


Abstract
Nep1 (Emg1) is a highly conserved nucleolar protein with an essential function in ribosome biogenesis. A mutation in the human Nep1 homolog causes Bowen–Conradi syndrome—a severe developmental disorder. Structures of Nep1 revealed a dimer with a fold similar to the SPOUT-class of RNA-methyltransferases suggesting that Nep1 acts as a methyltransferase in ribosome biogenesis. The target for this putative methyltransferase activity has not been identified yet. We characterized the RNA-binding specificity of Methanocaldococcus jannaschii Nep1 by fluorescence- and NMR-spectroscopy as well as by yeast three-hybrid screening. Nep1 binds with high affinity to short RNA oligonucleotides corresponding to nt 910–921 of M. jannaschii 16S rRNA through a highly conserved basic surface cleft along the dimer interface. Nep1 only methylates RNAs containing a pseudouridine at a position corresponding to a previously identified hypermodified N1-methyl-N3-(3-amino-3-carboxypropyl) pseudouridine (m1acp3-Ψ) in eukaryotic 18S rRNAs. Analysis of the methylated nucleoside by MALDI-mass spectrometry, HPLC and NMR shows that the methyl group is transferred to the N1 of the pseudouridine. Thus, Nep1 is the first identified example of an N1-specific pseudouridine methyltransferase. This enzymatic activity is also conserved in human Nep1 suggesting that Nep1 is the methyltransferase in the biosynthesis of m1acp3-Ψ in eukaryotic 18S rRNAs.
INTRODUCTION
Nucleotide modifications are widespread in functionally important classes of cellular RNA molecules including tRNAs, rRNAs and spliceosomal snRNAs (1). Presumably, the modifications are important for the proper folding, the stability and the function of these RNAs (2). The two most commonly observed modifications in rRNAs are 2′-O-methylations of ribose moieties and the conversion of uridine to pseudouridine but other modifications such as base methylations also exist. Clusters of modified rRNA nucleotides are found in functionally important domains such as rRNA regions associated with the peptidyltransferase center and the decoding center (3,4). Numerous dedicated enzymes acting on one or several specific nucleotides are responsible for the introduction of ribose 2′-O-methyl groups and pseudouridines in prokaryotic rRNA (5,6). In contrast, in eukaryotes and in archaea most 2′-O-methylations and all pseudouridinylations are catalyzed by small nucleolar ribonucleoprotein particles (snoRNPs) consisting of snoRNAs and associated proteins simultaneously with the processing of the 35S rRNA precursor. The snoRNAs act as guides for the snoRNPs to the specific modification sites by virtue of their sequence complementarity with the respective rRNA target sequences (7). C/D box snoRNAs in concert with the Nop1 methyltransferase introduce ribose 2′-O-methylations (8). H/ACA-box sno-RNAs together with pseudouridine kinase Cbf5 mediate the formation of pseudouridines [for a recent review, see (9)]. For other modifications, dedicated enzymes are required that modify specific nucleotides of the rRNA without the participation of snoRNPs (10). Examples include the universally conserved Dim1/Ksga methyltransferase responsible for the N6-dimethylations of A1781 and A1782 in 18S rRNA (11) and Bud23 catalyzing the N7-methylation of G1575 in 18S rRNA (12).
The Nep1 protein family, also referred to as Emg1, has been described as essential for ribosome biogenesis (13,14). Nep1 homologs are found in all eukaryotic genomes as well as in some archaea. It was demonstrated that the yeast and the human Nep1 proteins are localized in the nucleolus. Pre-rRNA processing of the 18S-rRNA precursor was blocked at sites A0, A1 and A2 in temperature sensitive Nep1 mutants at the restrictive temperature indicating that Nep1 plays a role in the biogenesis of the small ribosomal subunit (14). A mutation of a highly conserved aspartate to a glycine (D86G) in human Nep1 was shown to be the cause of the Bowen–Conradi syndrome—a severe developmental disorder resulting in early childhood death (15).
Recent crystal structures of yeast Nep1 (ScNep1) (16) and Nep1 from the archeon Methanocaldococcus jannaschii (MjNep1) (17) revealed a protein fold unique to members of the so called SPOUT-family of RNA methyltransferases (Figure 1A). Furthermore, bound S-adenosylmethionine and S-adenosylhomocysteine found in the two Nep1 crystal structures and a putative RNA-binding surface in close spatial proximity to the cofactor are in agreement with a putative RNA–methyltransferase function for Nep1. So far SPOUT-class RNA–methyltransferases have been shown to catalyze ribose 2′-OH, guanine N1, uridine N3 and pseudouridine N3 methylations (18–20). Numerous crystal structures have been reported for members of the SPOUT-family of RNA–methyltransferases (21–24). However, their mode of interaction with their target RNAs has not yet been characterized experimentally with atomic resolution. In some cases, a possible RNA-binding mode could be deduced by modeling studies for SPOUT-family members carrying an additional RNA-binding domain such as ribosomal protein L30e-like domains, OB-fold domains and PUA domains as an extension of the core fold (25,26). In other cases, bound sulfate ions in crystal structures revealed possible contact points for the phosphate backbone of the target RNA guiding subsequent modeling efforts and mutational studies (21,22). Nep1 contains two insertions with regard to the core fold of the SPOUT-class proteins (Figure 1A), a large, surface exposed loop and a β-α-β element with an antiparallel orientation of the two β-strands (17). The irregularly structured surface loop is rich in basic amino acids whereas the α -helix of the β-α-β element and its preceding loop contain three highly conserved arginine residues. This is suggestive of a role of the insertion elements in RNA-binding similar to other members of the family.
Figure 1.

3D structure of Nep1 and secondary structure and conservation of hypothetical Nep1 methyltransferase target sites in 18S rRNA. (A) Three-dimensional structure of the M. jannaschii Nep1 dimer in cartoon representation bound to the cofactor S-adenosylhomocysteine (3bbd). Insertion elements extending the SPOUT-class RNA methyltransferase core fold and unique to the Nep1 subfamily are colored gray in one monomer and red in the other monomer, respectively. Highly conserved arginine side chains implicated in RNA binding and Trp193 are shown in a stick representation. (B) Left: Secondary structure of a fragment of yeast 18S rRNA containing two examples of the yeast Nep1 binding RNA-consensus motif 5′-C/UUCAAC-3′ (red) in the proximity of putative methylation target sites. Modification sites are indicated as boxed nucleotides and the type of modification is given. Furthermore, the chemical structure of the hypermodified pseudouridine in position 1191 is shown in the inset. Right: Secondary structure and sequence of the same fragment of Methanocaldococcus jannaschii 16S rRNA with the location of the only conserved copy of the RNA consensus motif highlighted in red.
The actual RNA target for the putative methyltransferase activity of Nep1 is not known. However, yeast three-hybrid library screening revealed a 6-nt RNA consensus motif (5′-C/UUCAAC-3′) which binds to ScNep1 in vivo when it is presented in the context of hairpin loops or internal bulge structures. This motif occurs at three positions in the yeast 18S rRNA (nt 1566–1571 near helix 47, nt 1190–1195 near helix 35 and nt 349–354 near helix 12) (27). Interestingly, nt 1566–1571 are in proximity to two nucleotides that are modified through methylation (G1572, 2′-OH methylation and G1575, N7-methylation) whereas nt 1190–1195 include the hypermodified N1-Methyl-N3-(3-amino-3-carboxypropyl) pseudouridine [m1acp3-Ψ in position 1191 (Figure 1B)]. The 2′-OH methylation of G1572 is guided by the snoRNA snr57 (28). While earlier genetic data suggested a possible involvement of Nep1 in catalyzing the N7-methylation of G1575 (27), the Bud23 protein was recently identified as responsible for this reaction (12). The biogenesis of the hypermodified m1acp3-Ψ in position 1191 of 18S rRNA starts by a conventional U to Ψ conversion mediated by snoRNA snR35 (4). This pseudouridine then becomes N1-methylated in the nucleolus and the 3-amino-3-carboxypropyl modification at the N3-nitrogen is probably introduced after the export of the assembled ribosome subunit into the cytoplasm (29). The enzymes responsible for both reactions have not been identified yet.
