


Abstract
Location analysis for estrogen receptor-α (ERα)-bound cis-regulatory elements was determined in MCF7 cells using chromatin immunoprecipitation (ChIP)-on-chip. Here, we present the estrogen response element (ERE) sequences that were identified at ERα-bound loci and quantify the incidence of ERE sequences under two stringencies of detection: <10% and 10–20% nucleotide deviation from the canonical ERE sequence. We demonstrate that ∼50% of all ERα-bound loci do not have a discernable ERE and show that most ERα-bound EREs are not perfect consensus EREs. Approximately one-third of all ERα-bound ERE sequences reside within repetitive DNA sequences, most commonly of the AluS family. In addition, the 3-bp spacer between the inverted ERE half-sites, rather than being random nucleotides, is C(A/T)G-enriched at bona fide receptor targets. Diverse ERα-bound loci were validated using electrophoretic mobility shift assay and ChIP-polymerase chain reaction (PCR). The functional significance of receptor-bound loci was demonstrated using luciferase reporter assays which proved that repetitive element ERE sequences contribute to enhancer function. ChIP-PCR demonstrated estrogen-dependent recruitment of the coactivator SRC3 to these loci in vivo. Our data demonstrate that ERα binds to widely variant EREs with less sequence specificity than had previously been suspected and that binding at repetitive and nonrepetitive genomic targets is favored by specific trinucleotide spacers.
INTRODUCTION
Estrogens are steroid hormones that play critical roles in the initiation, development and metastasis of breast and uterine cancers (1). The estrogen (E2) response in breast cancer cells is predominantly mediated by the estrogen receptor-α (ERα/NR3A1), a ligand-activated transcription factor (2). ERα regulates the transcription of target genes through direct binding to its cognate recognition sites, known as estrogen response elements (EREs), or by modulating the activity of other DNA-bound transcription factors at alternative DNA sequences (3–10). These functions have recently been demonstrated to occur across great genomic distances (tens to hundreds of kilobases) and even across chromosomes (11–20). Rapid, ‘non-genomic’ estrogen effects have also been described, involving plasma membrane and cytosolic effects of the hormone that may or may not be mediated by the classical estrogen receptors and their various isoforms (11,12).
We and others have recently analyzed targets of estrogen signaling in MCF7 breast cancer cells by performing genome-wide binding site mapping of ERα (13–16). A significant fraction of ERα-bound sites that we identified (∼80%) were located >10 kb from any annotated transcription start site (TSS) (13). Nye and colleagues (17) demonstrated that ERα displays the ability to alter large-scale chromatin structure when tethered or directly bound to DNA. Together, these observations imply long-range interactions between ERα-bound regulatory regions and target promoters, closely tied to higher-order chromatin conformational changes such as chromatin decondensation, compaction, and territory formation (17–22). Some chromatin changes are initiated by unliganded ERα (Apo-ERα) and/or the pioneer factor forkhead box A1 (FOXA1/HNF3α) and may maintain gene targets in a state that is poised for rapid gene activation upon stimulation with estrogen (19,23,24).
Estrogen receptor dimers bind to the canonical 13-bp ERE, GGTCAnnnTGACC, a palindromic inverted repeat (IR) separated by any three nucleotides (nnn) originally identified from conserved sequence alignments of the estrogen-sensitive Xenopus laevis vitellogenin and the chicken apo-VLDL genes (25,26). The consensus ERE sequence was subsequently extended to a 15-bp palindrome (AGGTCAnnnTGACCT) when the flanking sequences were noted to contribute to dimer-binding affinity (27). Once full human genomic sequence data became available, several groups of investigators combined bioinformatic approaches (principally position weight matrices, or PWMs) with large-scale gene expression studies in order to identify E2-responsive and possibly ERα-regulated genes of interest (28–31). The PWMs were designed using fewer than 20 natural EREs. These EREs were all promoter-proximal elements located <∼2 kb from the transcription start sites for their respective genes (27).
The extent to which functional EREs might deviate from the known examples has remained uncertain. Although it was recognized that functional EREs generally did ‘not’ conform to the consensus sequence, in vivo (32,33), experimental data indicated decreased ERα binding to variant ERE sequences in vitro (28,34). In fact, single gene promoter analyses identified functional EREs containing single- double- and triple-nucleotide substitutions from the consensus ERE sequence (27,33).
Even when stringent nucleotide sequence criteria are applied, many more putative EREs exist in the human genome than are bound by ERα in any given cell type (13,14). For example, computational analysis of the human and mouse genomes, allowing up to 2-bp substitutions from the consensus ERE, revealed >17 000 and >15 000 possible EREs within 15 kb of annotated transcription start sites, respectively (28). An unbiased analysis of the published human genome reveals 2310 perfect EREs (13-bp core ERE sequences), 49 803 ERE sequences with only 1-bp deviation from the consensus sequence and 265 482 loci that deviate by only two mismatches. Yet, studies in MCF7 cells have indicated that only ∼1000–10 000 loci are bound by ERα in response to estrogen treatment (13–16,35). Importantly, there is substantial cell type-specific determination of ERα binding sites and this correlates with cell type-specific post-translational histone modifications at receptor-bound sites (36). Distinguishing histone modifications that are necessary for gene activation or repression from chromatin marks that are associated with these respective processes, but not necessarily causative, remains challenging.
Notably, DNA-binding affinity of transcription factors is not the sole determinant of transcription factor function. There is increasing evidence that multiple ERα-bound loci with varying DNA-binding affinities can cooperate to form a productive cis-regulatory module (28,35–39). Low-affinity receptor–DNA interactions may remain transcriptionally productive in some enhancer contexts. Additional determinants of receptor function may include local non-ERE DNA sequences, DNA methylation status, regional chromatin composition and post-translational modifications, cofactor concentrations and the nature of the receptor ligand that is engaged (13,19,24,35,40–42).
Once directly bound to a cis-regulatory element, the first zinc finger of the ERα DNA-binding domain (DBD) binds in the major groove of the DNA double helix and mediates the sequence-specific interaction of the receptor dimers at each half site of the ERE. Estrogen receptor amino acid residues interact with select DNA bases via hydrogen bonding and van der Waals interactions (43,44). The extent of receptor interactions with variant DNA sequences has never been determined on a large scale. Rather, transcription factor–chromatin association has traditionally been determined one element at a time, limiting the statistical power to characterize variations in cis-regulatory elements.
In order to comprehensively identify ERα-bound targets in MCF7 cells, and to address the question of ERE sequence specificity, we recently employed chromatin immunoprecipitation (ChIP) experiments with whole genome DNA arrays (i.e. ChIP-on-chip) (13). Here, these data are combined with data from a similar study conducted by the Brown lab (14) in order to develop a list of high-confidence ERα-bound loci. These immunoprecipitated chromatin fragments are likely to contain true estrogen responsive elements because (i) they were cross-linked to ERα in living cells (directly or via protein intermediaries), and (ii) they were detected by two independent laboratories.
We present the ERE sequences that were identified within 1017 high-confidence ERα-bound ChIP sites and quantify the prevalence of base-pair variations from the consensus ERE sequence. Approximately 50% of all ERα-bound loci do not have a discernable ERE and likely represent sites of ERα tethering via other transcription factors or contain atypical estrogen response elements (i.e. tandem half-ERE sites). Further, most ERα-bound cis-regulatory elements are not consensus EREs and the most commonly bound element in MCF7 cells is not a consensus ERE. We demonstrate that many ERα-bound sites have two or more ERE-like sequences within 2 kb of the center of the ChIP site, suggesting additive or synergistic potential of tandem ERE sequences. We demonstrate that the 3-bp spacer between the inverted ERE half-sites, rather than being random nucleotides, is enriched for selected sequences at in vivo receptor targets. Finally, many functional EREs reside within repetitive DNA elements, particularly of the Alu family of repetitive DNA sequences, and these sequences are likely to contribute to the estrogen-signaling cascade in MCF7 cells.
