


Abstract
Hybrids of RNA with arabinonucleic acids 2′F-ANA and ANA have very similar structures but strikingly different thermal stabilities. We now present a thorough study combining NMR and other biophysical methods together with state-of-the-art theoretical calculations on a fully modified 10-mer hybrid duplex. Comparison between the solution structure of 2′F-ANA•RNA and ANA•RNA hybrids indicates that the increased binding affinity of 2′F-ANA is related to several subtle differences, most importantly a favorable pseudohydrogen bond (2′F–purine H8) which contrasts with unfavorable 2′-OH–nucleobase steric interactions in the case of ANA. While both 2′F-ANA and ANA strands maintained conformations in the southern/eastern sugar pucker range, the 2′F-ANA strand’s structure was more compatible with the A-like structure of a hybrid duplex. No dramatic differences are found in terms of relative hydration for the two hybrids, but the ANA•RNA duplex showed lower uptake of counterions than its 2′F-ANA•RNA counterpart. Finally, while the two hybrid duplexes are of similar rigidities, 2′F-ANA single strands may be more suitably preorganized for duplex formation. Thus the dramatically increased stability of 2′F-ANA•RNA and ANA•RNA duplexes is caused by differences in at least four areas, of which structure and pseudohydrogen bonding are the most important.
INTRODUCTION
The arabinonucleic acids ANA and 2′F-ANA are very close cousins. Where ANA contains a hydroxyl group, 2′F-ANA contains fluorine (Figure 1A). The most extensive structural studies of ANA•RNA and 2′F-ANA•RNA duplexes to date, performed on short hairpins with modified stems, observed no major conformational differences between them—neither in sugar pucker nor hydrogen bonding nor steric effects (1,2). ANA and 2′F-ANA hybrid duplexes with RNA display structure and flexibility patterns that make them effective mimics of the DNA•RNA hybrid, even in terms of RNase H degradation (3–5).
Figure 1.

(A) Structures of ANA and 2′F-ANA in comparison with DNA and RNA. (B) Numbering scheme of the 10-mer hybrid duplexes used for this study. X indicates the DNA, 2′F-ANA or ANA strand. The corresponding hybrids are named DR, FR and AR, respectively. Thymines are replaced by uracils in the ANA strand. (Use of T instead of U does explain some of the increased duplex stability of the 2′F-ANA-containing hybrid. However, comparison of 2′F-ANA and ANA-containing hybrids where neither strand includes T/U and studies where both 2′F-ANA and ANA contained T confirm that the 2′F-ANA-containing hybrids are still significantly more stable than their ANA-containing counterparts.) (6,7).
And so it is a puzzle that their binding affinities are strikingly different: ANA has relatively low affinity for RNA (7), while 2′F-ANA binds to RNA with high affinity (8). Because of this high binding affinity, and many other favorable properties, 2′F-ANA has shown promise for applications as diverse as gene silencing therapeutics, diagnostics and aptamer design (9) and a 2′F-ANA-based antisense drug has received approval to begin clinical trials (10). The elusive origin of the difference in binding affinity is therefore of significant interest: it will deepen our understanding of nucleic acid structure and binding while potentially allowing the design of derivatives with improved properties.
Initially, it was supposed that an unfavorable steric interaction involving the 2′-OH of ANA was responsible for its low binding affinity (3,7,8). Other studies have suggested that there could be a major difference in sugar pucker or hydrogen bonding between the two analogues (11,12). However, these structural explanations were deemed unlikely based on the most significant structural studies of ANA•RNA and 2′F-ANA•RNA duplexes to date: high-resolution NMR studies that examined short hairpins containing a 4-bp hybrid stem and a 4-nt DNA loop (1,2). No major structural differences between ANA and 2′F-ANA were observed during this study, and the authors suggested differential hydration as a possible explanation for the greater stability of 2′F-ANA•RNA duplexes. However, we wondered if subtle differences might have been overlooked in the hairpin-based structural studies: while use of hairpins was convenient, the model contained only four hybrid base pairs, two of which might be affected by their proximity to the loop structure or the terminus.