Here we show that MjNep1 is capable of recognizing the yeast consensus RNA sequence. In addition, in vitro and in vivo approaches allowed us to identify high affinity RNA ligands for MjNep1. Their sequences include nt 913–918 (corresponding to nt 1190–1195 of yeast 18S rRNA) and proximal sequences of M. jannaschii 16S rRNA (Figure 1B). Structural insight into the basis of RNA recognition was gained from NMR-spectroscopic investigations of RNA binding to MjNep1 by using chemical shift perturbation (30) and paramagnetic relaxation enhancement experiments (31). Furthermore, the high affinity RNAs were used as substrates in in vitro methylation experiments with MjNep1. Only when a Ψ was present in position 914 corresponding to the m1acp3-Ψ containing position 1191 in yeast 18S rRNA (Figure 1B) MjNep1 was able to catalyze a RNA methylation. By combining HPLC and NMR-spectroscopy the modified nucleotide was identified as N1-methylpseudouridine. Thus, MjNep1 is a sequence specific pseudouridine N1-methyltransferase. The pseudouridine N1-methyltransferase activity is conserved in human Nep1 (HsNep1). Accordingly, Nep1 is most likely the methyltransferase responsible for the N1-methylation step in the biosynthesis of the hypermodified m1acp3-Ψ nucleotide conserved in eukaryotic 18S rRNA.
MATERIALS AND METHODS
Protein overexpression and purification
Full-length MjNep1 (aa1-205) was overexpressed and purified as described previously (17). Briefly, after induction with IPTG (250 mg/l) at an OD600 = 0.8 for 12 h at 24°C cells were harvested and resuspended in sonication buffer (25 mM Tris–HCl, pH 7.8, 500 mM NaCl, 2 mM β-mercaptoethanol). Following sonication, the supernatant was cleared by centrifugation and the NaCl-concentration adjusted to 250 mM. Nep1 was then purified from the supernatant by a cation-exchange step and a subsequent gel filtration step. 2H,15N- and 2H,13C,15N-labeled protein was obtained from cells grown in Escherichia coli-OD2 DN or CDN medium (Silantes GmbH, München, Germany). Proteins labeled selectively with either 15N-arginine or 15N-lysine were obtained from cells grown on unlabeled M9 minimal medium supplemented with either 300 mg/l of 15N-lysine or 250 mg/l of 15N-arginine, the remaining 19 amino acids in their unlabeled form and nucleotides according to the literature (32). Labeled MjNep1 protein for the NMR experiments was dialyzed overnight against 150 mM KCl, 25 mM Bis–Tris, pH 6.0, 2 mM β-mercaptoethanol at RT, concentrated to 400 µM (2H,13C,15N-labeled), 160 µM (2H,15N-labeled) or 80–200 µM (selectively labeled) in Vivaspin 2 concentrators (Sartorius Stedim, Aubagne, France) and adjusted to 10% (v/v) D2O.
For the production of human Nep1 (HsNep1), the human Nep1 gene was cloned into the recombinant expression plasmid pQE-9 (Quiagen, Hilden, Germany). The plasmid was transformed into E. coli M15(pREP4) by electroporation. HsNep1 was overexpressed in LB medium containing ampicillin (100 mg/l) and kanamycin (50 mg/l) after induction with IPTG (250 mg/l) at an OD600 of 0.8 for 12 h at 24°C. Cells were harvested by centrifugation and resuspended in sonication buffer (500 mM NaCl, 10 mM imidazole, 50 mM NaH2PO4, pH 7.5, 5 mM β-mercaptoethanol). After sonication cell debris was removed by centrifugation and the supernatant was applied to a Ni-NTA column (GE Healthcare, Chalfont St Giles, England). HsNep1 was eluted by a linear imidazole gradient (10 mM to 1 M imidazole in 500 mM NaCl, 50 mM NaH2PO4, pH 7.5, 5 mM β-mercaptoethanol) and dialyzed over night at 4°C against 250 mM NaCl, 50 mM NaH2PO4, pH 7.5, 5 mM β-mercaptoethanol.
RNA oligonucleotides
Unmodified and pseudouridine containing RNA oligonucleotides were purchased from Dharmacon (Lafayette, USA), deprotected following manufacturer’s instructions and used without further purification.
A 9mer RNA oligonucleotide with the sequence 5′-UUCAACGCC-3′ containing a nitroxyl spin-label at the 5′-uridine was synthesized and purified according to (33,34) as described in detail in the Supplementary Data.
Fluorescence quenching experiments
Fluorescence quenching experiments were performed at 27°C using a Fluorolog 3 spectrometer (Horiba Jobin Yvon, Unterhaching, Germany) and 2 ml polystyrene cuvettes. The MjNep1 concentration was 200 nM in 100 mM KCl, 25 mM Tris–HCl, pH 7.5. Excitation and emission wavelengths were set to 295 nm and 344 nm, respectively, except for RNAs 1 and 2 where a concentration of 2 µM and an excitation wavelength of 305 nM were used to reduce the inner filter effect and still attain a reasonable signal to noise ratio. RNA stock solutions were prepared in pure water at adequate concentrations to keep the final dilution of the MjNep1 solution below 2%. The measured fluorescence intensity after each titration was corrected for dilution and normalized to the fluorescence intensity of free MjNep1. Furthermore, for RNAs with a KD larger than 1 µM a correction for the inner filter effect was applied according to the equation:
(F0 fluorescence prior to inner filter effect, FIFE fluorescence after inner filter effect, c RNA concentration, ε absorption coefficient of RNA at excitation wavelength). The dissociation constant KD was obtained by fitting the binding curves to the following equation using the nonlinear least square algorithm of Origin 8 (OriginLab, Northampton, USA) treating a and KD as free parameters:
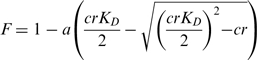 |
(c Nep1 dimer concentration, r RNA concentration, a fluorescence quenching constant, KD dissociation constant). All titrations were performed in triplicates, which were fitted independently.
Yeast three-hybrid screens
Yeast three-hybrid RNA-binding assays were essentially carried out as described previously (27). Briefly, plasmid libraries containing either random fragments of yeast genomic DNA (36) or the sequence NNNCAACNNN were expressed as hybrids with MS2-RNA in strain L40coat containing pGAD-MjNep1 encoding the M. jannaschii Nep1-protein fused to the Gal4 activation domain. For construction of the NNNCAACNNN-library, oligonucleotide (5′-GGC TAG AAC TAG TGG ATC CCC CGG GCG AGG CTT ATC CNN NCA CAN NNG GAT GTG CTG ACC CCG GGC AGC TTG CAT GCC TGC A-3′) was elongated and amplified with the primer pair (5′-TGG GAA CGA AAC TCT GGG AGC TGC GAT TGG CAG AAT TCC GGC TAG AAC TAG TGG ATC CCC C-3′) and (5′-CCT GCA GAC ATG GGT GAT CCT CAT GTT TTC TAG AGT CGA CCT GCA GGC ATG CAA GCT GCC C-3′). The resulting PCR product was integrated into pIII/MS2-2 by cotransformation together with the XmaI-linearized plasmid into yeast strain L40coat already containing pGAD-MjNep1. Quantification of ß-galactosidase activity in Miller-units was performed as previously described [(27) and references cited therein]. The given values are averaged activities from two independent transformants.