MATERIALS AND METHODS
Cell Culture
MCF7 cells (ATCC) were grown as described (45). Cells were changed to E2-depleted, phenol red-free media consisting of MEM alpha (Gibco) with 10% charcoal/dextran-stripped calf serum, insulin, penicillin G, streptomycin and L-glutamine (all Gibco), for 72 h prior to treatments. Where indicated, treatments included vehicle control (100% EtOH) and estradiol (10 or 100 nM, Sigma). Telomerase-immortalized Human Endometrial Stromal Cells (HESC cells), a generous gift from Dr. Graciela Krikun, were grown in the same media used for the MCF7 cells. HESC cells have normal chromosome numbers and structures (46).
Preparation of nuclear extracts and electrophoretic mobility shift assay
HESC nuclear extracts (NEs) were purified using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce), according to the manufacturer's; instructions. Human embryonic stem cells (HESCs) have no demonstrable ERα activity using sensitive luciferase reporter assays and no ERα protein detected by western blot analysis (data not shown). However, HESC cell nuclei have cofactors that promote the binding of recombinant ERα (rERα, Affinity Bioreagents) to target DNA in electrophoretic mobility shift assay (EMSA) and these factors enhance binding when compared to recombinant ERα alone. EMSA experiments were therefore conducted using HESC NEs combined with rERα. Protein determinations were performed using the Micro BCA assay (Pierce) and 5 μg of NE (with protease inhibitors, Roche) plus rERα (400 fmol) were run in each lane of a 5% acrylamide gel in TBE/glycerol buffer. Oligonucleotide probes were labeled using the Biotin 3′ End DNA Labeling Kit (Pierce). Each Biotin-labeled probe was used at 20 fmol/lane and binding reactions were performed per LightShift Chemiluminescent EMSA Kit instructions (Pierce). For super-shift assays, relevant antibody was used as indicated (400 ng/reaction): anti-ERa Ab-10 (LabVision) and anti Sp1 H-225 sc-14027 (Santa Cruz). A complete list of oligonucleotide sequences used as probes for EMSA is presented in the Supplementary Table S1.
ChIP—polymerase chain reaction
ChIP was performed as previously described (13). Briefly, MCF7 Cells were E2-deprived for 3 days and then treated with 100 nM E2 or vehicle for 45 min (19,41). Approximately 5 × 106 cells per ChIP were cross-linked with 1% formaldehyde for 10 min at 37°C then quenched with 125 mM glycine. The cells were washed with cold phosphate-buffered saline (PBS) and scraped into PBS with protease inhibitors (Roche). Cell pellets were resuspended in ChIP lysis buffer [1% sodium dodecyl sulfate (SDS), 10 mM ethylenediaminetetraacetic acid (EDTA), 50 mM Tris–HCl (pH 8.1)] and sonicated (Fisher Sonic Dysmembrinator) to produce sheared chromatin with average length 500 bp. The sheared chromatin was submitted to a clarification spin and the supernatant then used for ChIP or reserved as ‘Input’. Antibodies used were anti-ERα (Ab-1, Ab-3 and Ab-10 from Lab Vision and MC-20 from Santa Cruz) and anti-SRC3 (ab2831 Abcam). Quantitative PCR was performed using iQ-SYBR Green Master Mix (Biorad) in a Biorad Opticon 2 cycler. PCR reactions were assembled in triplicate and the enrichment of target sequences in ChIP material was calculated relative to 28S ribosomal RNA coding sequence as a reference. PCR values for E2-treated cells were then normalized to control (E2-depleted) cells. Forward and reverse primer sequences used for ChIP-PCR were designed to hybridize to unique nonrepetitive genomic sequences after excluding repetitive DNA sequences using RepeatMasker V3.1. Primers are listed in the Supplementary Table S1.
Luciferase reporter assays
Luciferase reporter assays were performed using the Luciferase Assay System (Promega) according to the manufacturer's; instructions. Potential ERE-containing regulatory elements were cloned into pGL2-Promoter (Promega) and transfected into MCF7 cells using the TransIT-LT1 Transfection Reagent (Mirus). Cotransfection with a β-galactosidase-expressing plasmid (Promega) enabled normalization of transfection efficiency across samples using a β-galactosidase assay kit (Promega) according to the manufacturer's; instructions.
Cloning and mutagenesis
PCR cloning was performed using PCR amplification of ERα-bound genomic loci from HESC cell genomic DNA which was prepared using the Genomic DNA Extraction kit (Qiagen) according to the manufacturer's; instructions. PCR products, average length 776 bp, were ligated into the reporter construct pGL2-promoter (Promega) at 5′-KpnI and 3′-XhoI sites, for use in Luciferase Reporter assays. Mutagenized reporter constructs were prepared using the Genetailor Site-Directed Mutagenesis System (Invitrogen) according to the manufacturer's; instructions. All clones and subclones were confirmed by DNA sequencing. Primers used for genomic locus amplification, subcloning, and site-directed mutagenesis are available upon request.
Computational detection of ERE aequences
Genome-wide location analysis for ERα, and E2-dependent gene expression profiling, were performed by two independent groups as previously described (13,14). The 1017 ERα-bound genomic loci common to both data sets (shared loci defined as falling within 1 kb of the center of each locus) were interrogated for ERE-like sequences. Starting at the center of each high-confidence ERα-bound locus, we extracted genomic sequences 1 kb in each direction (hg 18, build 36.1). As the chromatin shear size in ChIP experiments was optimized to average ∼500 bp, we estimated that interrogating sequences of average size 2 kb would have a reasonable likelihood of capturing most sequences directly bound by ERα in the ChIP assays. Transcription Element Search Software (TESS) was used to identify ERE sequences (47). Default settings were used with variation in Maximum Allowable String Mismatch of 10% and 20%. The TESS software will identify binding sites using consensus strings from the TRANSFAC, JASPAR, IMD and CBIL-GibbsMat databases. Repetitive DNA elements were determined using RepeatMasker V3.1 at the default settings (http://www.repeatmasker.org).
Statistics
Comparisons between two groups were made using a two-tailed Student's; t-test with P-values indicated. Statistical analysis of the base pair distributions in the ERE spacer sequences was performed using the Pearson's; chi-square to test for goodness of fit.
RESULTS
EREs found at 1017 high-confidence ERα-bound loci
We recently performed location analysis for ERα in MCF7 cells using ChIP-on-chip with whole genome tiling arrays and combined these data with gene expression profiling in response to E2 exposure (13). This work resulted in an expanded understanding of pathways involved in E2-mediated cellular proliferation and identified the E2-responsive chromatin protein, H2A.Z, as a predictor of breast cancer progression. The location analysis identified 1615 genomic targets of ERα and revealed that the majority (∼80%) of ERα-bound loci reside >10 kb from any annotated transcription start site. Further, of the E2-regulated genes that were identified, only 5.1% had an ERα-bound locus within 10 kb of the transcription start site, while 39% had a ChIP site within 200 kb of the TSS. Of 1615 loci that were bound by ERα in our analysis, 1017 (∼60%) were also detected by the Brown group (14). A list of the genomic coordinates for these highest-confidence ERα-bound loci appears in the Supplementary Table S2. ChIP-on-chip data were validated by ChIP-PCR for 17 sites and revealed E2-dependent recruitment (>2-fold) of ERα at all of the loci that were tested (Figure 1, for genomic coordinates see Table 2). All primers used for ChIP-PCR were designed to hybridize to unique nonrepetitive genomic sequences after excluding repetitive DNA sequences using RepeatMasker V3.1.
