as well as to identify simpler and synthetically more accessible IG receptor agonists that may have greater promise as drug candidates.
The basic chemistry and biology of the IGs remains a fascinating area for study. The ubiquitous presence of GPI-anchored proteins on eukaryotic cell surfaces, the highly conserved core structure of the GPI anchors, and the strong kinship in structure between the GPI protein anchors and the hormonally released IGs all suggest a significant role for this class of compounds in cell biology, certainly in hormone signal transduction, and possibly in autocrine and paracrine signaling. It is quite probable that in the next decade we will see an expanding appreciation of the role of IGs in controlling cellular function and that this will be accompanied by a renewal of interest in the use of IGs or their analogues in the treatment of human diseases.
Executive summary.
The inositol glycans (IGs) are small oligosaccharides released from insulin-sensitive cells upon stimulation by insulin. Isolated IGs are capable of activating insulin-sensitive cells.
The natural IGs are structurally microheterogeneous with representatives containing both myo- and chiro-inositol, both glucosamine and galactosamine, an oligomannose portion of varying length, and a variable number of phosphates and/or cyclic phosphates.
Elucidation of a relationship between IG structure and biological activity has been difficult with natural IGs because of the microheterogeneity.
Chemical synthesis has provided a large number of IGs with defined structures for biological evaluation. A comprehensive list of the structures and their activities is provided in this review.
The data suggest that the most important features that an IG should possess for high insulin-like activity are a cyclic phosphate on the inositol, a free (unacylated) amino group on the second sugar and a distal anionic group.
Table 2.
Disaccharides
| Study | Compound | Assays* | Activity | Lit. no.† | Ref. |
|---|---|---|---|---|---|
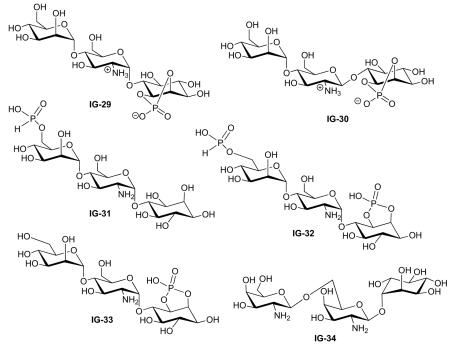
| Plourde (1992) | IG-2 | Lipogenesis | 30–40% MIR at 40 μM | 1 | [6] |
| Chakraborty (2005) | 18% MIR at 10.5 μM | IPG 1 | [49] | ||
| Frick (1998) | Activity ‘A’§ | 43 | [45] | ||
| Dietrich (1999) | Cell proliferation of otic vesicles |
Max. effect at 10 μM | 3 | [39] | |
| Fos oncoprotein expression Conformational study |
Max. effect at 2.2 ± μM | ||||
| cAMP phosphodiesterase | No activity | 1 | [6] | ||
| Plourde (1992) | Gluconeogenesis | No activity | |||
| PDH phosphatase | Active, but data not reported | 3 | [32] | ||
| McLean (2008) | PDK inhibition | ||||
|
| |||||
| Nestler (1998) | IG-3 | Thecal testosterone biosynthesis |
Effective range 1.0 -100 μM/L | INS-2 | [60] |
| Larner (2003) | Glucose incorporation | 40% decrease in hyperglycemia at 2 20 mg/Kg (6 mM–60 μM) |
1 | [43] | |
| PDH phosphatase | Effective at 100 μM range only when chelated with Mn2+ |
||||
| McLean (2008) | No activity with or without Mn2+ | 19 | [32] | ||
| Larner (2003) | Glucose incorporation in insulin-sensitive hepatoma H4IIE cells |
Effective from 0.1–100 μM in presence of insulin |
1 | [43] | |
|
| |||||
| Plourde (1992) | IG-4 | Lipogenesis | No activity | 5 | [6] |
| cAMP phosphodiesterase | No activity | ||||
| Gluconeogenesis | No activity | ||||
| Dietrich (1999) | Conformational study | 5 | [39] | ||
|
| |||||
| Turner (2005) | IG-5 | Lipogenesis | 47% MIR, Kd of 12.0 μM (R = 0.877) | 1 | [33] |
| Optical studies | Not transported across cell membranes | ||||
|
| |||||
| Reddy (1993) | IG-6 | No assay reported | 1 | [71] | |
|
| |||||
| Frick (1998) | IG-7 | Lipogenesis | Activity ‘A’§ | 42 | [45] |
|
| |||||
| Frick (1998) | IG-8 | Lipogenesis | Activity ‘A’§ | 44 | [45] |
|
| |||||
| Plourde (1992) | IG-9 | Lipogenesis cAMP phosphodiesterase |
No activity | 13 | [6] |
|
| |||||
| Plourde (1992) | IG-10 | Lipogenesis cAMP phosphodiesterase Gluconeogenesis |
No activity | 14 | [6] |
| McLean (2008) | PDH phosphatase | No activity | 1 | [32] | |
|
| |||||
| McLean (2008) | IG-11 | PDH phosphatase | No activity | 2 | [32] |
|
| |||||
| McLean (2008) | IG-12 | PDH phosphatase PDK inhibition |
0.125 nmol/min at 100 μM 0.08% inhibition of PDK at 1 mM |
4 | [32] |
|
| |||||
| McLean (2008) | IG-13 | PDH phosphatase PDK inhibition |
0.8 nmol/min at 100 μM 0.4% inhibition of PDK at 1 mM |
5 | [32] |
|
| |||||
| McLean (2008) | IG-14 | PDH phosphatase PDK inhibition |
2.7 nmol/min at 100 μM 75% inhibition of PDK at 1 mM |
6 | [32] |
|
| |||||