Therefore, to gain further insight on 2′F-ANA and ANA substitution in hybrid duplexes, we decided to carry out a combined biophysical and computational study of the decamer shown in Figure 1B. This allowed us to study flexibility, hydration and ion uptake together with structure and conformation, bringing together spectroscopic and computational data. The structure of the unmodified DNA•RNA duplex has been extensively studied by NMR and restrained molecular dynamics calculations, using conventional and time averaged constraints (13). This duplex is ideal for studying the origin of the difference in thermal stability between ANA•RNA and 2′F-ANA•RNA because it is representative of prototypical hybrids and because the difference in Tm values is nearly 18°C (1.8°C per base pair). We also verified that the duplex is a substrate of RNase H (Supplementary Figure S1) to ensure the highest applicability of the current study to the field of antisense therapeutics.
MATERIALS AND METHODS
Oligonucleotide synthesis and purification
Oligonucleotides were synthesized from phosphoramidite precursors using standard solid-phase methods. All masses were verified by ESI–MS. Purification of oligonucleotides was carried out using denaturing PAGE and ion-pairing HPLC. Small triethylammonium signals were visible in the NMR spectra from the ion-pairing reagent but did not interfere with an important region of the NMR spectrum.
UV melting and derivation of thermodynamic parameters
All UV spectroscopy was carried out in a Cary 300 or Cary 5000 UV spectrophotometer (Varian). Samples were kept under flowing nitrogen when below 15°C. Except where indicated, duplex concentration was 2 µM (4 µM total concentration of strands). For most of the Tm experiments, lower-melting samples DR, AR and DD were heated from 2°C to 60°C, while higher-melting samples FR and RR were heated from 15°C to at least 70°C. In this way, the spectra contained clear baseline regions while making the best use of spectrophotometer time.
Assignment of baselines was very clear in most cases. Curves with flaws in the baseline were discarded without analysis. For the lower-melting samples at low salt concentrations, baselines were of necessity short and somewhat ambiguous, and we chose a lower baseline that was parallel to the long upper baseline for the sake of consistency. For other curves, if there was ambiguity about the lower baseline, we chose the lowest linear region or in severe cases, discarded the run. We repeated the baseline analysis on a random sampling of 36 spectra including all duplexes, and found that the reanalyzed Tm values differed from the original values by 0.05°C on average. The standard deviation of the differences was 0.13°C, and this larger number was added as an additional uncertainty in the Tm values, and propagated through all subsequent calculations.
UV-based Tm experiments were carried out in phosphate (140 mM KCl, 5 mM Na2HPO4, 1 mM MgCl2, pH 7.2) or cacodylate (10 mM NaC2H6AsO2, 300 mM NaCl, 0.1 mM EDTA, pH 7.2) buffers. The Tm values of the five duplexes were essentially identical in these two buffers. The thermodynamic parameters of the melting of these duplexes were derived in two ways. A van’t Hoff plot, ln(K) versus 1/Tm, can be used to determine ΔH and ΔS, according to the equation ΔH = –R [d(lnK)/d(1/Tm)] (14). Since the equilibrium constant K can be written in terms of the mole fraction of duplex, α, it is straightforward to calculate the value of K at the Tm where α = 0.5. This calculation was carried out within the Cary software provided with the UV spectrophotometer.
This method is quite vulnerable to the choice of baseline, and an alternative method of calculating ΔH was also used which relies on the concentration dependence of the Tm. Starting with the well-known equation –RT[ln(K)] = ΔH° – TΔS°, for a non-self-complementary bimolecular equilibrium, this equation can be expanded and rearranged to give the following: 1/Tm = [R/ΔH°] ln(CT) + [(ΔS° – Rln(4))/ΔH°], where CT represents the total strand concentration. Thus, a plot of 1/Tm versus ln(CT) is linear with slope R/ΔH° (14). This plot is shown in Supplementary Figure S2. Both methods of calculating ΔH agreed within experimental error.
Ion uptake experiments
We studied the melting behavior of the duplexes in buffers containing 20–500 mM Na+, 20–500 mM K+ and 5–50 mM Mg2+ by UV spectroscopy as described above. For sodium buffers, 100 mM Na2HPO4 was adjusted to pH 7.2 using H3PO4; aliquots were then combined with the desired volume of 1 M NaCl (if needed) and diluted to a final concentration of 10 mM phosphate and 20, 50, 100 or 500 mM Na+. Potassium buffers were prepared in a similar fashion using K2HPO4, H3PO4 and KCl. Magnesium buffers were prepared using the 100 mM sodium buffer and MgCl2, resulting in a final concentration of 20 mM Na+ and either 5, 15 or 50 mM Mg2+. Duplex concentration was 2 µM (4 µM total strand concentration).