Figure 1.

ChIP-PCR validation of ERα-bound loci. ERα-bound loci determined by ChIP-on-chip were interrogated for E2-dependent ERα binding using quantitative ChIP-PCR (for genomic coordinates see Table 2). MCF7 cells were treated with E2 (100 nM) or vehicle (EtOH) for 45 min. ChIP was performed using antibodies against ERα. Quantitative PCR using genomic primers employed the 28S rRNA coding sequence as internal reference. Shown are E2-treated values normalized to control values. ERα was not recruited to an ERE-less and ChIP-on-chip negative locus dubbed ERE(–). Similar studies using genomic PCR primers for the known E2-responsive genes TFF1 and CTSD demonstrated rapid recruitment of ERα to these enhancer regions in response to E2. ChIP-PCR targeting 17 additional loci similarly revealed E2-dependent enrichment (>2-fold) of ERα at all sites. Values are the average of three experiments with SEM. Oligonucleotide sequences are presented in Supplementary Table S1.
Table 2.
ChIP sites cloned and employed in luciferase reporter assays
 |
aID assignments from Hua et al., Mol. Syst. Biol. 2008.
ChIP names were assigned and correspond to previously published ChIP ID numbers (13). ERE sequences detected within 2 kb of the center of each locus are indicated and mismatches from the consensus ERE sequence are highlighted in red. For ERE sequences residing within a repetitive DNA element, the type of element is indicated and its distance to the nearest E2-regulated gene is shown.
Using Transcription Element Search Software (TESS) (47), we performed an analysis of the 1017 ERα-bound loci (average length 2 kb) for the presence of ERE sequences. This analysis was performed at two stringencies of ERE detection: ≤ 10% nt deviation (≤2 mismatched residues within the core 15-bp ERE), and 10–20% nucleotide divergence (3–4 mismatched residues) from the 15-bp consensus ERE sequence (AGGTCAnnnTGACCT). We identified a total of 646 ERE sequences (Supplementary Table S3) from 509 ERα-bound loci, indicating that ∼50% of receptor-bound sites did not have a discernable ERE sequence; 391 (∼77%) of the ERE-containing ChIP sites contained a single ERE sequence, 101 (∼20%) contained two distinct ERE sequences and 17 loci (∼3%) contained three or more distinct ERE sequences within 2 kb of the center of their respective ChIP sites.
ERα-bound loci are highly enriched for non-consensus EREs
The sequence requirements for ERα binding to chromatin in vivo are surprisingly flexible. Table 1 demonstrates the base frequency at each position in the 15-bp ERE sequence for each stringency assayed (A and B in Table 1). The base frequencies are pooled for all EREs that were detected (0–20% nt divergence from consensus) (C in Table 1). At all stringencies assayed, the 13-bp core bases were more highly conserved than the flanking sequences located at positions 1 and 15 of the EREs. When we considered an additional 15 bp of flanking sequence in the 5′ and 3′ directions from the EREs no conservation of nucleotide sequences was found indicating that the ERE motif does not extend beyond these 15 bases (data not shown).
Table 1.
Base frequency matrix as a function of nucleotide position for ERE sequences detected
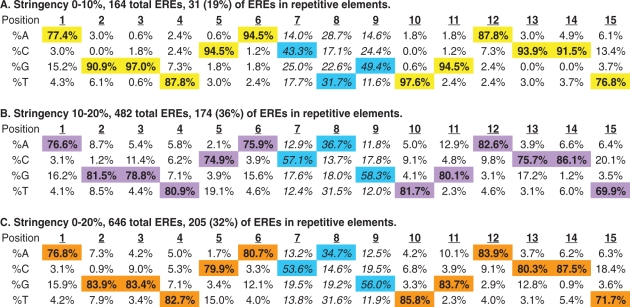 |
Transcription Element Search Software (TESS) was used to analyze 1017 ERα-bound loci at two different stringencies of detection. High stringency (0–10% deviation from the consensus ERE sequence) and low stringency (10–20% deviation from the consensus ERE) results are shown in A and B, respectively. These data are pooled (in C) to reveal a total of 646 ERE sequences residing within 509 receptor-bound loci. The distribution of ERE sequences within repetitive elements is indicated for each stringency. The trinucleotide spacer sequences are non-randomly distributed at all stringencies of detection (see main text) even when repetitive element EREs are excluded from the analysis (see Supplementary Table S5). Bold values (also highlighted) are the most common bases found at each position of the detected EREs and the trinucleotide spacer sequences are indicated in italics.
It is noteworthy that, even at high stringency of detection, most possible single base substitutions were detected in our dataset. These data are consistent with in vitro data (EMSAs) indicating that all single base-pair deviations from consensus are capable of binding to the estrogen receptor, although with variable affinity (28). When ERE detection criteria were relaxed to permit 10–20% base divergence from the consensus sequence, all forms of nucleotide substitutions were permissive for receptor binding though some substitutions were rarer than others (B and C in Table 1). For example, from all EREs that were detected, position 2 is rarely (<1%) cytosine, whereas position 12 is cytosine in 9.1% of EREs (C in Table 1).
Ignoring the trinucleotide spacer sequence, 348 different ERE sequences were detected from the group of 646 total EREs (Supplementary Table S4). There was equal representation of an imperfect ERE (16 examples of GGGTCAnnnTGACCT) and a perfect consensus ERE (16 examples of AGGTCAnnnTGACCT) (Supplementary Table S4). Excluding analysis of the less-conserved positions 1 and 15 in the 646 ERE sequences that we identified, we detected 51 (∼8%) perfect core consensus EREs (GGTCAnnnTGACC). Thus, of the 2310 perfect consensus ERE sequences detected in the published sequence of the human genome, our highest-confidence location analysis revealed ERα occupancy at only 51 (2.2%) of these sites in MCF7 cells. These data demonstrate that ERα binds to widely variant EREs in MCF7 cells and that many ‘perfect’ EREs are not receptor-bound in these cells under these E2-stimulated culture conditions.
The trinucleotide spacer sequences of ERα-bound EREs are conserved residues
The trinucleotide spacer sequence between the two ERE half sites does not make important base contacts with the estrogen receptor's; DBD (43,44,48) and has historically been described as nnn (meaning that any 3-bp sequence will suffice). Our data indicate that, at all stringencies of ERE detection, the trinucleotide spacer is conserved at receptor-bound EREs. Specifically, positions 7–9 are preferentially C(A/T)G at ERα-bound loci; this spacer sequence is found at more than 40% of EREs (Supplementary Table S4 and Table 1). When compared to the expected equal distribution of bases at each position, a statistically significant nonrandom distribution of sequences at positions 7–9 was indicated by the chi-square test with P-values of 1.9E–14, 2.04E–96 and 7.97E–106 for stringencies 0–10% (A), 10–20% (B) and 0–20% (C), respectively (Table 1). The observed conservation of the central triad sequence remained even when all repetitive element EREs were excluded from the analysis (Supplementary Table S5). While the molecular justification for this triad sequence preference is unclear, these data suggest that the 3-bp spacer has functional significance, possibly modulating ERα-ERE binding and subsequent transcriptional responses.
To identify ERE sequences which might distinguish between promoter-proximal and distal enhancer functions, we further analyzed our gene-expression data previously integrated with genome-wide location analysis for ERα (13). Of the E2-regulated genes that were identified, only 5.1% had an ERα-bound locus within 10 kb of the transcription start site (13). From our list of computationally detected ERE sequences (Supplementary Table S3), we identified 13 EREs that reside within the promoter regions of E2-regulated genes (defined as +/–1 kb from the annotated transcription start site for each gene). Analysis of EREs residing within E2-regulated gene promoters demonstrated similar enrichment for the C(A/T)G trinucleotide spacer sequence (data not shown). This observation suggests that the conservation of the C(A/T)G trinucleotide spacer sequence exists for both proximal promoter and distal enhancer EREs and does not distinguish between these functions in MCF7 cells.