| McLean (2008) | IG-15 | PDH phosphatase PDK inhibition |
30% activation of PDP at 100 μM 0.01% inhibition of PDK at 1 mM |
7 | [32] |
|
| |||||
| McLean (2008) | IG-16 | PDH phosphatase PDK inhibition |
130% activation of PDP at 100 μM 0.7% inhibition of PDK at 1 mM |
8 | [32] |
|
| |||||
| McLean (2008) | IG-17 | PDH phosphatase PDK inhibition |
320% activation of PDP at 100 μM 1.0% inhibition of PDK at 1 mM |
9 | [32] |
|
| |||||
| McLean (2008) | IG-18 | PDH phosphatase PDK inhibition |
0.125 nmol/min at 100 μM 0.02% inhibition of PDK at 1 mM |
10 | [32] |
|
| |||||
| McLean (2008) | IG-19 | PDH phosphatase PDK inhibition |
1.0 nmol/min at 100 μM 4.0% inhibition of PDK at 1 mM |
11 | [32] |
|
| |||||
| McLean (2008) | IG-20 | PDH phosphatase PDK inhibition |
1.0 nmol/min at 100 μM 9.5% inhibition of PDK at 1 mM |
12 | [32] |
|
| |||||
| McLean (2008) | IG-21 | PDH phosphatase PDK inhibition |
No activity | 13 | [32] |
|
| |||||
| McLean (2008) | IG-22 | PDH phosphatase PDK inhibition |
No activity | 14 | [32] |
|
| |||||
| McLean (2008) | IG-23 | PDH phosphatase PDK inhibition |
0.25 nmol/min at 100 μM 0.3% inhibition of PDK at 1 mM |
16 | [32] |
|
| |||||
| McLean (2008) | IG-24 | PDH phosphatase PDK inhibition |
2.2 nmol/min at 100 μM 0.4% inhibition of PDK at 1 mM |
17 | [32] |
|
| |||||
| McLean (2008) | IG-25 | PDH phosphatase PDK inhibition |
Significant activity | 18 | [32] |
|
| |||||
| McLean (2008) | IG-26 | PDH phosphatase PDK inhibition |
0.3 nmol/min at 100 μM 0.07% inhibition of PDK at 1 mM |
21 | [32] |
|
| |||||
| McLean (2008) | IG-27 | PDH phosphatase PDK inhibition |
0.25 nmol/min at 100 μM 0.6% inhibition of PDK at 1 mM |
22 | [32] |
|
| |||||
| McLean (2008) | IG-28 | PDH phosphatase | Significant activity | 23 | [32] |
Details of assays (in alphabetical order) are given in the text.
The number assigned to this compound in the cited publication.
Note: For Muller’s SAR study [45], aggregate activities were reported as follows: “A”, MIR < 20%; “B”, MIR 20–49%, EC20 25–200 μM; “C”, MIR 50–80%, EC50 10–100 μM; “D”, MIR > 80%, EC50 3–30 μM.
IG: Inositol glycan; Lit. no.: Literacy number; MIR: Maximal insulin response; PDH: Pyruvate dehydrogenase; PDK: Phosphoinositide-dependent kinase.
Table 3.
Trisaccharides.
| Study | Compound | Assays* | Activity | Lit. no.‡ | Ref. |
|---|---|---|---|---|---|
| Jaworek (2001) | IG-29 | Lipogenesis | Inactive | 1 | [72] |
| Dietrich (1999) | Conformational study | 4 | [39] | ||
|
| |||||
| Jaworek (2001) | IG-30 | Lipogenesis | Inactive | 2 | [72] |
| Frick (1998) | IG-31 | Lipogenesis | Inactive | 1 | [45] |
| Frick (1999) | 15% MIR EC50 = 25 μM | H | [101] | ||
| Glucose transport | 2% MIR, EC50 = 10 μM | ||||
|
| |||||
| Frick (1998) | IG-32 | Lipogenesis | 20% MIR at 100 μM | 7 | [45] |
| Frick (1999) | 25% MIR, EC50 = 15 μM | G | [101] | ||
| Frick (1998) | Glucose transport | 40% MIR at 20 μM | 7 | [45] | |
| Frick (1999) | 9% MIR, EC50 = 15 μM | G | [101] | ||
| Frick (1998) | GPAT | 25% MIR | 7 | [45] | |
| Glycogenesis | 33% MIR at 20 μM | ||||
| GLUT4 translocation | 50% MIR | ||||
| Lipolysis | 42% MIR | ||||
| Tyrosine phosphorylation of IRS-1 | 30% MIR at 10 μM | ||||
|
| |||||
| Frick (1998) | IG-33 | Lipogenesis | Activity ‘B’* | 8 | [45] |
|
| |||||
| McLean (2008) | IG-34 | PDH phosphatase | Negligible activity for PDP activation | 15 | [32] |
| PDK inhibition | 0.2% inhibition of PDK at 1 mM | ||||
Details of assays (in alphabetical order) are given in the text.
The number assigned to this compound in the cited publication.
Note: For Muller’s SAR study [45] aggregate activities were reported as follows: “A”, MIR < 20%; “B”, MIR 20–49%, EC20 25–200 μM; “C”, MIR 50–80%, EC50 10–100 μM; “D”, MIR > 80%, EC50 3–30 μM.
Lit. no.: Literacy number; MIR: Maximal insulin response; PDH: Pyruvate dehydrogenase; PDK: Phosphoinositide-dependent kinase.
Table 4.
Tetrasaccharides.
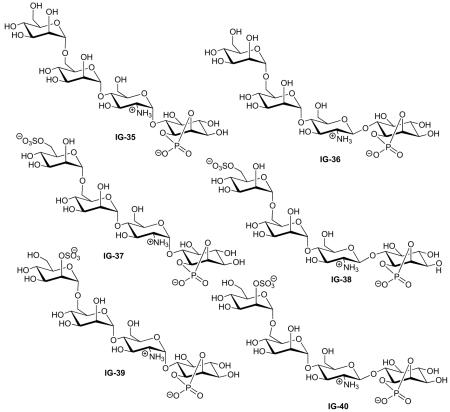
| Study | Compound | Assays* | Activity | Lit. no.‡ | Ref. |
|---|---|---|---|---|---|
| Chakraborty (2006) | IG-35 | Lipogenesis | Inactive | 3 | [73] |
| Chakraborty (2006) | IG-36 | Lipogenesis | Inactive | 4 | [73] |
| Chakraborty (2005) | IG-37 | Lipogenesis | 63% MIR, EC50 = 10.6 μM | 3α | [49] |
| Chakraborty (2005) | IG-38 | Lipogenesis | 51% MIR, EC50 = 9.8 μM | 3β | [49] |
| Chakraborty (2005) | IG-39 | Lipogenesis | 78% MIR, EC50 = 1.1 μM | 4α | [49] |
| Chakraborty (2005) | IG-40 | Lipogenesis | 47% MIR, EC50 = 9.5 μM | 4β | [49] |
Details of assays (in alphabetical order) are given in the text.