RNA-methyltransferase activity assays
The RNA methylation activity was tested in a reaction mixture (25 µl) containing 100 mM KCl, 40 mM NaCl, 25 mM Tris–HCl, pH 7.5, 1 mM MgCl2, 500 µM SAM, 250 µM RNA and 50 µM MjNep1 or 60 mM NaCl, 40 mM KCl, 50 mM NaH2PO4, pH 7.5, 1 mM MgCl2, 500 µM SAM, 250 µM RNA and 10 µM HsNep1. This mixture was incubated at 65°C for MjNep1 and at 37°C for HsNep1. Five-microliter samples were transferred into pre-cooled Eppendorf tubes after defined time intervals and immediately frozen at −20°C.
For MALDI mass spectrometry samples from the RNA methylation reaction were diluted 1:10 with pure water. For mass analysis 0.5-µl sample solution were mixed together with 2-µl matrix solution directly on the MALDI target and dried in a stream of air. 3-hydroxypicolinic acid (40 mg/ml in water) containing 5 mg/ml diammoniumhydrogencitrate was used as matrix. Mass spectra were recorded on a MALDI Orbitrap XL (Thermo Scientific, Waltham, USA) in positive ion mode. Resolution at this instrument was set to 30 000. Five to ten spectra were accumulated over a maximum of 80 laser shots, depending on the number of ions produced. Sequence analysis was performed by acid hydrolysis according to the procedure published recently (37).
RNA hydrolysis and HPLC analysis of nucleosides
For HPLC and NMR analysis of modified nucleosides RNA 12 (Table 1) was methylated on a preparative scale in a volume of 500 µl using the concentrations given above for MjNep1. Completion of the reaction was followed by MALDI mass spectrometry as described above. After incubation at 65°C for 20 min, the RNA was extracted with 500 µl of phenol/chloroform/isoamylalcohol (25/24/1 v/v/v), precipitated with 250 µl of 6 M NH4AcO and 2.25 ml abs. ethanol for 1 h at −20°C, washed with 75% ethanol and dissolved in pure water. The RNA was hydrolyzed and dephosphorylated using P1 nuclease (Sigma Aldrich, St. Louis, USA) and bacterial alkaline phosphatase (Sigma Aldrich, St. Louis, USA) following the protocols of Gehrke et al. (38). Analytical and preparative HPLC of the resulting nucleoside mixture was performed on a Supelcosil LC-18-S HPLC column (Sigma Aldrich, St. Louis, USA) equipped with a pre-column at 30°C on an Agilent 1200 HPLC system (Agilent, Santa Clara, USA) using buffers and gradients according to Ero et al. (19). Chemically synthesized N1-methylpseudouridine was purchased from Berry & Associates (Dexter, USA).
Table 1.
Binding affinities (mean value and standard deviation) and methylation of RNA-oligonucleotides used in this study
| Name | Sequence | KD (nM) | Methylation |
|---|---|---|---|
| RNA 1 | 5′-UUCAAC-3′ | 12 300 ± 1900 | n.d. |
| RNA 2 | 5′-UUUUUUUU-3′ | 25 600 ± 5100 | n.d. |
| RNA 3 | 5′-GGAUUCAAC-3′ | 1650 ± 380 | n.d. |
| RNA 4 | 5′-UUCAACGC-3′ | 77 ± 27 | n.d. |
| RNA 5 | 5′-UUCAACGCC-3′ | 8 ± 5 | n.d. |
| RNA 6 | 5′-GGAUUCAACGCC-3′ | 7 ± 2 | No |
| RNA 7 | 5′-UUGGAUUCAACGCCGG-3′ | 3 ± 1 | n.d. |
| RNA 8 | 5′-GAUΨCAACGCC-3′ | 11 ± 4 | Yes |
| RNA 9 | 5′-GAΨUCAACGCC-3′ | 2 ± 1 | No |
| RNA 10 | 5′-UΨCAACGCC-3′ | 14 ± 2 | Yes |
| RNA 11 | 5′-UΨCAACGC-3′ | 25 ± 4 | Yes |
| RNA 12 | 5′-ΨCAACGCC-3′ | 7 ± 2 | Yes |
n.d = not determined
NMR-spectroscopy
All NMR-spectra of MjNep1 and MjNep1/RNA-complexes were recorded at 37°C on Bruker AVANCE 600, 700, 800, 900 and 950 MHz spectrometers equipped with cryogenic triple resonance probes. In order to transfer the existing chemical shift assignments of MjNep1(39), which were obtained under less physiological conditions (50 mM KCl, 25 mM AcOH, pH 4.5, 47°C) to our sample conditions we recorded 2D 15N-HSQC, 3D HNCA, 3D HNCO and 3D HN(CA)CO spectra of the 2H,13C,15N-labeled sample in 150 mM KCl, 25 mM Bis–Tris, pH 6.0, 2 mM β-mercaptoethanol at 37°C using standard triple resonance pulse sequences (40).
1H,15N-HSQC-spectra for the complex of MjNep1 with spin-labeled RNA were recorded prior to and after reduction by the addition of sodium ascorbate to a final concentration of 500 µM and incubation for 2 h at RT directly in the NMR tube.
The modified nucleoside isolated by preparative HPLC as described above dissolved in 6 ml of HPLC buffer was freeze-dried and resuspended in 500 µl D2O for the NMR experiments. Chemically synthesized N1-methylpseudouridine resuspended in 6 ml of HPLC buffer was treated the same way to obtain identical buffer conditions for both samples. Final concentrations were 100 µM and 2 mM, respectively. Standard 2D DQF-COSY and 2D 1H,1H-NOESY spectra were recorded for the assignment of the sugar protons and to identify the position of the methyl group.
All spectra were processed using Bruker TOPSPIN 2.1 and analyzed using the program CARA (41).
RESULTS
Methanocaldococcus jannaschii Nep1 binds to the yeast Nep1 RNA consensus sequence
The RNA-consensus motif (5′-C/UUCAAC-3′) binding to yeast Nep1 (ScNep1) occurs three times in the yeast 18S rRNA sequence. In two instances, the motif either contains or is adjacent to base-methylated nucleotides m1acp3-ψ 1191 near helix 35 and N7-methylguanine 1575 near helix 47, respectively. Therefore, these nucleotides might represent possible targets for a putative methyltransferase activity of Nep1 in yeast.
Given the sequence and structural homology of ScNep1 and MjNep1 we asked if the RNA-binding specificity of the two proteins was also similar. The yeast consensus sequence occurs only once in M. jannaschii 16S rRNA as 5′-UUCAAC-3′ in the region corresponding to nt 913–918 (Figure 1B). This region of M. jannaschii 16S rRNA is equivalent to nt 1190–1195 in yeast 18S rRNA including the m1acp3-Ψ nucleotide at position 1191.
MjNep1 offers some advantages for biophysical investigations of RNA binding when compared to ScNep1 such as a non-conserved tryptophan (W193) located close to the sulfur atom of the bound cofactor and high-quality NMR-spectra despite its size of 48 kDa (39). Consequently, the binding of MjNep1 to a 6mer RNA containing the consensus sequence 5′-UUCAAC-3′ (RNA 1, Table 1) was probed in vitro by fluorescence quenching experiments as well as by NMR-spectroscopy.