We next focused our analysis on the 103 ERα-bound ERE sequences that reside within 100 kb of an E2-regulated gene (13). This analysis compared estrogen-stimulated versus estrogen-repressed genes and indicated similar enrichment of the C(A/T)G trinucleotide spacer sequence at the ERE sequences of these respective loci (data not shown). Thus, our data do not support a role for the trinucleotide spacer sequence in distinguishing between E2-stimulated and E2-repressed gene targets in MCF7 cells.
Many EREs reside within repetitive DNA elements
In our analysis, we found that a considerable proportion of EREs lay within repetitive element sequences (Supplementary Table S3). Depending upon the stringency of detection, between 19% and 36% of EREs resided within repetitive (i.e. repeat-masked) DNA elements. The most common repetitive element harboring ERE-like sequences was the Alu retrotransposable element (a member of the short interspersed element, or SINE, family). However, we detected EREs within a broad range of repetitive sequences including non-Alu SINE elements, long interspersed elements (LINEs), DNA transposable elements, long terminal repeat (LTR) retrotransposons, non-LTR retrotransposons and microsatellites [a.k.a. simple sequence repeats (SSRs)].
One group initially reported the existence of an ERE within an Alu sequence near the BRCA1 gene (49). Further study of their sequence revealed that this putative-ERE was non-functional in an ERα-containing and E2-responsive model cell system, calling into question their initial studies which used a hepatocyte cell system that was engineered to overexpress ERα (50). Additional findings in MCF7 cells ultimately lead the authors to conclude that this putative Alu-ERE did not function as a classical ERE and was unlikely to be an ERα-responsive enhancer (50). Our recent ChIP-on-chip data similarly do not support BRCA1 to be a direct ERα gene target in MCF7 cells (13).
The canonical 280-bp Alu sequence is composed of two monomers, derived from the 7SL RNA gene, separated by an adenosine rich connector (Figure 2A). Each monomer present in the progenitor Alu family sequences has an ERE-like sequence which, with mutation accumulation, might reasonably form a functional ERE (Figure 2B) (51–53). The majority of Alu-ERE elements that were detected from our ChIP sites resided within 3′ monomer sites of Alu elements (Supplementary Table S3).
Figure 2.

The canonical 280-bp Alu sequence is composed of two monomers, derived from the 7SL RNA gene, separated by an adenosine rich connector. The consensus AluSc sequence (A) contains an ERE-like sequence (red) identified in the 3′ monomer of the element. A similar element (blue) also occurs in the 5′ monomer of the Alu element. The monomers are separated by an A-rich connector sequence (underlined). The previously indentified Alu-Retinoic Acid Receptor response element (RARE) sequence is also indicated (purple). (B) Diverse ERE-like sequences located in the 3′-monomers of classical Alu element family sequences are indicated. Mismatches from the consensus ERE sequence are indicated by lower case letters.
Selected repetitive element EREs are capable of binding to ERα, in vitro
Given the widespread variations in ERE sequences detected from our ChIP-positive loci, we tested two ChIP-positive repetitive element EREs in order to illustrate that our variant ERE sequences (complete with their respective trinucleotide spacer sequences) are capable of binding to ERα in vitro. EMSAs indicated that the 3′ ERE sequence of locus B68 (a MIRb element) and the 5′ ERE sequence of locus D66 (also a MIRb element) bind specifically to ERα (Figure 3). Specific binding of DNA probes to ERα-containing protein complexes was indicated by anti-ERα antibody-mediated supershift (for the consensus ERE probe) or loss of binding (for the two repetitive DNA element probes). Nonspecific antibody (anti-Sp1) had no effect on ERα binding to consensus or B68 EREs but demonstrated some inhibition of protein binding to the D66-ERE, indicating that Sp1 may participate in the ERα-containing protein complexes that bind to this element. Sp1 is a ubiquitous transcription factor which can physically interact with ERα at imperfect and half-site EREs spaced near Sp1-REs (10,54). Such compound regulatory elements generally include GC-rich sequences similar to the probe for D66-ERE, which was designed entirely based upon the D66-ERE genomic sequence. Together, these data provide in vitro support for the conclusion that repetitive DNA element EREs are able to recruit ERα binding in vivo.
Figure 3.

Electrophoretic mobility shift assay (EMSA) of ERα binding to nonconsensus repetitive element ERE sequences. EMSA was performed using HESC cell nuclear lysates plus recombinant ERα and labeled consensus ERE probe (lanes 1–3) or repetitive DNA element ERE sequences as probes: B68 (lanes 4–6) and D66 (lanes 7–9). An ERα-containing complex bound to all three ERE sequences (arrowhead, lanes 1, 4 and 7) and was confirmed by supershift (bracket) using a monoclonal antibody that recognizes ERα (lane 2). Specific ERα-containing complexes were similarly shown by loss of bands for B68 (lane 5) and D66 (lane 8) when anti-ERα antibody was added. The nonspecific antibody recognizing Sp1 had no effect on the ERα-containing complexes bound to the classical ERE (lane 3) and B68 (lane 6), but displayed moderate effects on complexes bound to D66 (lane 9), indicating that Sp1 may cooperate with ERα in binding to this sequence. Mismatches from the consensus ERE sequence are indicated by lower case letters and red font. Oligonucleotide sequences are presented in Supplementary Table S1.
Selected repetitive element EREs mediate ERα-dependent transcriptional responses
Our observation that ERα-bound Alu elements represent a class of repetitive DNA elements that contain EREs prompted us to perform functional testing of these and other repetitive element EREs. We cloned a representative sampling of ChIP-positive, ERE-containing genomic loci (summarized in Table 2) to test these for E2-dependent enhancer function in luciferase reporter assays. Our loci were selected to include nonrepetitive element and repetitive-element ERE sequence(s). Some cloned loci contained more than one ERE sequence. The majority of loci resided <250 kb from at least one E2-responsive gene as determined by our prior gene expression profiling (13) and as reported in the Estrogen Responsive Genes Database (55).
Cloned loci demonstrated transcriptional responses that ranged from zero to strong E2-dependent responses in luciferase reporter assays. Figure 4 displays the responses of several cloned loci that demonstrated strong enhancer functions in response to E2. Locus D54 is near the E2-responsive gene MSX2 and harbors three ERE sequences (indicated above the bars in the graph). The third ERE at this locus resides in an MIR-element [SINE family of retrotransposable elements (Table 2)] and is indicated by the red font. Mismatches from consensus ERE sequences are indicated by lower case lettering. Site-directed mutagenesis of each ERE (wherein both half-sites are replaced with tttttt) indicated that the first and second EREs are each necessary for enhancer function as loss of either site (i.e. E1M and E2M) results in loss of reporter activity (Figure 4A). Mutation of the third site (E3M), the MIR-ERE, resulted in nearly 3-fold diminishment of enhancer function, indicating that it contributes to overall enhancer function of the cloned genomic fragment. We conclude that the MIR-ERE is necessary for full enhancer activity of the D54 locus in response to E2.
Figure 4.