The number assigned to this compound in the cited publication.
Lit. no.: Literacy number; MIR: Maximal insulin response.
Table 5.
Pentasaccharides.
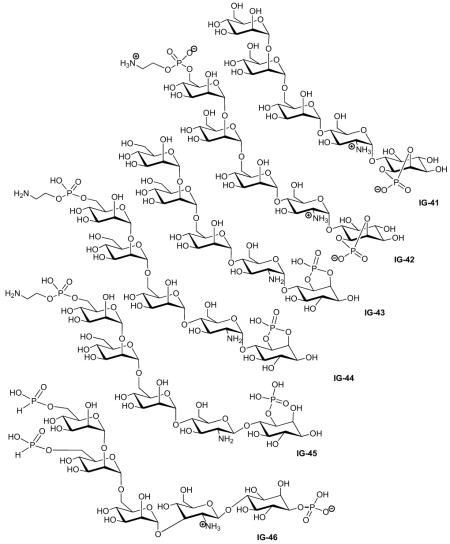
| Study | Compound | Assays* | Activity | Lit. no.‡ | Ref. |
|---|---|---|---|---|---|
| Martin-Lomas (2000) |
IG-41 | Lipogenesis PDH phosphatase cAMP-dependent protein kinase |
Inactive | 1a | [36] |
|
| |||||
| Misek (1992) | IG-42 | Lipolysis | 70% inhibition at 132 μM, IC50= 60 μM |
VSG-IPG | [4] |
| Glucose-6-phosphatase activity | 90% inhibitionat 160 μM, IC50 = 109 μM |
||||
| Fructose-1,6-bisphosphatase activity | 100% inhibition at 226 μM, IC50= 136 μM. |
||||
| Gluconeogenesis | 67% inhibitionat 102 μM, IC50= 84 μM |
||||
| Misek (1992) | Protein phosphorylation/ dephosphorylation |
Blocked IPT-dependent phosphorylation at 200 μM ~ 90% dephosphorylation at 150 μM, IC50 = 75 μM |
VSG-IPG | [4] | |
|
| |||||
| Frick (1998) | IG-43 | Lipogenesis | Activity‘A’§ | 20 | [45] |
|
| |||||
| Frick (1998) | IG-44 | Lipogenesis | Activity‘C’§ | 29 | [45] |
|
| |||||
| Frick (1998) | IG-45 | Lipogenesis | Activity‘B’§ | 23 | [45] |
|
| |||||
| Frick (1998) | IG-46 | Lipogenesis | Activity‘C’§ | 13 | [45] |
| Frick (1999) | 44% MIR, EC50 = 40 μM | Q | [101] | ||
| Glucose transport | 18% MIR, EC50 = 30 μM | ||||
Details of assays (in alphabetical order) are given in the text.
The number assigned to this compound in the cited publication.
Note: for Muller’s SAR study [45] aggregate activities were reported as follows: “A”, MIR < 20%; “B”, MIR 20–49%, EC20 25–200 μM; “C”, MIR 50–80%, EC50 10–100 μM; “D”, MIR > 80%, EC50 3–30 μM.
IPG: Inositol phosphate-glycan; Lit. no.: Literacy number; MIR: Maximal insulin response.
Table 6.
Hexasaccharides.
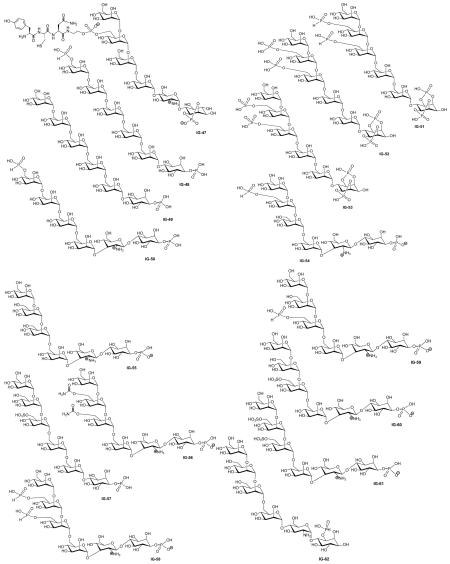

| Study | Compound | Assays* | Activity | Lit. no.‡ | Ref. |
|---|---|---|---|---|---|
| Muller (1997) | IG-47 | Lipogenesis | 15.5-fold stimulation, EC50 = 1.0 μM |
PIG-P | [37] |
| Muller (1998) | 65–75% MIR, EC50 = 2–5 μM | PIG-P | [51] | ||
| Muller (2002) | 22-fold stimulation at 10 μM | YCN-PIG | [48] | ||
| Muller (1997) | GPAT | 75–85% MIR, EC50 = 3–5 μM | PIG-P | [37] | |
| Muller (1998) | 85% MIR at 5 μM | PIG-P | [51] | ||
| Muller (1997) | Glucose transport | 15-fold increase, EC50 = 1.0–1.5 μM |
PIG-P | [37] | |
| Kessler (1998) | sixfold stimulation at 10 μM | PIG-P | [54] | ||
| Muller (1997) | GLUT4 translocation | 85% MIR, EC50 = 2–3 μM | PIG-P | [37] | |
| Muller (1997) | Glycogenesis | 75–90% MIR, EC50 = 8–10 μM | PIG-P | [37] | |
| Muller (2002) | eightfold stimulation at 10 μM | YCN-PIG | [48] | ||
| Muller (1997) | GS stimulation | 2.8-fold stimulation, EC50 = 8.0 μM |
PIG-P | [37] | |
| Muller (2002) | fourfold stimulation at 10 μM | YCN-PIG | [48] | ||
| Muller (2002) | Glycerol-3-phosphate acyltransferase activity |
sixfold stimulation at 10 μM | YCN-PIG | [48] | |
| Muller (1997) | cAMP-dependent protein kinase activity |
70% MIR, EC50 = 0.3–0.5 μM | PIG-P | [37] | |
| Muller (1997) | Lipolysis | 70% inhibition, EC50 = 0.5–0.8 μM |
PIG-P | [37] | |
| Muller (2002) | 95% inhibition at 10 μM | YCN-PIG | [48] | ||