Plotting Tm against the logarithm of ion concentration gave generally linear correlations in this range (Supplementary Figure S3, slopes are given in Supplementary Table S1). Using higher concentrations gave clear deviations from linearity; we tested up to 1 M Na+ and 150 mM Mg2+ but the data were not reliable in this range. The number of cations released upon duplex melting can then be obtained, as done previously (15), from Equation (1):
| (1) |
where δTm/δ(ln[ion]) represents the slope of a plot of Tm versus the natural logarithm of cation concentration, R is the ideal gas constant and 1.11 is a proportionality constant for converting between ionic activity and concentration. Enthalpy values were derived from the plot of 1/Tm versus ln(duplex concentration) discussed above (14).
Errors inherent in the ion uptake measurements were based on the method of Rozners and Moulder (16). Thus, the standard deviations of the Tm values for the extreme points on the Tm versus ln[ion] graph were used to calculate alternative minimum and maximum slopes δTm/δ(ln[ion]). Propagating the standard deviation in the slope through the subsequent calculations gave the uncertainty in the number of ions released upon melting.
NMR experiments
Samples of the three hybrid duplexes were suspended in 500 µl of either D2O or H2O/D2O 9:1 in phosphate buffer, 100 mM NaCl, pH 7. NMR spectra were acquired in Bruker Avance spectrometers operating at 600 or 800 MHz, and processed with Topspin software. DQF-COSY, hetero-COSY (1H–31P), TOCSY and NOESY experiments were recorded in D2O. The NOESY spectra were acquired with mixing times of 50, 150 and 250 ms, and the TOCSY spectra were recorded with standard MLEV-17 spin-lock sequence, and 80-ms mixing time. NOESY spectra in H2O were acquired with 50 and 150 ms mixing times. In 2D experiments in H2O, water suppression was achieved by including a WATERGATE (17) module in the pulse sequence prior to acquisition. Two-dimensional experiments in D2O were carried out at temperatures ranging from 5°C to 25°C, whereas spectra in H2O were recorded at 5°C to reduce the exchange with water. 31P resonances were assigned from proton-detected heteronuclear correlation spectra (18). 19F resonances were assigned from natural abundance 1H-19F HETCOR and 19F detected HOESY spectra (19). The spectral analysis program Sparky (20) was used for semiautomatic assignment of the NOESY cross-peaks and quantitative evaluation of the NOE intensities.
NMR constraints
Quantitative distance constraints were obtained from NOESY experiments by using a complete relaxation matrix analysis with the program MARDIGRAS (21). Error bounds in the interprotonic distances were estimated by carrying out several MARDIGRAS calculations with different initial models, mixing times and correlation times. Standard A- and B-form duplexes were used as initial models, and three correlation times (1.0, 2.0 and 4.0 ns) were employed, assuming an isotropic motion for the molecule. Experimental intensities were recorded at three different mixing times (50, 150 and 250 ms) for non-exchangeable protons. Final constraints were obtained by averaging the upper and lower distance bounds in all the MARDIGRAS runs. Qualitative limits of 1.8 and 5 Å were set in those distances where no quantitative analysis could be carried out, such as overlapping cross-peaks or those with a very weak intensity. 19F–1H distance constraints were extracted from a qualitative analysis of HOESY experiments. In addition to these experimentally derived constraints, Watson–Crick hydrogen bond restraints were used. Target values for distances and angles related to hydrogen bonds were set as described from crystallographic data. No backbone angle constraints were employed. Distance constraints with their corresponding error bounds were incorporated into the AMBER potential energy by defining a flat-well potential term.
1H–1H J-coupling constants could not be accurately measured due to the relatively high linewidths of the sugar proton signals. However, sums of J-coupling constants were roughly estimated from DQF-COSY cross-peaks. Loose values were set for the sugar dihedral angles δ, ν1 and ν2 to constrain ribose conformation of the RNA strand to the North domain, and the arabinose conformation to the East or South domain.
Structure determination from NMR restraints
Structures were calculated with the SANDER module of the molecular dynamics package AMBER 7.0 (22). Starting models of the hybrid duplexes were built in the A- and B- canonical structures using SYBYL. These structures were taken as starting points for the AMBER refinement, which started with an annealing protocol in vacuo (using hexahydrated Na+ counterions placed near the phosphates to neutralize the system). The temperature and the relative weights of the experimental constraints were varied during the simulations according to our standard annealing protocols (23).