Luciferase reporter assays of cloned genomic loci bound by ERα and displaying high enhancer activity in MCF7 cells. Genomic loci (average length 776 bp) containing one or more ERE sequences were cloned into a minimal promoter reporter construct driving luciferase gene expression. Specific wild-type ERE sequences and mutated (i.e. E1M) ERE sequences are indicated above each clone being tested (repetitive element EREs are indicated by red font, mismatches from the classical ERE sequence are indicated by lower case lettering, and mutated sequences were changed to tttttt where indicated). Basal and E2-stimulated luciferase values are shown normalized to co-transfected β-galactosidase-expressing plasmid. Clone D54 contains three ERE sequences and the third is in a repetitive element (see Table 2) which contributes to overall enhancer function (A). B68 contains two ERE sequences both of which reside within repetitive DNA elements and both contribute to enhancer function (B). D70 also contains two ERE sequences both of which reside within repetitive DNA elements (C). Values are the average of three experiments, performed in triplicate, with SEM indicated. *P < 0.01 comparing E2-treated wild type with E2-treated mutated reporter. §P < 0.01 comparing E2-treated empty vector with E2-treated mutated reporter. †P < 0.01 comparing vehicle control-treated with E2-treated reporter.
Figure 4B indicates the transcriptional response of locus B68 which harbors two repetitive element ERE sequences (indicated by red font), the first residing in an Alu element and the second in a MIRb element (Table 2). While mutagenesis of the MIRb-ERE (E2M) ablated enhancer function, loss of the Alu-ERE element (E1M) similarly resulted in ∼50% reduction in enhancer function of the cloned locus. These data support a model in which both repetitive element EREs contribute to B68-mediated enhancer function in response to E2. The B68 locus resides near the E2-responsive gene MREG and may contribute to E2-dependent enhancement of this gene (Table 2) (13,30).
D70 is a locus that contains two repetitive element EREs (indicated by red font), the first in an Alu sequence and the second in a simple repeat (microsatellite) sequence (Table 2). D70 resides near the E2-responsive genes IER3 and PRR3. While the relative contribution of each ERE to enhancer function was not assayed (both are in repetitive DNA elements), the combined effects of the EREs clearly indicate strong enhancer functions in response to E2 (Figure 4C).
Figure 5 displays the responses of additional cloned loci that demonstrated moderate, weak, and zero enhancer functions in luciferase reporter assays. The D75 locus, near the E2-responsive gene ELOVL5, has two ERE elements that reside within a non-LTR/CR1 repetitive sequence (a LINE element, Table 2). E2-dependent enhancer function of this locus was observed (Figure 5A). The C31 locus, near the E2-responsive gene SSR3, contains two ERE sequences neither of which resides within repetitive DNA elements. The C31 locus demonstrated E2-dependent enhancer function in which both ERE sequences contributed to overall transcriptional response and wherein the second ERE proved necessary for a response (Figure 5B).
Figure 5.

Luciferase reporter assays of cloned genomic loci bound by ERα and displaying moderate, low, and no enhancer activity in MCF7 cells. Genomic loci (average length 776 bp) containing one or more ERE sequences were cloned into a minimal promoter reporter construct driving luciferase gene expression. Specific wild-type ERE sequences and mutated (i.e. E1M) ERE sequences are indicated above each clone being tested (repetitive element EREs are indicated by red font, mismatches from the classical ERE sequence are indicated by lower case lettering, and mutated sequences were changed to tttttt where indicated). Basal and E2-stimulated luciferase values are shown normalized to co-transfected β-galactosidase-expressing plasmid. D75 contains two repetitive element ERE sequences responsible for enhancer function (A). C31 contains two, nonrepetitive element EREs that are functional (B). Additional ERE-containing clones with low or no enhancer function in luciferase assays are indicated (C). Values are the average of three experiments, performed in triplicate, with SEM indicated. *P < 0.01 comparing E2-treated wild-type with E2-treated mutated reporter. §P < 0.01 comparing E2-treated empty vector with E2-treated mutated reporter. †P < 0.01 comparing vehicle control-treated with E2-treated reporter.
Additional repetitive and nonrepetitive ERE-containing genomic sequences were tested for enhancer function, revealing modest or no transcriptional enhancer function for this set of cloned fragments (Figure 5C). Importantly, all of the cloned loci for these studies were first identified using ChIP-on-chip and ERα recruitment to all sites was subsequently confirmed using ChIP-PCR (Figure 1). Despite confidence that ERα is recruited to all of these sites in response to E2, not all of these sites behaved as transcriptional enhancers in reporter assays. It remains possible that, in their native chromatin contexts with appropriate regional cis- and trans-acting factors, these elements could participate in E2-mediated transcriptional responses. Alternatively, some of these weak/nonacting loci may not serve as enhancer elements, in vivo, despite their recruitment of ERα. Our reporter assays indicate that arbitrarily sized genomic fragments, whether containing repetitive element EREs (i.e. I47 and K31), nonrepetitive element EREs (i.e. K32, F78 and G99), or combinations of each (i.e. J58 and I20), will not predictably demonstrate in vitro transcriptional responses. Nevertheless, in aggregate, our data provide evidence supporting a functional role for repetitive DNA element EREs in ERα-mediated transcriptional responses.
Loci with repetitive element EREs demonstrate E2-dependent recruitment of SRC3
In response to estrogen exposure, functional EREs recruit ERα-bound cofactors leading to enhanced or repressed target gene expression. In MCF-7 cells, an important coactivator for the estrogen receptor is SRC3 (NCOA3) (56). We performed ChIP-PCR using an antibody directed against SRC3 and tested a selection of loci for hormone-dependent recruitment of SRC3. Focusing principally on loci which were shown to contain repetitive element EREs that contribute to overall luciferase reporter activity (Figures 4 and 5), we demonstrated greater than 2-fold enhanced recruitment of SRC3 to these genomic sites in response to estrogen when compared to vehicle-treated cells (Figure 6). In addition, a control locus that did not recruit ERα in our original studies (i.e. an ERα ChIP-negative site), dubbed ERE(–), also did not recruit SRC3 in response to estrogen. Interestingly, two loci that were ChIP-positive using antibodies against ERα but were inactive in our luciferase reporter assays, G99 and K31 (Figure 5C), were nonetheless sites in which E2 exposure resulted in the recruitment of SRC3 (Figure 6). This observation suggests that the in vivo function of a cis-regulatory element may not always be recapitulated using assays in vitro, as mentioned above. In aggregate, the hormone-dependent recruitment of both ERα and its coactivator to repetitive element ERE-containing loci, combined with functional studies in enhancer-reporter assays, strongly support a role for repetitive element ERE sequences in mediating ERα-dependent transcriptional responses.
Figure 6.

ChIP-PCR confirms estrogen-dependent recruitment of SRC3 to repetitive element ERE-containing genomic loci. MCF7 cells were treated with E2 (100 nM) or vehicle (EtOH) for 45 min. ChIP was performed using antibodies against SRC3. Quantitative PCR using genomic primers employed the 28S rRNA coding sequence as internal reference. Shown are E2-treated values normalized to control values. SRC3 was not recruited to an ERE-less and ERα ChIP-negative locus dubbed ERE(–). Similar studies using genomic PCR primers for the known E2-responsive gene TFF1 demonstrated rapid recruitment of SRC3 to this enhancer region in response to E2. ChIP-PCR targeting seven additional loci revealed E2-dependent enrichment (>2-fold) of SRC3 at all sites. B68, D54, D66, D70, D75 and K31 are all genomic loci that demonstrate repetitive element ERE sequences. Values are the average of four experiments with SEM. Oligonucleotide sequences are presented in Supplementary Table S1.