| Kessler (1998) | PI-3 kinase activity | 2.3-fold stimulation, | 76% MIR PIG-P | [54] | |
| Muller (2002) | Radiolabeled study with hcDIGs of adipocyte plasma membranes |
Specific interaction with hcDIGs, Kd 30–100 nM, Bmax = 5–10 pmol/mg hcDIGs |
YCN-PIG | [74] | |
| Muller (1998) | Tyrosine phosphorylation of caveolin |
80% MIR at 10 μM, EC50 = 1–2 μM |
PIG-P | [51] | |
| Kessler (1998) | Tyrosine | 2.3-fold stimulation, 44% MIR | PIG-P | [54] | |
| Muller (2002) | phosphorylation of IRS-1 | 24-fold stimulation at 5 nM | YCN-PIG | [48] | |
|
| |||||
| Frick (1998) | IG-48 | Lipogenesis | Activity ‘A’§ | 2 | [45] |
| Frick (1999) | 10% MIR, EC50 = 20 μM | O | [101] | ||
| Glucose transport | 5% MIR, EC50 = 50 μM | ||||
|
| |||||
| Frick (1998) | IG-49 | Lipogenesis | Activity ‘A’§ | 3 | [45] |
|
| |||||
| Frick (1998) | IG-50 | Lipogenesis | Activity ‘A’§ | 4 | [45] |
| Frick (1999) | 18% MIR, EC50 = 20 μM | 1 | [101] | ||
| Glucose transport | 14% MIR, EC50 = 50 μM | ||||
|
| |||||
| Frick (1998) | IG-51 | Lipogenesis | Activity ‘B’§ | 5 | [45] |
|
| |||||
| Frick (1998) | IG-52 | Lipogenesis | Activity ‘B’§ | 6 | [45] |
|
| |||||
| Frick (1998) | IG-53 | Lipogenesis | Activity ‘B’§ | 9 | [45] |
|
| |||||
| Frick (1998) | IG-54 | Lipogenesis | Activity ‘B’§ | 10 | [45] |
| Frick (1999) | 33% MIR, EC50 = 20 μM | D | [101] | ||
| Glucose transport | 18% MIR, EC50 = 18 μM | ||||
|
| |||||
| Frick (1998) | IG-55 | Lipogenesis | Activity ‘B’§ | 11 | [45] |
|
| |||||
| Frick (1998) | IG-56 | Lipogenesis | Activity ‘C’§ | 14 | [45] |
| Frick (1999) | 48% MIR, EC50 = 15 μM | R | [101] | ||
| Glucose transport | 19% MIR, EC50 = 10 μM | ||||
|
| |||||
| Frick (1998) | IG-57 | Lipogenesis | Activity ‘C’§ | 15 | [45] |
| Frick (1999) | 50% MIR, EC50 = 15 μM | S | [101] | ||
| Glucose transport | 34% MIR, EC50 = 20 μM | ||||
|
| |||||
| Frick (1998) | IG-58 | Lipogenesis | Activity ‘C’§ | 16 | [45] |
| Frick (1999) | 72% MIR, EC50 = 12 μM | T | [101] | ||
| Glucose transport | 30% MIR, EC50 = 10 μM | ||||
|
| |||||
| Frick (1998) | IG-59 | Lipogenesis | Activity ‘D’§ | 17 | [45] |
| Frick (1999) | 80% MIR, EC50 = 15 μM | U | [101] | ||
| Glucose transport | 34% MIR, EC50 = 15 μM | ||||
|
| |||||
| Frick (1998) | IG-60 | Lipogenesis | Activity ‘D’§ | 18 | [45] |
| Frick (1999) | 83% MIR, EC50 = 15 μM | V | [101] | ||
| Glucose transport | 48% MIR, EC50 = 15 μM | ||||
|
| |||||
| Frick (1998) | IG-61 | Lipogenesis | Activity ‘D’§ | 19 | [45] |
| Frick (1999) | 86% MIR, EC50 = 10 μM | W | [101] | ||
| Glucose transport | 42% MIR, EC50 = 10 μM | ||||
|
| |||||
| Frick (1998) | IG-62 | Lipogenesis | Activity ‘B’§ | 21 | [45] |
|
| |||||
| Frick (1998) | IG-63 | Lipogenesis | Activity ‘B’§ | 22 | [45] |
|
| |||||
| Frick (1998) | IG-64 | Lipogenesis | Activity ‘B’§ | 25 | [45] |
|
| |||||
| Frick (1998) | IG-65 | Lipogenesis | Activity ‘B’§ | 26 | [45] |
|
| |||||
| Frick (1998) | IG-66 | Lipogenesis | Activity ‘B’§ | 27 | [45] |
|
| |||||
| Frick (1998) | IG-67 | Lipogenesis | Activity ‘C’§ | 30 | [45] |
| Frick (1999) | 48% MIR, EC50 = 60 μM | E | [101] | ||
| Glucose transport | 34% MIR, EC50 = 30 μM | ||||
|
| |||||
| Frick (1998) | IG-68 | Lipogenesis | Activity ‘C’§ | 31 | [45] |