The resulting structures from in vacuo calculations were refined including explicit solvent, periodic boundary conditions and the Particle–Mesh–Ewald method to evaluate long-range electrostatic interactions (24). Thus, the structures obtained in the previous step were placed in the center of a water-box with around 4000 water molecules and 16 sodium counterions to obtain electroneutral systems. We used the parmbsc0 (25) revision of the parm99 force field (26,27) including suitable parameters for the arabino and 2′F-arabino derivatives extracted from our previous work (5). The TIP3P model was used to describe water molecules (28). The protocol for the constrained molecular dynamics refinement in solution consisted of an equilibration period of 160 ps using a standard equilibration process (29), followed by 10 independent 500 ps runs. Averaged structures were obtained by averaging the last 250 ps of individual trajectories and further relaxation of the structure.
In order to complement information derived from NMR data and, in particular, to get more insight in the effect of water on the structure and stability of the hybrids, longer molecular dynamics simulations were performed both with and without NMR restraints. For reasons of computational efficiency these calculations were performed with the Gromacs-4 software (30) using the same force field described above. In the first case trajectories extended for 4 ns, while in the second were extended for 35 ns (after the 2 ns equilibration). The SETTLE algorithm (31) was used to constrain bond lengths and angles of water molecules, and P-LINCS (32) was used for all other bond lengths. The temperature of the simulation was kept constant at 300 K by the use of the canonical ensemble-preserving thermostat proposed by Bussi et al. (33), using separate coupling groups for the nucleic acid and for the ions-solvent. The pressure of the systems was kept constant by weak isotropic coupling to a pressure bath of 1 atm (34). As in AMBER simulations, periodic boundary conditions and Particle–Mesh–Ewald were used.
Analysis of the representative structures as well as the MD trajectories was carried out with the programs CURVES V5.1 (35), MOLMOL (36), the analysis tools of AMBER and GROMACS, and other ‘in house’ programs.
Free energy calculations
Free energy calculations were carried out by using a standard thermodynamic cycle, where the initial systems (for ANA and 2′F-ANA) were neutralized, solvated, optimized, thermalized and pre-equilibrated using a standard protocol (29), followed by an extensive 2 ns re-equilibration. Mutations were performed both in the arabino→2′F-arabino and 2′F-arabino→arabino directions following the thermodynamic integration algorithm (TI) as implemented in Gromacs-4 (30), using 21 windows of 2 ns (0.5 ns equilibration and 1.5 ns for collection). For each window we collected two independent estimates of ΔG/Δλ by using two blocks of 750 ps, which were then integrated through the entire mutation pathway to obtain mutation free energies (with associated errors).
Rough estimates of the changes in intra-duplex stability related to the ANA→2′F-ANA change were estimated by computing the change in the electrostatic and van der Waals interaction energies between –OH and –F groups with the rest of the oligo in the single-stranded and duplex forms. The change in solvation free energy due to the –OH → –F change was determined from discrete linear response calculations (37) in single strand and duplex. The global estimate of –F and –OH relative stability was determined by adding the intramolecular and solvation terms. This type of simple analysis is less accurate than MD/TI free energy values computed as above, but provides qualitatively useful information on the nature of the stabilizing/destabilizing effect of the ANA→2′F-ANA change.
All calculations were performed using the Mare Nostrum Supercomputer at the Barcelona Supercomputing Center and on local computers in our laboratories.
RESULTS
Duplex formation and thermodynamics
Duplex formation and melting was monitored by UV and NMR spectroscopy. Besides the three hybrid duplexes used for NMR structural studies (Figure 1B), isosequential RNA•RNA (RR) and DNA•DNA (DD) controls were included in the UV melting experiments. Tm values are shown in Table 1, and sample Tm curves for all five duplexes are shown in Figure 2. The 2′F-ANA•RNA (FR) duplex had the highest thermal stability of the five (Tm = 51.2°C) while the ANA•RNA (AR) duplex was the least stable (Tm = 32.4°C).
Table 1.