Both fluorescence quenching experiments utilizing the tryptophan fluorescence of W193 and chemical shift perturbation experiments revealed that RNA 1 binds to MjNep1 albeit with low affinity (Supplementary Figure S1). The differences in the 15N-HSQC-spectra of free MjNep1 and MjNep1 in the presence of increasing amounts of the RNA are indicative of binding in the medium to fast-exchange regime on the NMR time scale corresponding to dissociation constants larger than 1 µM (Supplementary Figure S1). Importantly, NMR signals that change their chemical shifts during the titration with RNA 1 correspond to amino acids located close to the postulated RNA-binding area in a positively charged groove at the dimer interface (17). The fluorescence quenching data yield a KD of 12 µM (Supplementary Figure S1) in agreement with the NMR-data. This KD is only ∼2-fold lower than the KD for a longer oligoU sequence (RNA 2, Table 1).
RNA-binding specificity by in vitro using fluorescence quenching assays and yeast three-hybrid screening
The rather poor binding affinity of the consensus RNA-oligonucleotide might indicate that it is either not the proper target of MjNep1 or that additional RNA recognition elements either close in sequence or in the 3D structure of 16S rRNA are important for RNA-binding. Thus, we extended the sequence by including nucleotides of M. jannaschii 16S rRNA 5′ to nt 913 or 3′ to nt 918. While a 5′-extension by 3 nt including nt G910–A912 of 16S rRNA (RNA 3, Table 1) only lead to an 7-fold enhancement in binding affinity (Figure 2A), the 3′-extension by 2 or 3 nt (RNA4, RNA5) lead to a dramatic improvement in binding affinity by a factor of up to ∼1600 (Figure 2B, Table 1) yielding a KD of 8 nM for RNA5. Combination of the 5′- and 3′-extensions e.g. in a sequence including the complete hairpin loop 35 (RNA 7) of M. jannaschii 16S rRNA only slightly improved the affinity (Figure 2B).
Figure 2.

Identification of sequence determinants required for high affinity binding by fluorescence quenching experiments and yeast three-hybrid library screening. (A) Fluorescence titration curve of MjNep1 with an RNA oligonucleotide containing a 5′-extension of the 5′-UUCAAC-3′ consensus motif (RNA 3, Table 1). (B) Fluorescence titration curves of MjNep1 with RNA oligonucleotides containing either 3′-end extensions (RNA 5, Table 1) or 5′-end and 3′-end extensions (RNA 7,8) and a pseudouridine modification at a position corresponding to nucleotide 914 in M. jannaschii 16S rRNA (RNA 8). (C) Results of the yeast three-hybrid screening against a focused library of 5′-NNNCAACNNN-3′ containing RNAs displayed as a sequence logo.
Yeast 18S rRNA includes the hypermodified m1acp3-Ψ at position 1191 whereas it is unclear if M. jannaschii 16S rRNA contains a similar modification at the equivalent position 914. Other archea e.g. Haloferax volcanii contain a 3-amino-3-carboxypropyl uridine (acp3U) at this position (42). We therefore tested the binding of RNAs where the uridine at position 914 was replaced with a pseudouridine (RNA 8, 10–12). As a control the first uridine of the consensus sequence (position 913) was replaced with a pseudouridine (RNA 9). The presence of pseudouridine instead of uridine in either of those two positions had only a minor influence on the observed KD. Even a 5′-truncated RNA lacking the first uridine of the consensus sequence (RNA 12) was bound with comparable affinity.
The high affinity of the extended RNAs containing nt 910–921 of M. janaschii 16S rRNA for MjNep1 strongly suggests that they resemble the natural MjNep1 target RNA.
To probe RNA-binding specificity of MjNep1 under in vivo conditions and with a more diverse set of sequences we resorted to yeast three-hybrid screening against an RNA fragment library consisting of random fragments of yeast genomic DNA. While the identified binding clones did not reveal an extended consensus sequence many of them displayed the sequence 5′-CAAC-3′ partially reminiscent of the consensus sequence found to bind yeast Nep1. Thus in a second selection round we constructed a focused library randomizing 3 nt 5′ and 3′ of a constant 5′-CAAC-3′ sequence (5′-NNNCAACNNN-3′). Using this library 18 clones with a specific affinity to MjNep1 were isolated. Most of them displayed a yeast three-hybrid activation (ß-galactosidase activities of 2.1–17.3 U/mg) similar or higher than the yeast consensus sequence 5′-UUCAAC-3′ (2.5 U/mg) itself. Sequence analysis revealed a strong preference for an extension of the 5′-CAAC-3′ motif at its 3′-end with the sequence 5′-GCG-3′. All three positions were found to be conserved in more than 50% of the isolated clones (Figure 2C). In contrast A and U are favoured in the two positions 5′ of 5′-CAAC-3′, but less stringently as nucleotides extending the motif in the 3′-region. Thus, the sequences of the artificial binders for MjNep1 identified in the yeast three-hybrid screen reflect the sequence of the M. jannaschii 16S rRNA loop closing helix 35 and fit well to the in vitro binding data, in which the 3′-terminal extension of the 5′-UUCAAC-3′ motif by G and C lead to a dramatic improvement in binding affinity.
RNA-methyltransferase activity of Nep1 in vitro
A subset of RNA-oligonucleotides binding MjNep1 with high affinity were used as substrates in RNA-methylation assays. To this end, 6.25 nmol RNA was incubated with 1.25 nmol MjNep1 and 12.5 nmol S-adenosylmethionine at 65°C. The reaction products were then analyzed with MALDI-mass spectrometry. The results of the methylation assays for different RNAs are shown in Table 1. Remarkably, only oligonucleotides containing a pseudouridine at the position corresponding to nt 914 in M. jannaschii 16S rRNA were methylated as evidenced by a signal for the reaction product with a mass increased by 14 Da as expected for the addition of a methyl group (Figure 3). Neither RNAs with an U at position 914 nor those with the pseudouridine shifted to the position corresponding to nt 913 showed evidence of methylation (Table 1, Figure 3). In addition, no reaction was observed in the absence of S-adenosylmethionine or MjNep1 or in the presence of S-adenosylhomocysteine (data not shown). On the other hand, even the 5′-truncated oligonucleotide starting directly with Ψ914 (RNA 12, Table 1) was efficiently methylated by MjNep1 (Supplementary Figure S2) in the presence of S-adenosylmethionine.
Figure 3.

RNA-methyltransferase activity of MjNep1. (A) MALDI mass spectra of a reaction mixture of S-adenosylmethionine, MjNep1 and high affinity RNAs containing a pseudouridine either at the position corresponding to nt 914 (highlighted in red) or scrambled to the preceding position. The formation of a reaction product with a 14 Da increase in mass corresponding to the addition of a methyl group is observable for 5′-GAUΨCAACGCC-3′ but not for 5′-GAΨUCAACGCC-3′.
The position of the methylation was then verified by subjecting the methylated RNA 8 (5′-GAUΨCAACGCC-3′) to limited acid hydrolysis and analyzing the resulting fragments with MALDI-MS (37). Mass analysis of the resulting fragment ions unambiguously indicated that the modified nucleotide carrying the additional methyl group must be the Ψ at the position equivalent to nt 914 in the M. jannaschii 16S rRNA (Supplementary Table 1).