| Frick (1999) | 53% MIR, EC50 = 15 μM | F | [101] | ||
| Glucose transport | 26% MIR, EC50 = 25 μM | ||||
|
| |||||
| Frick (1998) | IG-69 | Lipogenesis | Activity ‘C’ § | 32 | [45] |
|
| |||||
| Frick (1998) | IG-70 | Lipogenesis | Activity ‘C’§ | 33 | [45] |
| Frick (1999) | 59% MIR, EC50 = 7 μM | A | [101] | ||
| Glucose transport | 28% MIR, EC50 = 8 μM | ||||
|
| |||||
| Frick (1998) | IG-71 | Lipogenesis | Activity ‘C’§ | 34 | [45] |
| Frick (1999) | 63% MIR, EC50 = 8 μM | J | [101] | ||
| Glucose transport | 21% MIR, EC50 = 3 μM | ||||
|
| |||||
| Frick (1999) | IG-72 | Lipogenesis | Activity ‘C’§ | 35 | [45] |
| Frick (1999) | 64% MIR, EC50 = 7 μM | C | [101] | ||
| Glucose transport | 29% MIR, EC50 = 10 μM | ||||
|
| |||||
| Frick (1998) | IG-73 | Lipogenesis | Activity ‘C’§ | 36 | [45] |
| Frick (1999) | 72% MIR, EC50 = 8 μM | K | [101] | ||
| Glucose transport | 23% MIR, EC50 = 15 μM | ||||
|
| |||||
| Frick (1998) | IG-74 | Lipogenesis | 80% MIR at 20 μM, IC50 = 2.2 ± 0.2 μM |
37 | [45] |
| Frick (1999) | 77% MIR, EC50 = 8.2 μM | B | [101] | ||
| Frick (1998) | GPAT | 90% MIR, IC50 = 8.0 ± 1.1 μM | 37 | [45] | |
| Glucose transport 90–95% MIR at 20 μM, IC50 = 8.3 ± 1.5 μM |
|||||
| Frick (1999) | 41% MIR, EC50 = 8.1 μM | B | [101] | ||
| Frick (1998) | GLUT4 translocation | 50% MIR | 37 | [45] | |
| Glycogenesis 65% MIR at 20 μM, IC50 = 7.1 ± 2.0 μM |
|||||
| Lipolysis | 95% MIR, IC50 = 2.5 ± 0.5 μM | ||||
| Tyrosine phosphorylation of IRS-1 | 65% MIR at 10 μM | ||||
|
| |||||
| Frick (1998) | IG-75 | Lipogenesis | Activity ‘D’§ | 38 | [45] |
| Frick (1999) | 90% MIR, EC50 = 7 μM | M | [101] | ||
| Glucose transport | 39% MIR, EC50 = 6 μM | ||||
|
| |||||
| Frick (1998) | IG-76 | Lipogenesis | Activity ‘D’§ | 39 | [45] |
| Frick (1999) | 84% MIR, EC50 = 7 μM | L | [101] | ||
| Glucose transport | 36% MIR, EC50 = 8 μM | ||||
|
| |||||
| Frick (1998) | IG-77 | Lipogenesis | Activity ‘D’§ | 40 | [45] |
|
| |||||
| Frick (1998) | IG-78 | Lipogenesis | 90% MIR at 20 μM, IC50 = 2.5 ± 0.9 μM |
41 | [45] |
| Frick (1999) | 96% MIR, EC50 = 3 μM | N | [101] | ||
| Frick (1998) | GPAT | 90% MIR, IC50 = 3.5 ± 0.8 μM | 41 | [45] | |
| Frick (1999) | Glucose transport | 90–95% MIR at 20 μM, IC50 = 3.5 ± 0.6 μM |
|||
| Frick (1998) | 52% MIR, EC50 = 5 μM | N | [101] | ||
| GLUT4 translocation 50% MIR | 41 | [45] | |||
| Glycogenesis 70% MIR at 20 μM, IC50 = 3.3 ± 0.5 μM |
|||||
| Lipolysis | 95% MIR, IC50 = 1.6 ± 0.5 μM | ||||
|
| |||||
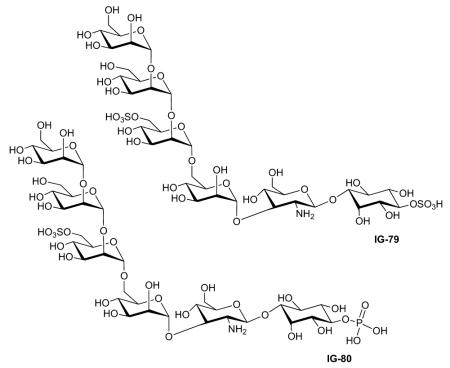
| Frick (1998) | IG-79 | Tyrosine phosphorylation of IRS-1 |
95% MIR at 10 μM | 45 | [45] |
| Lipogenesis | Activity ‘C’§ | ||||
| Tyrosine phosphorylation of IRS-1 |
55% MIR at 10 μM | ||||
|
| |||||
| Frick (1998) | IG-80 | Lipogenesis | Activity ‘C’§ | 46 | [45] |
Details of assays (in alphabetical order) are provided in the text.
The number assigned to this compound in the cited publication.
Note: for Muller’s SAR study [45] aggregate activities were reported as follows: “A”, MIR < 20%; “B”, MIR 20–49%, EC20 25–200 μM; “C”, MIR 50–80%, EC50 10–100 μM; “D”, MIR > 80%, EC50 3–30 μM.
Lit. no.: Literacy number; MIR: Maximal insulin response.