DISCUSSION
Recent location analysis for ERα-binding sites throughout the human genome revealed evidence for substantial long-range (>10 kb) enhancer functions of the receptor (13,14). Here, we reported the presence of full ERE sequences at approximately half of all receptor-bound loci. The absence of ERE sequences at many ERα-bound genomic loci may reflect widespread tethering of ERα to DNA targets via alternative transcription factors [i.e. AP-1, Sp1 (10)] or the presence of widely divergent ERα-binding motifs not detected using our motif searching software. With regard to tethering to genomic targets via protein–protein interactions with transcription factors such as AP-1 and Sp1, there exist examples where the estrogen receptor does not directly interact with DNA sequences and others where ERE half-sites (i.e. AGGTCA) enable estrogen receptor dimers to bind to DNA while stabilized by a cooperating transcription factor that is proximally bound to DNA (10). We, and others, have demonstrated significant enrichment of binding site motifs for Sp1 and AP-1 at ERα-bound genomic loci (13–15).
Although estrogen receptor dimers do not bind to isolated half-ERE sequences in vitro (27), and receptor dimerization is an important requisite for stable interaction with target DNA sequences (44), there are reports of receptor-mediated transcriptional responses at tandem half-ERE sites (57,58). The extent to which such ERE half-sites actually represent degenerate full EREs or composite elements (cooperating with alternative transcription factors in vivo) remains uncertain. It is noteworthy, however, that our dataset of 1017 high-confidence ChIP sites is significantly enriched for ERE half-sites when compared to permutations of 1017 randomly selected genomic sequences of the same size. We observed an average of 2.2 half-EREs per ChIP site in our data set, compared with an average of 1.1 half-EREs in the randomly selected genomic sequences in our permutation analysis (P = 0.000004 from 100 000 permutations). An in vivo role for tandem ERE half-sites in ERα-mediated transcriptional responses merits further study.
Unexpectedly, we observed that ∼20–30% of predicted EREs reside within repetitive DNA sequences, principally within the 3′ monomers of Alu elements. We found only one prior report of an Alu-ERE which was later demonstrated to be nonfunctional (49,50). Our data indicate considerable potential for repetitive element EREs, and Alu-EREs in particular, to contribute to ERα-mediated gene regulation. We tested several Alu-EREs, and EREs that reside in alternative repetitive DNA elements, and found that these contribute to enhancer function and are located near E2-regulated genes.
Alu elements expanded extensively throughout primate evolution and now occupy ∼10% of the human genome (59). Their expansion throughout primate genomes depended upon the transposition machinery encoded by L1 retrotransposons, which are LINEs (long interspersed elements) (52). Increasing evidence suggests that Alu repeats are a source of diverse cis-regulatory elements that regulate transcriptional initiation by RNA polymerase II (60–62). The Mader group recently reported the presence of diverse Retinoic Acid Receptor (RAR) response elements (RAREs) residing within the 5′ monomers of Alu elements (Alu-RAREs) (63). There is emerging evidence that E2 and RA can exert opposing gene-regulatory effects in breast cancer cells via receptor-mediated functions at neighboring cis-regulatory loci (64). It is possible that some of these competing gene-regulatory effects occur via ERα and RAR functioning at neighboring 3′ monomer Alu-ERE and 5′ monomer Alu-RARE sequences, respectively.
The expansion of Alu elements through Alu transposition may have distributed gene regulatory elements which were later evolutionarily maintained (61,63). In particular, this process may have contributed to the evolution of novel E2-dependent gene regulatory networks. Given the wide spectrum of regulatory sites present in Alus, it remains important to include these loci in genome-wide screening for transcription-factor response elements and to consider these loci, which are often repeat-masked and excluded, when performing studies pertaining to identification of disease predisposition markers.
Our study comprises the largest collection of ERE sequences published to date (Supplementary Table S3) and is supported by evidence of in vivo ERα binding at these sites. Our observations indicate that most ERα-bound cis-regulatory elements are not consensus EREs and that considerable deviation from the consensus sequence can be permissive for receptor binding in vivo. Low-affinity interactions between transcription factors and imperfect DNA binding sites have historically been difficult to detect in vivo. Recent data in yeast suggest that such interactions may be more common, and provide more biological impact, than had previously been suspected (65). In addition, promiscuity of DNA-binding sequences for a multitude of transcription factors in mammals was recently suggested based upon a systems approach to binding site detection in the mouse (66). Together with our data, these observations suggest numerous low-affinity or transient transcription-factor–DNA interactions occurring with diverse DNA sequences, some of which may modulate transcriptional responses.
We found that 23% of ERE-containing sites have two or more ERE-like sequences within 2 kb of the center of the ChIP site. This finding indicates that, in addition to distantly spaced enhancers contributing to E2-mediated transcriptional responses (28,35–39), some enhancer regions may be composed of multiple, imperfect, ERE sequences that provide additive or synergistic cis-regulatory potential. This conclusion is supported by the function of selected tandem EREs interrogated in our luciferase reporter assays.
We found that the trinucleotide spacer sequences between ERE half-sites is non-random at ERα-bound loci, wherein C(A/T)G spacers are favored. This finding held true even when repetitive element (i.e. repeat-masked) EREs were excluded. It has been suggested that DNA sequence serves as an additional ‘ligand’ for transcription factor-containing protein complexes and can contribute to receptor dimerization at imperfect EREs, alter receptor conformation, and influence the net transcriptional response of an ERE (67–71). Our data suggest that the effects of the ERE sequence on ERα-mediated transcriptional responses may be influenced by the trinucleotide spacer sequence, a hypothesis that we are currently testing. In addition, the trinucleotide spacer sequence may add predictive value when scoring sequences for functional ERE motifs.
Full function of repetitive element and nonrepetitive element EREs may depend upon the presence and distribution of additional cis-regulatory elements which work cooperatively in order to promote a transcriptional response (28,36–39). Even high-confidence ERα-bound loci with nearly perfect consensus ERE sequences demonstrate unpredictable cis-regulatory function in reporter assays, a feature, which seems to reflect the influence of surrounding DNA sequences. Such observations have been noted by many authors when performing promoter–reporter analyses using serially truncated promoter sequences: function can be enhanced, lost, and then enhanced again, with successive promoter truncations/mutations. These findings are consistent with a model in which DNA sequence and context will dictate the repertoire of inhibitory and activating co-factors, which, collectively, determine the net transcriptional response of any cloned DNA fragment. Whether genomic loci containing multiple EREs represent stronger enhancers or repressors of target gene expression, regulate multiple gene targets, or recruit different cofactor complexes when compared to single ERE-containing loci remains to be studied.
The chromatin modifications that are necessary for ERα-mediated transcriptional responses remain incompletely described. We recently reported that the gene for the variant histone H2A.Z is E2-responsive in MCF7 cells and found that H2A.Z protein expression is an independent predictor of breast cancer survival (13). We also showed that H2A.Z is necessary for the E2-stimulated proliferative response in MCF7 cells. Gevry et al. (42) recently demonstrated that H2A.Z is cyclically incorporated into the enhancer and promoter regions of ERα gene targets and is important to gene induction by the liganded receptor. Combined, these data argue for a feed-forward loop in breast cancer cells in which E2 stimulates H2A.Z production, which in turn maximizes ERα-mediated transcriptional responses.
Interestingly, H2A.Z is also highly enriched at both glucocorticoid-inducible and constitutively nuclease accessible glucocorticoid receptor-bound sites, suggesting a shared chromatin remodeling mechanism modulating the transcriptional responses of these nuclear hormone receptors (72). Indeed, increasing evidence indicates that the recruitment of H2A.Z to active promoters and enhancers may represent a general mechanism permitting or promoting gene activation in mammalian cells (73). Given that a minority of predicted ERE sequences is operative in any given cell type (36) and that diverse repetitive element and nonrepetitive element ERE sequences are capable of recruiting ERα in vivo, understanding which ERE sequences are functional in a given cell type and milieu remains challenging. The cell-type-specific determinants of ERE utilization remain to be fully understood as are the mechanisms by which these determinants are maintained or modulated by the cellular milieu.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Research Scientist Development Program (to C.B.K.); and the National Institutes of Health (NIH-5K12HD00849 to C.B.K., and R01-CA129424 to N.S. and C.B.K.). Funding for open access charge: National Institutes of Health ((NIH-5K12HD00849).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Drs. Graciela Krikun and Charles Lockwood for providing the immortalized human endometrial stromal cell line, and to Drs. Joshua R. Friedman and Robert N. Taylor for scientific discussions.