Identification of the methylated nucleotide as N1-methylpseudouridine
Ψ nucleotides may in principle become methylated at three different positions—the N1 and N3 nitrogen atoms of the base or the 2′-OH group of the ribose. To identify the chemical nature of the modified pseudouridine we resorted to nucleoside analysis of the modified RNA oligonucleotides by reversed phase HPLC. To minimize possible peak overlap in HPLC chromatograms RNA 12 (5′-ΨCAACGCC-3′) was used as a substrate. It contains only three other nucleotide species besides the pseudouridine but is still quantitatively modified (Supplementary Figure S2). Unmodified and MjNep1 treated RNA 12 were first hydrolyzed to mononucleotides by RNAse P1 and then dephosphorylated by alkaline bacterial phosphatase. The resulting nucleoside mixtures were analyzed by reversed phase (RP)-HPLC according to established protocols (19). Comparison of the HPLC-chromatograms for the unmodified RNA 12 and the MjNep1 treated RNA 12 revealed the disappearance of a peak with a retention time of 5.5 min in the MjNep1 treated RNA which can be assigned to Ψ (38) and the appearance of a peak with a retention time of 10.4 min not present in the chromatogram of the unmodified oligonucleotide (Figure 4A). Under our HPLC conditions commercially obtained N1-methylpseudouridine has a retention time of 10.3 min suggesting that this peak represents N1-methylpseudouridine (Figure 4A).
Figure 4.

Identification of the modified nucleoside as N1-methylpseudouridine by RP-HPLC and NMR-spectroscopy. (A) HPLC-chromatograms of hydrolyzed and dephosphorylated RNA 12 prior to (top) and after treatment with MjNep1 in the presence of S-adenosylmethionine (middle). The signal of the pseudouridine nucleoside disappears and a novel signal (indicated by an asterisk) appears (middle) at a retention time very similar to that of commercially available N1-methylpseudouridine (bottom). (B) 2D-1H,1H-NOESY (top) and 1D-1H- (middle) NMR-spectra of the modified nucleoside purified by preparative RP-HPLC from MjNep1 treated RNA 12 compared to a 1D 1H-NMR-spectrum of commercially available N1-methylpseudouridine (bottom). NMR-signal assignments are indicated. Signals of an impurity are marked by asterisks. The section of the 2D-1H,1H-NOESY-spectrum (top) of the modified nucleoside reveals an NOE between the aromatic H6 proton of the pyrimdine ring and the methyl group. Such an NOE is only observable in N1-methylpseudouridine but not in N3-methylpseudouridine (see inset).
To unambiguously establish the chemical identity of the modified nucleoside with N1-methylpseudouridine the modified nucleoside was isolated from a preparative scale methylation reaction with RNA 12 using the same HPLC-method and analyzed by 1H-NMR-spectroscopy (Figure 4B). For N1-methylpseudouridine a significant NOE is expected between the N1-methyl group and the H6 of the base whereas this NOE should be absent in N3-methylpseudouridine. This NOE was readily observable in a 2D-1H,1H-NOESY-spectrum of the isolated modified nucleoside (Figure 4B). The chemical shifts of the methyl group and the H6 proton of 3.4 ppm and 7.8 ppm, respectively, correspond to those expected for a methyl group bound to a nitrogen atom and the aromatic H6 proton of a pyrimidine base. In addition, these chemical shifts are similar to those reported under slightly different NMR-conditions for chemically synthesized N1-methylpseudouridine (42). Furthermore, the 1D-1H spectra of commercially obtained N1-methylpseudouridine and the isolated modified nucleoside are identical except for a stronger signal of uncharacterized impurities in the latter one (Figure 4B).
Thus, MjNep1 selectively methylates RNA-oligonucleotides with sequences resembling the 910–921 region of M. jannaschii 16S rRNA but only when they contain a pseudouridine at the position corresponding to nucleotide 914. The methyl group is transferred to the N1 nitrogen of the pseudouridine base. Therefore, MjNep1 is a pseudouridine N1 methyltransferase.
The pseudouridine N1-Methyltransferase activity is conserved in human Nep1
To investigate the conservation of the Nep1 pseudouridine N1 methyltransferase activity in higher eukaryotes the enzymatic activity of human Nep1 (HsNep1) was tested. To this end RNA-oligonucleotides were designed that corresponded to nucleotides 1245–1255 of human 18S rRNA and contained either a pseudouridine (5′-GACΨCAACACG-3′) or a uridine at position 1248 (5′-GACUCAACACG-3′). This position corresponds to nucleotide 914 of M. jannaschii 16S rRNA and to nt 1191 of yeast 18S rRNA. It is known to be N1-methylated and to possess an N3-3-amino-3-carboxypropyl modification in native human 18S rRNA (44). MALDI-mass spectrometrical analysis of the reaction products revealed that only the RNA containing the pseudouridine becomes methylated by HsNep1 (Figure 5). Therefore, the specific pseudouridine N1 methyltransferase activity of Nep1 is apparently conserved in the human enzyme.
Figure 5.

Conserved pseudouridine dependent RNA-methyltransferase activity of human Nep1. MALDI mass spectra of oligonucleotides corresponding to nt 1245–1255 of human 18S rRNA incubated with HsNep1 and SAM containing either a Ψ (top and middle) or a U (bottom) residue at a position equivalent to nt 1248 modified to m1acp3-Ψ in human 18S rRNA. The appearance of a reaction product with an additional mass of 14 Da corresponding to an additional methyl group is only observed in the case of the pseudouridine containing oligonucleotide.
Identification of the binding site for methylation target RNAs by NMR-spectroscopy
Complete NMR-signal assignments of the backbone amide groups were obtained previously for the apo state of MjNep1 under conditions taking advantage of the thermostability of MjNep1 (39) and thus favorable for NMR-spectroscopy in solution (47°C, 25 mM acetate, pH 4.5, 50 mM KCl). These NMR-signal assignments were transferred to conditions more suitable for NMR-based RNA-binding assays (37°C, 25 mM Bis–Tris, pH 6.0, 150 mM KCl) by carrying out temperature and pH-titrations as well as recording a subset of the standard 3D assignment experiments (data not shown). RNAs 10 and 11 (Table 1) were used for mapping the RNA-binding site of MjNep1 by chemical shift perturbation experiments due to their high affinity.
15N-HSQC-spectra of uniformly 15N,2H-labeled MjNep1 revealed a number of signals losing intensity and novel signals appearing and increasing in intensity upon titration with the 8mer RNA 11 (Figure 6A). However, many other signals were unaffected by the titration. This indicates the formation of a high-affinity RNA–protein complex with a dissociation rate which is slow on the NMR-time scale as well as specific binding of the RNA to a defined binding site on MjNep1. Interestingly, many but not all signals affected by binding of RNA 11 also showed gradual chemical shift changes upon titration with the low affinity RNA 1 containing only the consensus sequence 5′-UUCAAC-3′ (see above). Quantification of chemical shift changes in the case of slow exchange between the bound and the free state is difficult without a complete reassignment of the NMR-signals for the bound state. Therefore, only signals that disappeared upon RNA binding and had novel peaks appearing at least one line-width apart from the old signal position were considered as indicating proximity of the corresponding amino acid to the RNA-binding site. Due to the large number of signals present this is difficult in the crowded central regions of the 15N-HSQC-spectra. Thus, we used selectively 15N-lysine and 15N-arginine labeled Nep1 samples. Although lysine is a very abundant amino acid in MjNep1 (29 lysines), the signals of the lysine amide groups are well dispersed in 15N-HSQC-spectra of the selectively lysine-labeled protein (Figure 6B). The result of the titration of selectively 15N-lysine-labeled MjNep1 with the RNA 11 is shown in Figure 6B. Whereas the lysines are rather evenly distributed over the entire surface of the protein (Figure 6C) only a subset of the signals corresponding to lysines in the vicinity of the bound cofactor or located in the extension elements of the SPOUT-class fold on the same side of the protein (Figure 6C) is affected by RNA binding. This is in agreement with a site specific RNA-binding event and in contrast to what would be expected in the case of an unspecific electrostatically driven interaction between the positively charged lysine side chains and the negatively charged RNA-backbone. Nep1 contains four highly conserved arginines implicated in RNA binding by mutational studies. Since the signals corresponding to these arginines could not be tracked in the spectra of the uniformly 15N,2H-labeled protein titrations were repeated with selectively 15N-arginine-labeled MjNep1 and RNA 11. For four of the eight arginine residues strongly shifted or disappearing signals were observed upon addition of the RNA. These four arginines correspond to the four highly conserved arginines R54, R95, R98 and R102 that line a basic groove along the dimer interface in the vicinity of the bound SAH (Figure 6E).