Tm values and thermodynamic parameters for the duplexes (buffer conditions 10 mM sodium cacodylate, 300 mM NaCl, 0.1 mM EDTA, pH 7.2)
| Name | Tm (°C)a | −ΔH (van’t Hoff) (kcal/mol)a | −ΔH (1/Tm versus. ln(c)) (kcal/mol)b | −ΔS (van’t Hoff) (cal/mol/K)a | −ΔG300 (kcal/ mol)c |
|---|---|---|---|---|---|
| DR | 33.7 ± 0.2 | 76.7 ± 2.4 | 74.1 ± 8.0 | 222 ± 8 | 7.5 |
| AR | 32.4 ± 0.3 | 86.0 ± 1.8 | 80.9 ± 10 | 254 ± 6 | 4.7 |
| FR | 51.2 ± 0.5 | 88.7 ± 5.9 | 89.2 ± 5.8 | 246 ± 18 | 15.4 |
| DD | 37.1 ± 0.3 | 75.7 ± 1.8 | 72.4 ± 5.2 | 216 ± 6 | 7.6 |
| RR | 46.9 ± 0.2 | 86.8 ± 3.6 | 93 ± 17 | 244 ± 11 | 19.8 |
aStandard deviations are given.
bErrors are estimated from the 1/Tm versus ln(c) plot.
cCalculated using (1/Tm versus ln(c)) values for ΔH and van’t Hoff values for ΔS.
Figure 2.

Melting of AR (ANA•RNA), DR (DNA•RNA), DD (DNA•DNA), RR (RNA•RNA) and FR (2′F-ANA•RNA), as followed by observing the change in A260 of duplex samples upon heating from 5 to 65°C. Duplexes were 20 µM in phosphate buffer (140 mM KCl, 5 mM Na2HPO4, 1 mM MgCl2, pH 7.2) Data derived from UV melting experiments such as these are given in Table 1. Starting values of A260 were 0.89 ± 0.04.
Thermodynamic parameters for duplex dissociation were derived from UV-monitored melting curves and are shown in Table 1. Values of enthalpy were calculated using two independent methods: van’t Hoff analysis of the melting curves, and a plot of the concentration dependence of the Tm. Both methods gave results for ΔH that agreed within experimental error, and both are listed in Table 1. The free energy of binding (ΔG) was also calculated and is given in Table 1. The free energy of binding for FR was 10.7 kcal/mol greater (i.e. more favorable) than that of AR.
NMR spectra are also consistent with duplex formation in all cases. Melting temperature experiments carried out on the NMR samples (by observing signals from imino protons upon increasing temperature) confirmed that the FR duplex is much more stable than DR or AR under the conditions used for NMR experiments (Supplementary Figure S4). The imino regions of the NOESY spectra are similar in the three duplexes, and their NOE cross-peak patterns are typical of Watson-Crick base pairs (Supplementary Figure S5).
NMR assignment
Sequential assignments of exchangeable and non-exchangeable proton resonances were conducted following standard methods for right-handed, double-stranded nucleic acids using DQF-COSY, TOCSY and 2D NOESY spectra. The assignment pathways could be followed in the base-H1′ (Supplementary Figures S6 and S7), and in the base-H2′ or base-H2″ regions. Assignment of 2′F-ANA and ANA strands was more complicated than the control duplex because of the low spectral dispersion of the arabinose proton resonances. In spite of this, complete assignments could be carried out with the exception of H5′/H5″ protons.
Exchangeable protons were assigned with the NOESY spectra recorded in H2O (Supplementary Figure S5). Most of the labile protons were assigned following standard methods, except some amino resonances of the terminal guanines that were not detected. The cross-peak patterns observed for the exchangeable protons indicate that all bases are forming Watson-Crick pairs throughout the duplex (Supplementary Figure S5). Chemical shifts of exchangeable protons are almost identical in the FR and DR duplexes, and exhibit small changes in the AR duplex.
Assignment of 19F resonances was carried out through their heteronuclear correlations with the adjacent H2′, H3′ and H1′ protons. Sequential and intra-residual 19F–1H6/1H8 cross-peaks, along with three sequential 19F–thymine methyl cross-peaks, were observed in the HOESY spectrum (Figure 3).
Figure 3.

Heteronuclear 19F–1H correlation HOESY spectrum of FR in D2O. Sequential assignment pathway between intra-residual and sequential 19F–1H6/1H8 cross-peaks is shown. Strong sequential 19F(T)–H8(A/G) cross-peaks (at TA and TG steps) are indicated with arrows.
Full assignments are given in Supplementary Tables S2 (FR) and S3 (AR).
Experimental constraints and structure calculations
Quantitative distance constraints were obtained from NOESY experiments by using a complete relaxation matrix analysis with the program MARDIGRAS. The total number of experimental distance constraints was around 200 (summarized in Supplementary Table 4). Only structurally relevant distance constraints were included. Intra-residual constraints involving protons of the same sugar are not included, with the exception of H1′–H4′ and H2″–H4′. Considering these constraints, the average number per base pair is around 20. In addition, a total of 21 19F – 1H distance constraints were obtained from heteronuclear dipolar correlation experiments. Sequential HOE cross-peaks were particularly intense for the thymine-adenine and thymine-guanine steps (Figure 3).