REFERENCES
- 1.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 2.Smith CL, O'M;alley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 3.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 4.Cerillo G, Rees A, Manchanda N, Reilly C, Brogan I, White A, Needham M. The oestrogen receptor regulates NFkappaB and AP-1 activity in a cell-specific manner. J. Steroid Biochem. Mol. Biol. 1998;67:79–88. doi: 10.1016/s0960-0760(98)00078-8. [DOI] [PubMed] [Google Scholar]
- 5.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 6.Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999;13:1672–1685. doi: 10.1210/mend.13.10.0357. [DOI] [PubMed] [Google Scholar]
- 7.Khan S, Abdelrahim M, Samudio I, Safe S. Estrogen receptor/Sp1 complexes are required for induction of cad gene expression by 17beta-estradiol in breast cancer cells. Endocrinology. 2003;144:2325–2335. doi: 10.1210/en.2002-0149. [DOI] [PubMed] [Google Scholar]
- 8.Kim K, Barhoumi R, Burghardt R, Safe S. Analysis of estrogen receptor alpha-Sp1 interactions in breast cancer cells by fluorescence resonance energy transfer. Mol. Endocrinol. 2005;19:843–854. doi: 10.1210/me.2004-0326. [DOI] [PubMed] [Google Scholar]
- 9.Krishnan V, Wang X, Safe S. Estrogen receptor-Sp1 complexes mediate estrogen-induced cathepsin D gene expression in MCF-7 human breast cancer cells. J. Biol. Chem. 1994;269:15912–15917. [PubMed] [Google Scholar]
- 10.Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J. Mol. Endocrinol. 2008;41:263–275. doi: 10.1677/JME-08-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2001;12:152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- 12.Bjornstrom L, Sjoberg M. Estrogen receptor-dependent activation of AP-1 via non-genomic signalling. Nucl. Recept. 2004;2:3. doi: 10.1186/1478-1336-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua S, Kallen CB, Dhar R, Baquero MT, Mason CE, Russell BA, Shah PK, Liu J, Khramtsov A, Tretiakova MS, et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 2008;4:188. doi: 10.1038/msb.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 15.Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon YS, Garcia-Bassets I, Hutt KR, Cheng CS, Jin M, Liu D, Benner C, Wang D, Ye Z, Bibikova M, et al. Sensitive ChIP-DSL technology reveals an extensive estrogen receptor alpha-binding program on human gene promoters. Proc. Natl Acad. Sci. USA. 2007;104:4852–4857. doi: 10.1073/pnas.0700715104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nye AC, Rajendran RR, Stenoien DL, Mancini MA, Katzenellenbogen BS, Belmont AS. Alteration of large-scale chromatin structure by estrogen receptor. Mol. Cell. Biol. 2002;22:3437–3449. doi: 10.1128/MCB.22.10.3437-3449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collingwood TN, Urnov FD, Wolffe AP. Nuclear receptors: coactivators, corepressors and chromatin remodeling in the control of transcription. J. Mol. Endocrinol. 1999;23:255–275. doi: 10.1677/jme.0.0230255. [DOI] [PubMed] [Google Scholar]
- 19.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Carroll JS, Brown M. Estrogen receptor target gene: an evolving concept. Mol. Endocrinol. 2006;8:1707–1714. doi: 10.1210/me.2005-0334. [DOI] [PubMed] [Google Scholar]
- 21.Lupien M, Eeckhoute J, Meyer CA, Krum SA, Rhodes DR, Liu XS, Brown M. Coactivator function defines the active estrogen receptor alpha cistrome. Mol. Cell. Biol. 2009;29:3413–3423. doi: 10.1128/MCB.00020-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lupien M, Brown M. Cistromics of hormone-dependent cancer. Endocr. Relat. Cancer. 2009;16:381–389. doi: 10.1677/ERC-09-0038. [DOI] [PubMed] [Google Scholar]
- 23.Metivier R, Penot G, Carmouche RP, Hubner MR, Reid G, Denger S, Manu D, Brand H, Kos M, Benes V, et al. Transcriptional complexes engaged by apo-estrogen receptor-alpha isoforms have divergent outcomes. EMBO J. 2004;23:3653–3666. doi: 10.1038/sj.emboj.7600377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laganiere J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguere V. Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc. Natl Acad. Sci. USA. 2005;102:11651–11656. doi: 10.1073/pnas.0505575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC. A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res. 1988;16:647–663. doi: 10.1093/nar/16.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker P, Germond JE, Brown-Luedi M, Givel F, Wahli W. Sequence homologies in the region preceding the transcription initiation site of the liver estrogen-responsive vitellogenin and apo-VLDLII genes. Nucleic Acids Res. 1984;12:8611–8626. doi: 10.1093/nar/12.22.8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol. Endocrinol. 2004;18:1411–1427. doi: 10.1210/me.2003-0441. [DOI] [PubMed] [Google Scholar]
- 29.Tang S, Tan SL, Ramadoss SK, Kumar AP, Tang MH, Bajic VB. Computational method for discovery of estrogen responsive genes. Nucleic Acids Res. 2004;32:6212–6217. doi: 10.1093/nar/gkh943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bourdeau V, Deschenes J, Laperriere D, Aid M, White JH, Mader S. Mechanisms of primary and secondary estrogen target gene regulation in breast cancer cells. Nucleic Acids Res. 2007;35:3465–3477. doi: 10.1093/nar/gkm945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamalakaran S, Radhakrishnan SK, Beck WT. Identification of estrogen-responsive genes using a genome-wide analysis of promoter elements for transcription factor binding sites. J. Biol. Chem. 2005;280:21491–21497. doi: 10.1074/jbc.M409176200. [DOI] [PubMed] [Google Scholar]
- 32.Gruber CJ, Gruber DM, Gruber IM, Wieser F, Huber JC. Anatomy of the estrogen response element. Trends Endocrinol. Metab. 2004;15:73–78. doi: 10.1016/j.tem.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 33.O'L;one R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol. Endocrinol. 2004;18:1859–1875. doi: 10.1210/me.2003-0044. [DOI] [PubMed] [Google Scholar]
- 34.Driscoll MD, Sathya G, Muyan M, Klinge CM, Hilf R, Bambara RA. Sequence requirements for estrogen receptor binding to estrogen response elements. J. Biol. Chem. 1998;273:29321–29330. doi: 10.1074/jbc.273.45.29321. [DOI] [PubMed] [Google Scholar]
- 35.Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009;28:1418–1428. doi: 10.1038/emboj.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krum SA, Miranda-Carboni GA, Lupien M, Eeckhoute J, Carroll JS, Brown M. Unique ERalpha cistromes control cell type-specific gene regulation. Mol. Endocrinol. 2008;22:2393–2406. doi: 10.1210/me.2008-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deschenes J, Bourdeau V, White JH, Mader S. Regulation of GREB1 transcription by estrogen receptor alpha through a multipartite enhancer spread over 20 kb of upstream flanking sequences. J. Biol. Chem. 2007;282:17335–17339. doi: 10.1074/jbc.C700030200. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Nawaz Z, Slingerland JM. Long-range activation of GREB1 by estrogen receptor via three distal consensus estrogen-responsive elements in breast cancer cells. Mol. Endocrinol. 2007;21:2651–2662. doi: 10.1210/me.2007-0082. [DOI] [PubMed] [Google Scholar]