Figure 6.

Chemical shift mapping of the RNA-binding surface of MjNep1 for its high affinity substrates and orientation of the RNA in the binding pocket. (A) 1H,15N-HSQC-spectra of uniformly 2H,15N-labeled MjNep1 in the absence (black) and the presence (red) of RNA 11 indicate the formation of a specific RNA–protein complex. (B) 1H,15N-HSQC-spectra of selectively 15N-lysine labeled MjNep1 in the absence (black) and the presence (red) of RNA 11. (C) Distribution of lysines across the surface of the MjNep1 dimer (amino acids in blue and orange) and location of lysines affected by the addition of RNA 11 (highlighted in orange). Unaffected lysines are coloured blue and the bound cofactor SAH is coloured yellow. (D) Mapping of RNA 11 induced chemical shift changes in uniformly and selectively labeled MjNep1 samples on the surface of the MjNep1 dimer. Amino acids with significant chemical shift changes are coloured in orange, unaffected amino acids are colored in blue, amino acids with untraceable signals are coloured black and the bound cofactor SAH is colored yellow. (E) Localization of the 3′-end of the bound RNA by investigating differential chemical shift changes induced by RNA 11 and RNA 10 carrying an extra nucleotide at its 3′-end. Left: Overlay of 1H,15N-HSQC-spectra of selectively 15N-arginine labeled free MjNep1 (black), MjNep1 (labelled with an *) bound to RNA 11 (red) and MjNep1 bound to the extended RNA 10 (blue). Two signals are only affected by the presence of RNA 10 indicating a location of the extra 3′-nucleotide close to R24 (red orange) and R27 (red orange) in the large irregularly structured extension loop (right). Arginine residues with signals affected by both RNAs are shown in orange. Those with chemical shifts unaffected by either RNA are coloured blue. (F) Localization of the 5′-end of the bound RNA using paramagnetic relaxation enhancement induced by spin labeled RNA. Left: Overlay of 1H,15N-HSQC-spectra of selectively 15N-lysine labeled MjNep1 in the presence of an RNA containing a spin-label at position 5 of the 5′-terminal uridine residue (5′-UUCAACGCC-3′, the spin-labeled nucleotide is highlighted in italics) when the spin-label is oxidized and paramagnetic (black) and upon reduction to its diamagnetic state (red). Right: Mapping of the amino acids showing no signals in the presence of the oxidized spin-label due to spatial proximity to the 5′-end of the bound RNA on the structure of the MjNep1 dimer. Affected lysines are shown in orange, unaffected lysines are colored blue. The signals of K143 and K30 are too weak to be reliably tracked in these experiments.
When the chemical shift perturbation data from the experiments with the uniformly and selectively labeled samples are taken together and the affected amino acids are plotted on the surface of the protein a contiguous RNA-binding surface is mapped out (Figure 6D). This surface extends from the vicinity of the bound cofactor along the basic groove at the dimer interface including α-helix α4 of the β−α−β insertion element to the extended irregularly structured surface loop. Thus, the two insertion elements unique for Nep1 when compared to the canonical SPOUT-class core fold are both involved in RNA binding along with amino acids found in the core elements of Nep1. The RNA-binding site appears to be extended along the dimer interface spanning a length of ∼45 Å. Notably, the RNA-binding site also includes the highly conserved aspartate (D56 in MjNep1) which is mutated in Bowen–Conradi syndrome in humans. It forms a salt bridge with and orients the side chain of the similarly conserved R54 which is also affected by RNA binding.
To define the orientation of the RNA in the binding site we took advantage of the high resolution available in the spectra of the selectively 15N-lysine and 15N-arginine-labeled samples. Chemical shift perturbation effects were compared for RNA 11 and RNA 10 which contains an extra C at its 3′-end. Most signals behaved similarly in titration experiments with either RNA 10 or RNA 11 indicating a very similar binding mode for the two RNAs. However, a subset of signals not or only slightly affected in titrations using RNA 11 showed chemical shift changes upon titration with the 3′-extended RNA 10. The signals only affected by the presence of RNA 10 correspond to K25 in the selectively 15N-lysine-labeled sample (data not shown) and R24 and R27 in the 15N-arginine-labeled samples (Figure 6E). All three amino acids are located next to each other in the irregularly structured surface loop and are apparently involved in binding to the extra 3′-nt of RNA 10.
Addition of nt 5′ to the position corresponding to nt 1190 in the 18S rRNA only lead to moderate increases in the binding affinity indicating that these nucleotides play only a minor role for RNA binding and might not be specifically recognized by the protein. Accordingly we employed a different technique to map the location of the 5′-end of the RNA. A version of RNA 10 containing a nitroxyl spin label attached to the base of the 5′-terminal uridine (equivalent to nt 913 in 16S rRNA) and a uridine instead of the pseudouridine in the adjacent position due to synthetic reasons was synthesized. The nitroxyl spin label is paramagnetic and NMR-signals corresponding to amino acids in close spatial vicinity of the spin label are reduced in intensity due to enhanced relaxation caused by the presence of the unpaired electron in the spin label. Titration of 15N-lysine labeled MjNep1 with this spin-labeled RNA 10 lead to the disappearance of the signals corresponding to K47, K49, K168, K170 and K188. These amino acids are all located close to the cofactor-binding site. Accordingly, the 5′-end of RNA 10 must bind in the vicinity of the bound SAH. This immediately places the nucleotide corresponding to the modified nt 914 next to the active site of MjNep1 as expected since this nucleotide is the methyl group acceptor in the reaction. Thus, MjNep1 binds its RNA target along an extended basic surface cleft at the dimer interface that involves both insertions to the SPOUT-class core fold in an orientation consistent with the proposed methylation of nt 914 in M. jannaschii 16S rRNA (Supplementary Figure S3).
DISCUSSION
Structural studies of Nep1 suggested that Nep1 might be a SPOUT-class RNA methyltransferase (16,17). Our in vitro investigations of the RNA-binding specificity and enzymatic activity of MjNep1 described here demonstrate that MjNep1 is a specific pseudouridine N1-methyltransferase. To our knowledge, MjNep1 is the first enzyme described to have such an activity and extends the known catalytic repertoire of SPOUT-class methyltransferases. The only other pseudouridine modifying methyltransferase identified so far is the bacterial protein and SPOUT-class family member YbeA (RlmH). In contrast to MjNep1, this enzyme transfers the methyl group to the N3 nitrogen of pseudouridine (19,20).