Acknowledgments
Financial & competing interests disclosure The authors gratefully acknowledge the NIH (DK-44589 and GM-84819) for support of the work in Marc d’Alarcao’s laboratory cited in this review. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Glossary
- Inositol glycan
A member of a family of inositol-containing oligosaccharides, some of which are thought to be involved in regulating various cellular processes
- Signal transduction
The process of relaying an extracellular biological stimulus to the interior of a cell
- Diabetes
A disease characterized by an elevated concentration of glucose in the blood
- Lipogenesis
The synthesis of lipids, usually triglycerides, occurring in specialized fat cells
- Anabolic stimulation
The activation of biochemical pathways leading to the synthesis of larger molecules such as proteins, lipids, and polysaccharides
Bibliography
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Saltiel AR, Cuatrecasas P. Insulin stimulates the generation from hepatic plasma membranes of modulators derived from an inositol glycolipid. Proc. Natl Acad. Sci. USA. 1986;83(16):5793–5797. doi: 10.1073/pnas.83.16.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larner J. Insulin mediator – fact or fancy. J. Cyclic Nucleotide Res. 1982;8(5):289–296. [PubMed] [Google Scholar]
- 3.Larner J, Huang LC, Schwartz CF, et al. Rat liver insulin mediator which stimulates pyruvate dehydrogenase phosphate contains galactosamine and D-chiroinositol. Biochem. Biophys. Res. Commun. 1988;151(3):1416–1426. doi: 10.1016/s0006-291x(88)80520-5. [DOI] [PubMed] [Google Scholar]
- 4.Misek DE, Saltiel AR. An inositol phosphate glycan derived from a Trypanosoma brucei glycosylphosphatidylinositol mimics some of the metabolic actions of insulin. J. Biol. Chem. 1992;267(23):16266–16273. [PubMed] [Google Scholar]
- 5.Misek DE, Saltiel AR. An inositol phosphate glycan derived from a Trypanosoma brucei glycosyl phosphatidylinositol promotes protein dephosphorylation in rat epididymal adipocytes. Endocrinology. 1994;135(5):1869–1876. doi: 10.1210/endo.135.5.7956908. [DOI] [PubMed] [Google Scholar]
- 6.Plourde R, d’Alarcao M, Saltiel AR. Synthesis and characterization of an insulin-mimetic disaccharide. J. Org. Chem. 1992;57(9):2606–2610. [Google Scholar]
- 7.Romero G, Larner J. Insulin mediators and the mechanism of insulin action. Adv. Pharmacol. 1993;24:21–50. doi: 10.1016/s1054-3589(08)60932-1. [DOI] [PubMed] [Google Scholar]
- 8.Varela-Nieto I, Leon Y, Caro HN. Cell signalling by inositol phosphoglycans from different species. Comp. Biochem. Physiol. B, Biochem. Mol. Biol. 1996;115(2):223–241. doi: 10.1016/0305-0491(96)00087-9. [DOI] [PubMed] [Google Scholar]
- 9.Jones DR, Varela-Nieto I. Diabetes and the role of inositol-containing lipids in insulin signaling. Mol. Med. 1999;5(8):505–514. [PMC free article] [PubMed] [Google Scholar]
- 10.Stralfors P. Insulin second messengers. Bioessays. 1997;19(4):327–335. doi: 10.1002/bies.950190410. [DOI] [PubMed] [Google Scholar]
- 11.Field MC. Is there evidence for phosphooligosaccharides as insulin mediators? Glycobiology. 1997;7(2):161–168. doi: 10.1093/glycob/7.2.161-d. [DOI] [PubMed] [Google Scholar]
- 12.Larner J, Huang LC. Identification of a novel inositol glycan signaling pathway with significant therapeutic relevance to insulin resistance: an insulin signaling model using both tyrosine kinase and G-proteins. Glycobiology. 1999;7(3):217–231. [Google Scholar]
- 13.Rademacher TW, Caro H, Kunjara S, Wang DY, Greenbaum AL, McLean P. Inositolphosphoglycan 2nd messengers. Braz. J. Med. Biol. Res. 1994;27(2):327–341. [PubMed] [Google Scholar]
- 14.Gigg R, Gigg J. Synthesis of glycosylphoshatidylinositol anchors. In: Large D, Warren CD, editors. Glycolipids and Related Compounds. Marcel Dekker; NY, USA: 1997. [Google Scholar]
- 15.Helmreich EJM. Regulation by a hormone: the insulin response. Oxford University Press; Oxford, UK: 2001. [Google Scholar]
- 16.Saltiel AR. New perspectives into the molecular pathogenesis and treatment of Type 2 diabetes. Cell. 2001;104(4):517–529. doi: 10.1016/s0092-8674(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 17.Myers MG, White MF. Insulin signal transduction and the IRS proteins. Ann. Rev. Pharmacol. Toxicol. 1996;36:615–658. doi: 10.1146/annurev.pa.36.040196.003151. [DOI] [PubMed] [Google Scholar]
- 18.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 19.Kaburagi Y, Okochi H, Satoh S, et al. Role of IRS and PHIP on insulin-induced tyrosine phosphorylation and distribution of IRS proteins. Cell Struct. Funct. 2007;32(1):69–78. doi: 10.1247/csf.07003. [DOI] [PubMed] [Google Scholar]
- 20.Hresko RC, Mueckler M. mTOR-RICTOR is the Ser(473) kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 2005;280(49):40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 21.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998;10(2):262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 22.Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 2002;277(37):33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- 23.Watson RT, Pessin JE. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem. Sci. 2006;31(4):215–222. doi: 10.1016/j.tibs.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Saltiel AR, Pessin JE. Insulin signaling in microdomains of the plasma membrane. Traffic. 2003;4(11):711–716. doi: 10.1034/j.1600-0854.2003.00119.x. [DOI] [PubMed] [Google Scholar]
- 25.Ahmad F, Lindh R, Tang Y, et al. Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem. J. 2007;404:257–268. doi: 10.1042/BJ20060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Degerman E, Belfrage P, Manganiello VC. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3) J. Biol. Chem. 1997;272(11):6823–6826. doi: 10.1074/jbc.272.11.6823. [DOI] [PubMed] [Google Scholar]
- 27.Omori K, Kotera J. Overview of PDEs and their regulation. Circ. Res. 2007;100(3):309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 28.Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Ann. Rev. Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- 29.Schinner S, Scherbaum WA, Bornstein SR, Barthel A. Molecular mechanisms of insulin resistance. Diabet. Med. 2005;22(6):674–682. doi: 10.1111/j.1464-5491.2005.01566.x. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez JF, Sanchez-Arias JA, et al. Transport in isolated rat hepatocytes of the phospho-oligosaccharide that mimics insulin action. Effects of adrenalectomy and glucocorticoid treatment. Biochem. J. 1991;274(Pt 2):369–374. doi: 10.1042/bj2740369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larner J, Huang LC, Suzuki S, et al. Insulin mediators and the control of pyruvate dehydrogenase complex. Ann. N.Y. Acad. Sci. 1989;573:297–305. doi: 10.1111/j.1749-6632.1989.tb15006.x. [DOI] [PubMed] [Google Scholar]
- 32.McLean P, Kunjara S, Greenbaum AL, et al. Reciprocal control of pyruvate dehydrogenase kinase and phosphatase by inositol phosphoglycans dynamic state set by ‘push–pull’ system. J. Biol. Chem. 2008;283(48):33428–33436. doi: 10.1074/jbc.M801781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner DI, Chakraborty N, d’Alarcao M. A fluorescent inositol phosphate glycan stimulates lipogenesis in rat adipocytes by extracellular activation alone. Bioorg. Med. Chem. Lett. 2005;15(8):2023–2025. doi: 10.1016/j.bmcl.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 34.Muller G. Dynamics of plasma membrane microdomains and cross-talk to the insulin signalling cascade. FEBS Lett. 2002;531(1):81–87. doi: 10.1016/s0014-5793(02)03402-6. ■■ Presents one of the best current hypotheses for the mechanism of inositol glycan (IG) action.