- 39.Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 40.Wu F, Khan S, Wu Q, Barhoumi R, Burghardt R, Safe S. Ligand structure-dependent activation of estrogen receptor alpha/Sp by estrogens and xenoestrogens. J. Steroid Biochem. Mol. Biol. 2008;110:104–115. doi: 10.1016/j.jsbmb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 42.Gevry N, Hardy S, Jacques PE, Laflamme L, Svotelis A, Robert F, Gaudreau L. Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev. 2009;23:1522–1533. doi: 10.1101/gad.1787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwabe JW, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell. 1993;75:567–578. doi: 10.1016/0092-8674(93)90390-c. [DOI] [PubMed] [Google Scholar]
- 44.Schwabe JW, Chapman L, Finch JT, Rhodes D, Neuhaus D. DNA recognition by the oestrogen receptor: from solution to the crystal. Structure. 1993;1:187–204. doi: 10.1016/0969-2126(93)90020-h. [DOI] [PubMed] [Google Scholar]
- 45.Wang C, Yu J, Kallen CB. Two estrogen response element sequences near the PCNA gene are not responsible for its estrogen-enhanced expression in MCF7 cells. PLoS ONE. 2008;3:e3523. doi: 10.1371/journal.pone.0003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krikun G, Mor G, Alvero A, Guller S, Schatz F, Sapi E, Rahman M, Caze R, Qumsiyeh M, Lockwood CJ. A novel immortalized human endometrial stromal cell line with normal progestational response. Endocrinology. 2004;145:2291–2296. doi: 10.1210/en.2003-1606. [DOI] [PubMed] [Google Scholar]
- 47.Schug J. Using TESS to Predict Transcription Factor Binding Sites in DNA Sequence. 2008. Curr Protoc Bioinformatics. Chapter 2:Unit 2.6. J. Wiley and Sons, Malden, MA, U.S.A. [DOI] [PubMed] [Google Scholar]
- 48.Schwabe JW, Fairall L, Chapman L, Finch JT, Dutnall RN, Rhodes D. The cocrystal structures of two zinc-stabilized DNA-binding domains illustrate different ways of achieving sequence-specific DNA recognition. Cold Spring Harb. Symp. Quant. Biol. 1993;58:141–147. doi: 10.1101/sqb.1993.058.01.018. [DOI] [PubMed] [Google Scholar]
- 49.Norris J, Fan D, Aleman C, Marks JR, Futreal PA, Wiseman RW, Iglehart JD, Deininger PL, McDonnell DP. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J. Biol. Chem. 1995;270:22777–22782. doi: 10.1074/jbc.270.39.22777. [DOI] [PubMed] [Google Scholar]
- 50.Marks JR, Huper G, Vaughn JP, Davis PL, Norris J, McDonnell DP, Wiseman RW, Futreal PA, Iglehart JD. BRCA1 expression is not directly responsive to estrogen. Oncogene. 1997;14:115–121. doi: 10.1038/sj.onc.1200808. [DOI] [PubMed] [Google Scholar]
- 51.Jurka J. Evolutionary impact of human Alu repetitive elements. Curr. Opin. Genet. Dev. 2004;14:603–608. doi: 10.1016/j.gde.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 52.Jurka J, Kapitonov VV, Kohany O, Jurka MV. Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genomics Hum. Genet. 2007;8:241–259. doi: 10.1146/annurev.genom.8.080706.092416. [DOI] [PubMed] [Google Scholar]
- 53.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 54.Safe S, Kim K. Nuclear receptor-mediated transactivation through interaction with Sp proteins. Prog. Nucleic Acid Res. Mol. Biol. 2004;77:1–36. doi: 10.1016/S0079-6603(04)77001-4. [DOI] [PubMed] [Google Scholar]
- 55.Tang S, Han H, Bajic VB. ERGDB: Estrogen Responsive Genes Database. Nucleic Acids Res. 2004;32:D533–D536. doi: 10.1093/nar/gkh083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 57.Pethe V, Shekhar PV. Estrogen inducibility of c-Ha-ras transcription in breast cancer cells. Identification of functional estrogen-responsive transcriptional regulatory elements in exon 1/intron 1 of the c-Ha-ras gene. J. Biol. Chem. 1999;274:30969–30978. doi: 10.1074/jbc.274.43.30969. [DOI] [PubMed] [Google Scholar]
- 58.Watanabe T, Inoue S, Hiroi H, Orimo A, Muramatsu M. NMDA receptor type 2D gene as target for estrogen receptor in the brain. Brain Res. Mol. Brain Res. 1999;63:375–379. doi: 10.1016/s0169-328x(98)00304-0. [DOI] [PubMed] [Google Scholar]
- 59.Hedges DJ, Batzer MA. From the margins of the genome: mobile elements shape primate evolution. Bioessays. 2005;27:785–794. doi: 10.1002/bies.20268. [DOI] [PubMed] [Google Scholar]
- 60.Grover D, Majumder PP, C BR, Brahmachari SK, Mukerji M. Nonrandom distribution of alu elements in genes of various functional categories: insight from analysis of human chromosomes 21 and 22. Mol. Biol. Evol. 2003;20:1420–1424. doi: 10.1093/molbev/msg153. [DOI] [PubMed] [Google Scholar]
- 61.Shankar R, Grover D, Brahmachari SK, Mukerji M. Evolution and distribution of RNA polymerase II regulatory sites from RNA polymerase III dependant mobile Alu elements. BMC Evol. Biol. 2004;4:37. doi: 10.1186/1471-2148-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laperriere D, Wang TT, White JH, Mader S. Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genomics. 2007;8:23. doi: 10.1186/1471-2164-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hua S, Kittler R, White KP. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell. 2009;137:1259–1271. doi: 10.1016/j.cell.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanay A. Extensive low-affinity transcriptional interactions in the yeast genome. Genome Res. 2006;16:962–972. doi: 10.1101/gr.5113606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loven MA, Likhite VS, Choi I, Nardulli AM. Estrogen response elements alter coactivator recruitment through allosteric modulation of estrogen receptor beta conformation. J. Biol. Chem. 2001;276:45282–45288. doi: 10.1074/jbc.M106211200. [DOI] [PubMed] [Google Scholar]
- 68.Loven MA, Wood JR, Nardulli AM. Interaction of estrogen receptors alpha and beta with estrogen response elements. Mol. Cell. Endocrinol. 2001;181:151–163. doi: 10.1016/s0303-7207(01)00491-9. [DOI] [PubMed] [Google Scholar]
- 69.Wood JR, Likhite VS, Loven MA, Nardulli AM. Allosteric modulation of estrogen receptor conformation by different estrogen response elements. Mol. Endocrinol. 2001;15:1114–1126. doi: 10.1210/mend.15.7.0671. [DOI] [PubMed] [Google Scholar]
- 70.Wood JR, Greene GL, Nardulli AM. Estrogen response elements function as allosteric modulators of estrogen receptor conformation. Mol. Cell. Biol. 1998;18:1927–1934. doi: 10.1128/mcb.18.4.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nardulli AM, Romine LE, Carpo C, Greene GL, Rainish B. Estrogen receptor affinity and location of consensus and imperfect estrogen response elements influence transcription activation of simplified promoters. Mol. Endocrinol. 1996;10:694–704. doi: 10.1210/mend.10.6.8776729. [DOI] [PubMed] [Google Scholar]
- 72.John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, Voss TC, Davis SR, Meltzer PS, Stamatoyannopoulos JA, et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol. Cell. 2008;29:611–624. doi: 10.1016/j.molcel.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 73.Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark 'nucleosome-free regions' of active promoters and other regulatory regions. Nat. Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.