MjNep1 methylates RNA sequences resembling nt 910–921 of M. jannaschii 16S rRNA but only when they contain a pseudouridine in position 914. While it is known that the equivalent position contains a hypermodified pseudouridine (m1acp3-Ψ) in yeast and human 18S rRNA (29) and a 3-amino-3-carboxypropyl-uridine in the archeon Haloferax volcanii (42) it is not known whether and how this nucleotide is modified in M. jannaschii 16S rRNA. Our results now suggest that M. jannaschii 16S rRNA contains an at least N1-methylated pseudouridine at position 914. The pseudouridine N1-methyltransferase activity of Nep1 is conserved in the human enzyme where m1acp3-Ψ in position 1248 of human 18S rRNA is the only known methylated pseudouridine. Therefore, Nep1 apparently catalyzes the S-adenosylmethionine dependent N1-methylation step during the conversion of uridine to N1-methyl-N3-(3-methyl-3-carboxypropyl)-pseudouridine in 18S rRNA.
MjNep1 binds its substrate RNAs with a surprisingly high affinity which might be detrimental for an enzyme optimized towards substrate turnover. Thus, the high substrate affinity of MjNep1 might be either due to the extreme requirements of the thermophilic environment of its host organism or hint at an additional function of Nep1 besides its methyltransferase activity requiring this high substrate affinity. Such an additional function might be the replacement of the snoRNA required for the introduction of the pseudouridine [snR35 in the case of yeast (4)] from its substrate. Indeed, genetic experiments in yeast suggest a role for Nep1 in the replacement of snR57 from the 18S rRNA precursor which binds to the second copy of the Nep1-binding RNA consensus motif (27). Alternatively, one can imagine a scenario where Nep1 keeps its RNA target effectively in a single stranded conformation and thereby prevents non-native interactions with other sequence elements during the folding of the 18S rRNA precursor and small ribosomal subunit biogenesis.
The RNA-binding mode of Nep1 as deduced from our NMR-studies resembles the RNA-binding mode of other SPOUT-class family members with respect to the utilization of extension elements not part of the core fold for RNA interactions. However, the structure of these extensions is unique to Nep1 homologs and does not resemble known RNA-binding domains. Notably, the experimentally mapped RNA-binding surface of MjNep1 includes the highly conserved amino acid D56. A mutation of the corresponding amino acid D86 in HsNep1 to a glycine has been recently identified as the cause for Bowen–Conradi syndrome (15). Since the aspartate side chain of this residue orients the side chain of an arginine residue also shown to be involved in RNA binding it is conceivable that this mutation leads to a protein with an altered RNA affinity and/or specificity and consequently with an altered methylation activity. Fournier and coworkers recently analyzed the consequences of a knock-out of snR35 in yeast (4). This knock-out prevents the conversion of U1191 to a pseudouridine and consequently the methylation of this nucleotide by Nep1 but not the subsequent addition of the 3-amino-3-carboxypropyl group to the N3 occuring in the cytosol. They showed that the loss of methylation but not the loss of snoRNA binding in this region results in a reduced overall growth rate and the accumulation of 18S rRNA precursors in yeast. Thus, it is conceivable that an altered methylation activity in the Nep1 mutant might cause imbalances in ribosome biogenesis and thereby contribute to the symptoms of Bowen–Conradi syndrome.
In any case, the techniques and results described in this article for the identification and characterization of the enzymatic activity of Nep1 set the stage for detailed investigations of the functional, structural and dynamical consequences of the Nep1 mutation linked to Bowen–Conradi syndrome.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Aventis Foundation Endowed Professorship (to J.W.); Center for Biomolecular Magnetic Resonance (BMRZ); Cluster of Excellence ‚Macromolecular complexes’, Johann-Wolfgang-Goethe-University Frankfurt, and the Deutsche Forschungsgemeinschaft (DFG) through the SFB 579 ‘RNA-ligand interactions’ (to J.W., J.W.E. and K-D.E.). Funding for open access charge: Aventis Foundation.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to M. Görlach, H. Schwalbe, M. Bohnsack, and E. Duchardt for helpful discussions and their critical reading of the manuscript.
REFERENCES
- 1.Rozenski J, Crain PF, McCloskey JA. The RNA modification database: 1999 update. Nucleic Acids Res. 1999;27:196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Decatur WA, Fournier MJ. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002;27:344–351. doi: 10.1016/s0968-0004(02)02109-6. [DOI] [PubMed] [Google Scholar]
- 3.King TH, Liu B, McCully RR, Fournier MJ. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell. 2003;11:425–435. doi: 10.1016/s1097-2765(03)00040-6. [DOI] [PubMed] [Google Scholar]
- 4.Liang XH, Liu Q, Fournier MJ. Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA. 2009;15:1716–1728. doi: 10.1261/rna.1724409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caldas T, Binet E, Bouloc P, Costa A, Desgres J, Richarme G. The FtsJ/RrmJ heat shock protein of Escherichia coli is a 23 S ribosomal RNA methyltransferase. J. Biol. Chem. 2000;275:16414–16419. doi: 10.1074/jbc.M001854200. [DOI] [PubMed] [Google Scholar]
- 6.Ofengand J, Malhotra A, Remme J, Gutgsell NS, Del Campo M, Jean-Charles S, Peil L, Kaya Y. Pseudouridines and pseudouridine synthases of the ribosome. Cold Spring Harb. Symp. Quant. Biol. 2001;66:147–159. doi: 10.1101/sqb.2001.66.147. [DOI] [PubMed] [Google Scholar]
- 7.Reichow SL, Hamma T, Ferré-D'A;maré AR, Varani G. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. doi: 10.1093/nar/gkl1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tollervey D, Lehtonen H, Jansen R, Kern H, Hurt EC. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell. 1993;72:443–457. doi: 10.1016/0092-8674(93)90120-f. [DOI] [PubMed] [Google Scholar]
- 9.Ye K. H/ACA guide RNAs, proteins and complexes. Curr. Opin. Struct. Biol. 2007;17:287–292. doi: 10.1016/j.sbi.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 10.Lapeyre B, Purushothaman SK. Spb1p-directed formation of Gm2922 in the ribosome catalytic center occurs at a late processing stage. Mol. Cell. 2004;16:663–669. doi: 10.1016/j.molcel.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 11.Lafontaine D, Delcour J, Glasser AL, Desgrès J, Vandenhaute J. The DIM1 gene responsible for the conserved m6(2)Am6(2)A dimethylation in the 3'-terminal loop of 18 S rRNA is essential in yeast. J. Mol. Biol. 1994;241:492–497. doi: 10.1006/jmbi.1994.1525. [DOI] [PubMed] [Google Scholar]
- 12.White J, Li Z, Sardana R, Bujnicki JM, Marcotte EM, Johnson AW. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol. Cell. Biol. 2008;28:3151–3161. doi: 10.1128/MCB.01674-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu PC, Thiele DJ. Novel stress-responsive genes EMG1 and NOP14 encode conserved, interacting proteins required for 40S ribosome biogenesis. Mol. Biol. Cell. 2001;12:3644–3657. doi: 10.1091/mbc.12.11.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eschrich D, Buchhaupt M, Kotter P, Entian K.-D. Nep1p (Emg1p), a novel protein conserved in eukaryotes and archaea, is involved in ribosome biogenesis. Curr. Genet. 2002;40:326–338. doi: 10.1007/s00294-001-0269-4. [DOI] [PubMed] [Google Scholar]