- 35.Muller G, Schulz A, Wied S, Frick W. Regulation of lipid raft proteins by glimepiride- and insulin-induced glycosylphosphatidylinositol-specific phospholipase C in rat adipocytes. Biochem. Pharmacol. 2005;69(5):761–780. doi: 10.1016/j.bcp.2004.11.014. ■■ Presents one of the best current hypotheses for the mechanism of IG action.
- 36.Martin-Lomas M, Khiar N, Garcia S, et al. Inositolphosphoglycan mediators structurally related to glycosyl phosphatidylinositol anchors: synthesis, structure and biological activity. Chemistry. 2000;6(19):3608–3621. doi: 10.1002/1521-3765(20001002)6:19<3608::aid-chem3608>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 37.Muller G, Wied S, Crecelius A, Kessler A, Eckel J. Phosphoinositolglycan-peptides from yeast potently induce metabolic insulin actions in isolated rat adipocytes, cardiomyocytes, and diaphragms. Endocrinology. 1997;138(8):3459–3475. doi: 10.1210/endo.138.8.5308. [DOI] [PubMed] [Google Scholar]
- 38.Thompson WJ, Appleman MM. Multiple cyclic nucleotide phosphodiesterase activities from rat brain. Biochemistry. 1971;10(2):311–316. [PubMed] [Google Scholar]
- 39.Dietrich H, Espinosa JF, Chiara JL, et al. Glycosyl inositol derivatives related to inositolphosphoglycan mediators: synthesis, structure, and biological activity. Chem. Eur. J. 1999;5(1):320–336. [Google Scholar]
- 40.Represa JJ, Miner C, Barbosa E, Giraldez F. Bombesin and other growth-factors activate cell-proliferation in chick-embryo otic vesicles in culture. Development. 1988;103(1):87–96. doi: 10.1242/dev.103.1.87. [DOI] [PubMed] [Google Scholar]
- 41.Fleischer S, Kervina M. Sub cellular fractionation of rat liver. In: Fleischer S, Packer L, editors. Methods in Enzymology. (Volume 31) Academic Press; NY, USA: 1974. Biomembranes, Part a. [DOI] [PubMed] [Google Scholar]
- 42.Soley M, Hollenberg MD. Epidermal growth-factor (urogastrone)-stimulated gluconeogenesis in isolated mouse hepatocytes. Arch. Biochem. Biophys. 1987;255(1):136–146. doi: 10.1016/0003-9861(87)90303-1. [DOI] [PubMed] [Google Scholar]
- 43.Larner J, Price JD, Heimark D, et al. Isolation, structure, synthesis, and bioactivity of a novel putative insulin mediator. A galactosamine chiro-inositol pseudo-disaccharide Mn2+ chelate with insulin-like activity. J. Med. Chem. 2003;46(15):3283–3291. doi: 10.1021/jm030071j. [DOI] [PubMed] [Google Scholar]
- 44.Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for ortho-phosphate determination and its use in alkaline phosphatase-based enzyme-immunoassay. Anal. Biochem. 1988;171(2):266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 45.Frick W, Bauer A, Bauer J, Wied S, Muller G. Structure–activity relationship of synthetic phosphoinositolglycans mimicking metabolic insulin action. Biochemistry. 1998;37(38):13421–13436. doi: 10.1021/bi9806201. ■■ The most comprehensive structure–activity study on IGs published to date is this tour de force.
- 46.Muller G, Wied S. The sulfonylurea drug, glimepiride, stimulates glucose-transport, glucose-transporter translocation, dephosphorylation in insulin-resistant rat adipocytes in vitro. Diabetes. 1993;42(12):1852–1867. doi: 10.2337/diab.42.12.1852. [DOI] [PubMed] [Google Scholar]
- 47.Vila MD, Milligan G, Standaert ML, Farese RV. Insulin activates glycerol-3-phosphate acyltransferase (de novo phosphatidic-acid synthesis) through a phospholipid-derived mediator – apparent involvement of Gi-α and activation of a phospholipase-C. Biochemistry. 1990;29(37):8735–8740. doi: 10.1021/bi00489a033. [DOI] [PubMed] [Google Scholar]
- 48.Muller G, Jung C, Frick W, Bandlow W, Kramer W. Interaction of phosphatidylinositolglycan(-peptides) with plasma membrane lipid rafts triggers insulin-mimetic signaling in rat adipocytes. Arch. Biochem. Biophys. 2002;408(1):7–16. doi: 10.1016/s0003-9861(02)00450-2. [DOI] [PubMed] [Google Scholar]
- 49.Chakraborty N, d’Alarcao M. An anionic inositol phosphate glycan pseudotetrasaccharide exhibits high insulin-mimetic activity in rat adipocytes. Bioorg. Med. Chem. Lett. 2005;13(24):6732–6741. doi: 10.1016/j.bmc.2005.07.020. ■■ The smallest IG with high insulin-like activity is the tetrasaccharide prepared in this study.