Some structural information can be readily determined from these distances. For example, intra-residual and sequential H1′–base and H2′–base NOEs in the RNA strand are consistent with a standard A-form duplex. However, many of the inter-proton distances in the 2′F-ANA and ANA strands are not consistent with a pure A-form conformation in the modified strand. This is particularly clear in the strong sequential H2″–H6/8 NOEs of the ANA strand in the AR hybrid, indicating significant B-like conformation in the ANA strand. In both 2′F-ANA and ANA sugar moieties, H1′-H4′ NOEs are strong, indicating a high population of an East-type sugar conformation, as expected (1,2,5,9).
In addition to the NOE-derived information, a qualitative analysis of the J-coupling constants obtained from DQF-COSY spectra was carried out. J1′2′ are undetectable in most of the riboses of the RNA strand of all the duplexes, indicating that riboses are in a pure North-type conformation. In the 2′F-ANA and ANA strands, however, J1′2′ are medium or weak. This, together with the small J3′2′ and the large J4′3′ coupling constants (∼8–10 Hz) point towards a higher population of East conformation for these sugars. According to this information, torsion angle constraints were set for the dihedral angles of the sugars. The riboses of the RNA strand were constrained to a North conformation. In the 2′F-ANA and ANA strands, sugar dihedrals were constrained to avoid North conformations and allow the East and South regions. Backbone dihedral angles were not constrained since the 31P–1H correlation spectra were not of high enough quality to estimate coupling constants. No scalar coupling was detected between fluorine atoms and H6/H8 protons.
All the distance and torsion angle constraints were used to calculate the structure of both hybrid duplexes by restrained molecular dynamics as described in the ‘Materials and Methods’ section. The 10 final structures of both hybrids resulting from restrained molecular dynamics calculation including the solvent explicitly are displayed in Figure 4. As can be seen in this figure (and also in Supplementary Table S4), the two duplexes are well defined, with an RMSD of 0.7 Å (excluding the terminal residues). RMSD values are in the same range for the FR and AR duplexes. The final AMBER energies and NOE terms are reasonably low in all the structures, with no distance constraint violation greater than 0.4 Å.
Figure 4.

(A) Superposition of the 10 refined structures of FR (left) and AR (right). Stereoscopic views of the average structures of the three hybrids FR (B), AR (C) and DR (D). Blue and magenta indicate different strands. Fluorine atoms are shown in green.
Relaxation data
Proton spin-lattice relaxation times (T1) and spin-spin relaxation times (T2) were measured with 1D NMR techniques. T1 was determined for isolated base protons with the inversion-recovery method, and T2 was estimated by spin-echo experiments. The T1 and T2 relaxation times are listed in Supplementary Table S5. There is not a large difference between spin-lattice relaxation times between protons in the RNA and in the 2′F-ANA or ANA strand. This result suggests that 2′F-ANA, ANA and RNA strands have similar dynamics. In contrast, significant differences are observed between protons in the two strands of DR (38). Such a difference seems to be a general feature of DNA•RNA hybrids; it is partly due to the presence of the H2″ proton and partly to the enhanced flexibility of the DNA strand.
Description of the structures
The overall shapes of FR and AR hybrids are intermediate between canonical A-form and B-form duplexes, but are closer to the A-form, as found for DR hybrids. The RMSD between the average structure of FR and AR hybrids and canonical A- and B-form duplexes are around 2.5 and 3.5 Å, respectively. RMSDs comparing the average structure of the FR, AR and DR duplexes are shown in Supplementary Table S6. The RMSD between the FR and AR average structures is 1.5 Å for heavy atoms in the non-terminal residues. The deviation with the average structure of the control duplex is in the same range (1.8 Å).
The geometry of the RNA strand is very similar in the three hybrids, with most of the riboses in the C3′-endo conformation and glycosidic angles around −160° (the only exception is U14). Thus the RNA strand in hybrids is quite rigid and conformationally similar to the RNA homoduplex. In the FR and AR hybrids, pseudorotation phase angles of arabinoses and 2′F-arabinoses are between 100° and 150°, in the southeast, and the glycosidic torsion angles range from −100° to −140°. These values tend to be higher than the corresponding ones for the deoxyriboses in the control duplex DR. This reflects an average between northern and southern conformations for DR, whereas for FR and AR the average occurs between eastern and southern puckers. Complete tables of geometrical parameters are shown in Supplementary Tables S7–S8.