- 15.Armistead J, Khatkar S, Meyer B, Mark BL, Patel N, Coghlan G, Lamont RE, Liu S, Wiechert J, Cattini PA, et al. Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am. J. Hum. Genet. 2009;84:728–739. doi: 10.1016/j.ajhg.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leulliot N, Bohnsack MT, Graille M, Tollervey D, Van Tilbeurgh H. The yeast ribosome synthesis factor Emg1 is a novel member of the superfamily of alpha/beta knot fold methyltransferases. Nucleic Acids Res. 2008;36:629–639. doi: 10.1093/nar/gkm1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor AB, Meyer B, Leal BZ, Kötter P, Schirf V, Demeler B, Hart PJ, Entian K.-D, Wöhnert J. The crystal structure of Nep1 reveals an extended SPOUT-class methyltransferase fold and a pre-organized SAM-binding site. Nucleic Acids Res. 2008;36:1542–1554. doi: 10.1093/nar/gkm1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tkaczuk KL, Dunin-Horkawicz S, Purta E, Bujnicki JM. Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinformatics. 2007;8:73. doi: 10.1186/1471-2105-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ero R, Peil L, Liiv A, Remme J. Identification of pseudouridine methyltransferase in Escherichia coli. RNA. 2008;14:2223–2233. doi: 10.1261/rna.1186608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purta E, Kaminska KH, Kasprzak JM, Bujnicki JM, Douthwaite S. YbeA is the m3Psi methyltransferase RlmH that targets nucleotide 1915 in 23S rRNA. RNA. 2008;14:2234–2244. doi: 10.1261/rna.1198108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn HJ, Kim H.-W, Yoon H.-J, Lee BI, Suh SW, Yang JK. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. EMBO J. 2003;22:2593–2603. doi: 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nureki O, Watanabe K, Fukai S, Ishii R, Endo Y, Hori H, Yokoyama S. Deep knot structure for construction of active site and cofactor binding site of tRNA modification enzyme. Structure. 2004;12:593–602. doi: 10.1016/j.str.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Kim DJ, Kim HS, Lee SJ, Suh SW. Crystal structure of Thermotoga maritima SPOUT superfamily RNA methyltransferase Tm1570 in complex with S-adenosyl-L-methionine. Proteins. 2009;74:245–249. doi: 10.1002/prot.22249. [DOI] [PubMed] [Google Scholar]
- 24.Kuratani M, Bessho Y, Nishimoto M, Grosjean H, Yokoyama S. Crystal structure and mutational study of a unique SpoU family archaeal methylase that forms 2'-O-methylcytidine at position 56 of tRNA. J. Mol. Biol. 2008;375:1064–1075. doi: 10.1016/j.jmb.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 25.Mosbacher TG, Bechthold A, Schulz GE. Structure and function of the antibiotic resistance-mediating methyltransferase AviRb from Streptomyces viridochromogenes. J. Mol. Biol. 2005;345:535–545. doi: 10.1016/j.jmb.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 26.Dunstan MS, Hang PC, Zelinskaya NV, Honek JF, Conn GL. Structure of the thiostrepton resistance methyltransferase S-Adenosyl-L-methionine complex and its interaction with ribosomal RNA. J. Biol. Chem. 2009;284:17013–17020. doi: 10.1074/jbc.M901618200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchhaupt M, Meyer B, Kötter P, Entian K.-D. Genetic evidence for 18S rRNA binding and an Rps19p assembly function of yeast nucleolar protein Nep1p. Mol. Genet. Genomics. 2006;276:273–284. doi: 10.1007/s00438-006-0132-x. [DOI] [PubMed] [Google Scholar]
- 28.Lowe TM, Eddy SR. A computational screen for methylation guide snoRNAs in yeast. Science. 1999;283:1168–1171. doi: 10.1126/science.283.5405.1168. [DOI] [PubMed] [Google Scholar]
- 29.Brand RC, Klootwijk J, Planta RJ, Maden BE. Biosynthesis of a hypermodified nucleotide in Saccharomyces carlsbergensis 17S and HeLa-cell 18S ribosomal ribonucleic acid. Biochem. J. 1978;169:71–77. doi: 10.1042/bj1690071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Görlach M, Wittekind M, Beckman RA, Mueller L, Dreyfuss G. Interaction of the RNA-binding domain of the hnRNP C proteins with RNA. EMBO J. 1992;11:3289–3295. doi: 10.1002/j.1460-2075.1992.tb05407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos A, Varani G. A new method to detect long-range protein RNA contacts: NMR detection of electron proton relaxation induced by nitroxide spin-labeled RNA. J. Am. Chem. Soc. 1998;120:10992–10993. [Google Scholar]
- 32.Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW. Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Meth. Enzymol. 1989;177:44–73. doi: 10.1016/0076-6879(89)77005-1. [DOI] [PubMed] [Google Scholar]
- 33.Piton N, Mu Y, Stock G, Prisner TF, Schiemann O, Engels JW. Base-specific spin-labeling of RNA for structure determination. Nucleic Acids Res. 2007;35:3128–3143. doi: 10.1093/nar/gkm169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schiemann O, Piton N, Plackmeyer J, Bode BE, Prisner TF, Engels JW. Spin labeling of oligonucleotides with the nitroxide TPA and use of PELDOR, a pulse EPR method, to measure intramolecular distances. Nat. Protoc. 2007;2:904–923. doi: 10.1038/nprot.2007.97. [DOI] [PubMed] [Google Scholar]
- 35.Mertens ML, Kägi JH. A graphical correction procedure for inner filter effect in fluorescence quenching titrations. Anal. Biochem. 1979;96:448–455. doi: 10.1016/0003-2697(79)90605-5. [DOI] [PubMed] [Google Scholar]
- 36.Sengupta DJ, Wickens M, Fields S. Identification of RNAs that bind to a specific protein using the yeast three-hybrid system. RNA. 1999;5:596–601. doi: 10.1017/s1355838299002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bahr U, Aygün H, Karas M. Sequencing of single and double stranded RNA oligonucleotides by acid hydrolysis and MALDI mass spectrometry. Anal. Chem. 2009;81:3173–3179. doi: 10.1021/ac900100x. [DOI] [PubMed] [Google Scholar]
- 38.Gehrke CW, Kuo KC. Ribonucleoside analysis by reversed-phase high-performance liquid chromatography. J. Chromatogr. 1989;471:3–36. doi: 10.1016/s0021-9673(00)94152-9. [DOI] [PubMed] [Google Scholar]
- 39.Wurm JP, Duchardt E, Meyer B, Leal BZ, Kötter P, Entian K.-D, Wöhnert J. Backbone resonance assignments of the 48 kDa dimeric putative 18S rRNA-methyltransferase Nep1 from Methanocaldococcus jannaschii. Biomol. NMR Assign. 2009;3:251–254. doi: 10.1007/s12104-009-9187-z. [DOI] [PubMed] [Google Scholar]
- 40.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Progress in Nuclear Magnetic Resonance Spectroscopy. 1999;34:93–158. [Google Scholar]
- 41.Keller R. The Computer Aided Resonance Assignment Tutorial. Goldau: Cantina Verlag; 2004. [Google Scholar]
- 42.Kowalak JA, Bruenger E, Crain PF, McCloskey JA. Identities and phylogenetic comparisons of posttranscriptional modifications in 16 S ribosomal RNA from Haloferax volcanii. J. Biol. Chem. 2000;275:24484–24489. doi: 10.1074/jbc.M002153200. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharya BK, Devivar RV, Revankar GR. A practical synthesis of N1-Methyl-2'-deoxy-ψ-uridine (ψ-Thymidine) and its incorporation into G-rich triple helix forming oligonucleotides. Nucleosides, Nucleotides and Nucleic Acids. 1995;14:1269–1287. [Google Scholar]
- 44.Maden BE. Identification of the locations of the methyl groups in 18 S ribosomal RNA from Xenopus laevis and man. J. Mol. Biol. 1986;189:681–699. doi: 10.1016/0022-2836(86)90498-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}