- 50.Cobb JE, Johnson MR. Synthesis of 6-O-2 aminoethyl-D L-myo-inositol 1 2 cyclic phosphate a model of a putative insulin second messenger. Tetrahedron. 1991;47(1):21–30. [Google Scholar]
- 51.Muller G, Wied S, Piossek C, et al. Convergence and divergence of the signaling pathways for insulin and phosphoinositolglycans. Mol. Med. 1998;4(5):299–323. [PMC free article] [PubMed] [Google Scholar]
- 52.Rodbell M. Metabolism of isolated fat cells. I. Effects of hormones on glucose metabolism + lipolysis. J. Biol. Chem. 1964;239(2):375–384. [PubMed] [Google Scholar]
- 53.Wieland O. Eine Enzymatische Methode Zur Bestimmung Von Glycerin. Biochem. J. 1957;329(4):313–319. [PubMed] [Google Scholar]
- 54.Kessler A, Muller G, Wied S, Crecelius A, Eckel J. Signalling pathways of an insulin-mimetic phosphoinositolglycan-peptide in muscle and adipose tissue. Biochem. J. 1998;330(Pt 1):277–286. doi: 10.1042/bj3300277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caro HN, Kunjara S, Rademacher TW, et al. Isolation and partial characterisation of insulin-mimetic inositol phosphoglycans from human liver. Biochem. Mol. Med. 1997;61(2):214–228. doi: 10.1006/bmme.1997.2607. [DOI] [PubMed] [Google Scholar]
- 56.Lilley K, Zhang C, Villar-Palasi C, Larner J, Huang L. Insulin mediator stimulation of pyruvate dehydrogenase phosphatases. Arch. Biochem. Biophys. 1992;296(1):170–174. doi: 10.1016/0003-9861(92)90559-f. [DOI] [PubMed] [Google Scholar]
- 57.Damuni Z, Reed LJ. Purification and characterization of a divalent cation-independent, spermine-stimulated protein phosphatase from bovine kidney mitochondria. J. Biol. Chem. 1987;262(11):5133–5138. [PubMed] [Google Scholar]
- 58.Newman JD, Armstrong JM, Bornstein J. Assay of insulin mediator activity with soluble pyruvate-dehydrogenase phosphatase. Endocrinology. 1985;116(5):1912–1919. doi: 10.1210/endo-116-5-1912. [DOI] [PubMed] [Google Scholar]
- 59.Teague WM, Pettit FH, Wu TL, Silberman SR, Reed LJ. Purification and properties of pyruvate-dehydrogenase phosphatase from bovine heart and kidney. Biochemistry. 1982;21(22):5585–5592. doi: 10.1021/bi00265a031. [DOI] [PubMed] [Google Scholar]
- 60.Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J. Clin. Endocrinol. Metab. 1998;83(6):2001–2005. doi: 10.1210/jcem.83.6.4886. [DOI] [PubMed] [Google Scholar]
- 61.Represa J, Avila MA, Miner C, et al. Glycosylphosphatidylinositol/inositol phosphoglycan: a signaling system for the low-affinity nerve growth factor receptor. Proc. Natl Acad. Sci. USA. 1991;88(18):8016–8019. doi: 10.1073/pnas.88.18.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tachado SD, Gerold P, Schwarz R, et al. Signal transduction in macrophages by glycosylphosphatidylinositols of Plasmodium, Trypanosoma, Leishmania: activation of protein tyrosine kinases and protein kinase C by inositolglycan and diacylglycerol moieties. Proc. Natl Acad. Sci. USA. 1997;94(8):4022–4027. doi: 10.1073/pnas.94.8.4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shapiro E, Brown SD, Saltiel AR, Schwartz JH. Short-term action of insulin on aplysia neurons – generation of a possible novel modulator of ion channels. J. Neurobiol. 1991;22(1):55–62. doi: 10.1002/neu.480220106. [DOI] [PubMed] [Google Scholar]
- 64.Robinson PJ. Phosphatidylinositol membrane anchors and T-cell activation. Immunol. Today. 1991;12(1):35–41. doi: 10.1016/0167-5699(91)90110-F. [DOI] [PubMed] [Google Scholar]
- 65.Eardley DD, Koshland ME. Glycosylphosphatidylinositol – a candidate system for interleukin-2 signal transduction. Science. 1991;251(4989):78–81. doi: 10.1126/science.1824727. [DOI] [PubMed] [Google Scholar]
- 66.Merida I, Pratt JC, Gaulton GN. Regulation of interleukin-2-dependent growth-responses by glycosylphosphatidylinositol molecules. Proc. Natl Acad. Sci. USA. 1990;87(23):9421–9425. doi: 10.1073/pnas.87.23.9421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobson PRM, Brown BL. The mechanism of action of interleukin-1 – a cytokine acting through glycosyl-phosphatidylinositol hydrolysis. Adv. Second Messenger Phosphoprotein Res. 1990;24:170–175. [PubMed] [Google Scholar]
- 68.Vivien D, Petitfrere E, Martiny L, et al. IPG (inositolphosphate glycan) as a cellular signal for TGF-β 1 modulation of chondrocyte cell cycle. J. Cell. Physiol. 1993;155(3):437–444. doi: 10.1002/jcp.1041550302. [DOI] [PubMed] [Google Scholar]
- 69.Fanjul LF, Marrero I, Gonzalez J, et al. Does oligosaccharide-phosphatidylinositol (glycosyl-phosphatidylinositol) hydrolysis mediate prolactin signal transduction in granulosa cells? Eur. J. Biochem. 1993;216(3):747–755. doi: 10.1111/j.1432-1033.1993.tb18194.x. [DOI] [PubMed] [Google Scholar]
- 70.Gaulton GN, Kelly KL, Pawlowski J, Mato JM, Jarett L. Regulation and function of an insulin-sensitive glycosyl-phosphatidylinositol during T lymphocyte activation. Cell. 1988;53(6):963–970. doi: 10.1016/s0092-8674(88)90509-0. [DOI] [PubMed] [Google Scholar]
- 71.Reddy KK, Falck JR, Capdevila J. Insulin second messengers – synthesis of 6-O-(2-amino-2-deoxy-α-D-glucopyranosyl)-D-chiroo-inositol-1-phosphate. Tetrahedron Lett. 1993;34(49):7869–7872. [Google Scholar]
- 72.Jaworek CH, Iacobucci S, Calias P, d’Alarcao M. Synthesis of inositol glycan cyclic phosphates. Carbohydr. Res. 2001;331(4):375–391. doi: 10.1016/s0008-6215(01)00047-7. [DOI] [PubMed] [Google Scholar]
- 73.Chakraborty N, d’Alarcao M. 2006. Unpublished results.
- 74.Muller G, Hanekop N, Kramer W, Bandlow W, Frick W. Interaction of phosphoinositolglycan(-peptides) with plasma membrane lipid rafts of rat adipocytes. Arch. Biochem. Biophys. 2002;408(1):17–32. doi: 10.1016/s0003-9861(02)00451-4. [DOI] [PubMed] [Google Scholar]
- 101.Frick W, Mueller G. Office, U. S. P., editor. Inositolglycans having insulin-like action. Patent Number: 6004938 Hoescht Aktiengesellschaft. 1999 ■■ Describes the synthesis and biological activity of 47 IG analogues.