The minor groove width in all three hybrids is intermediate between those of standard A-form and B-form helices (Figure 5). Of the three hybrids, however, AR features the narrowest minor groove width (Figure 5). This is consistent with a more B-like conformation for the AR hybrid.
Figure 5.

(A) Values of minor groove width along the sequence of the three hybrids, and (B) view of the hybrids from their minor groove side. Standard A-form and B-form duplexes are shown for comparison.
Helical parameters are relatively dispersed and reflect that the hybrid structures are intermediate between A- and B-form. Whereas rise values are around 3.1 Å, typical of B-form helices, twist angles are around 32°, which is characteristic of A-form helices. Significant roll values are observed in all pyrimidine–purine steps in the FR duplex, probably related to the presence of F•H pseudohydrogen bonds (see below). A summary of important helical parameters is shown in Supplementary Tables S9 and S10.
Hydration at fluorine atoms
NMR spectroscopy can give information about hydration at fluorine atoms by examining the chemical shift of fluorine nuclei in H2O and D2O buffers. For the FR duplex, the 19F chemical shift was relatively constant in both H2O and D2O (differences are lower than 0.01 ppm in all cases), implying that the fluorine atoms are not well hydrated. In contrast, nucleosides such as 2′-deoxy-2′-fluorocytidine and oligonucleotides containing solvent-exposed 2′-fluorine groups can show chemical shift changes up to 0.2 ppm (39).
Other biophysical experiments were carried out to confirm whether the modified duplexes could release water molecules to different degrees upon melting. The results of osmotic stressing experiments (15,40) and melting curves in the presence of D2O (41) are described in the Supplementary Data. These experiments did not give useful data on our system (for example, different results were obtained with different osmolytes). Since evidence from our computational work and NMR experiments indicated that hydration was relatively unimportant, we did not pursue these methods further.
Ion uptake studies by thermal melting
The ordered water structure around nucleic acids also contains ordered cations which can have a significant effect on duplex structure (42,43). Using Tm studies at various ionic strengths, several groups have examined the ion dependence of duplex melting (15,40). Because the 2′-substituents of arabinonucleic acids point into the major groove, specific ion interactions in the major groove could easily be different between ANA and 2′F-ANA.
Melting of the three hybrid duplexes, along with A-form RNA (RR) and B-form DNA (DD) controls, was studied under a variety of buffer conditions. Keeping the concentrations of duplex and other buffer components constant, the concentrations of Na+, K+ and Mg2+ were varied one at a time from 20 to 500 mM (Na+ and K+) or from 5 to 50 mM (Mg2+). As expected, increasing the concentration of any of the salts led to large increases in Tm values, however the magnitude of the increase varied significantly from duplex to duplex. The dependence of Tm values on ion concentrations is shown in Supplementary Figure S3.
The slopes of these ion-dependence graphs were used to calculate the number of cations released upon melting of the various duplexes (Table 2; details of calculations given in Materials and Methods section). These numbers provide a picture of the relative importance of ion uptake (and release) by the modified duplexes.
Table 2.
Number of ions released upon duplex melting
| Name | DR | AR | FR | RR | DD |
|---|---|---|---|---|---|
| Na+ | 2.6 ± 0.3 | 2.0 ± 0.4 | 2.5 ± 0.2 | 2.9 ± 0.6 | 2.6 ± 0.2 |
| K+ | 2.5 ± 0.3 | 1.9 ± 0.3 | 2.3 ± 0.2 | 2.8 ± 0.5 | 2.5 ± 0.2 |
| Mg2+ | 1.1 ± 0.2 | 0.5 ± 0.2 | 0.9 ± 0.1 | 1.1 ± 0.3 | n.d. |
Changes in the uptake of the monovalent cations were relatively small between the various modified duplexes, but some meaningful differences were observed. The pure A-form duplex RR had the highest ion uptake upon duplex formation, while duplex AR had the lowest. DR and FR hybrids, along with the B-form DNA duplex DD, had an intermediate level of ion uptake. Much larger differences were observed for AR in its Mg2+ uptake, which was about half that of the other duplexes, which all showed comparable levels of Mg2+ uptake. This suggests that ANA•RNA duplexes may have a much smaller affinity for divalent cations, and to some extent for counterions in general.
Theoretical calculation of binding free energies and associated energy terms
The relative free energy of binding of the arabinonucleic acid derivatives towards a RNA substrate (ΔΔGbind) was determined by using a standard thermodynamic cycle (Figure 6). In this computational technique, differences in the free energies of the vertical processes (duplex formation) are derived from analysis of the reversible work associated with the horizontal processes, which correspond to the interconversion of arabino and 2′F-arabino derivatives in an isolated single strand or hybrid duplex. All mutations were smooth, without apparent discontinuities that could signal the existence of hysteresis effects (Supplementary Figure 8). Replacing all 2′-OH groups of ANA with fluorine (2′F-ANA) in a 12-nt sequence leads to a free energy difference of −16.2 ± 0.6 kcal/mol favoring the stability of 2′F-ANA over ANA (see ‘Materials and Methods’ section). This corresponds to a free energy difference per nucleotide of −1.3 ± 0.6 kcal/mol (see Table 3), in good agreement with the experimental difference of around −1.1 kcal/mol (derived from numbers in Table 1). Very interestingly, almost the same value per sugar (−1.2 ± 0.5 kcal/mol; see Table 3) is found when the simulation is repeated for a 5-nt sequence, which suggests that the stabilization free energy induced by the arabino→2′F-arabino mutation runs parallel with the length of the oligo. The agreement with experimental results support the validity of these theoretical calculations and demonstrates that the larger stability of 2′F-ANA-modified hybrids can be fully rationalized under the ideal conditions considered by our calculations: low duplex concentrations and low ionic strengths.
Figure 6.

Details of the thermodynamic cycle in MD/TI free energy calculations.
Table 3.
Free energy differences from MD/TI calculationsa
| ΔG duplex (12-mer) RNA·ANA→ RNA·2′F-ANA | ΔG single strand (12-mer) ANA→2′F-ANA | ΔΔG(binding) ANA→2′F-ANA | ΔG single strand (5-mer) ANA→2′F-ANA | ΔΔG(binding) ANA→2′F-ANA | |
|---|---|---|---|---|---|
| Entire oligo | −204.3 ± 0.3 | −188.1 ± 0.3 | −16.2 ± 0.6 | −78.8 ± 0.2 | – |
| Per substituted sugar | −17.0 ± 0.3 | −15.7 ± 0.3 | −1.3 ± 0.6 | −15.8 ± 0.2 | −1.2 ± 0.5 |
aFree energy differences of the ANA→2′F-ANA mutation in a 12-mer and 5-mer. Values were computed for both duplex and single strand forms (12-mer) or single strand form only (5-mer) and subtracted to obtain ΔΔG. All values are in kcal/mol.
MD/TI calculations are very powerful to predict changes in stability due to small chemical modifications, but the global character of a free energy difference precludes a straightforward decomposition scheme (44–46) and so the origin of the stabilization produced by the change from arabinose to 2′F-arabinose is not obvious. Encouraged that the stability difference was computationally reproducible, we performed a systematic computational study on the ANA•RNA and 2′F-ANA•RNA duplexes as well as the single-stranded ANA and 2′F-ANA oligos and examined several possible reasons for the higher stability of 2′F-ANA•RNA duplexes.
Analysis of the equilibrium samplings did not reveal any significant structural change that could justify the greater stability of the 2′F-ANA derivative, in agreement with the high-resolution NMR data (see above). In general the 2′F-ANA hybrids appear more rigid than the ANA ones (Supplementary Figure S9), as is usually found in more stable structures, but the differences are small as suggested by NMR experiments. Analysis of the impact of the arabino→2′F-arabino change in sugar puckering also failed to detect any dramatic conformational difference between the two molecules which could explain the difference in stability (5).
The analysis of global hydration reveals that the arabinose 2′OH is well solvated in both the single strand and duplex. In addition, the solvation free energy (in the duplex versus single strand) is slightly better for ANA than for 2′F-ANA (Table 4) arguing that hydration cannot explain the increased stability of 2′F-ANA-containing hybrids.
Table 4.
Theoretical binding free energiesa
| 2′-substituent | ΔE binding total | ΔE binding intra-duplex | ΔE binding solvation |
|---|---|---|---|
| F | –0.9 | –1.2 | 0.3 |
| OH | 1.7 | 1.8 | –0.1 |
aAll values are in kcal/mol.
Detailed energy calculation of the internal hybrid duplex interaction energy (i.e. intra-duplex energy) shows that the major reason for the increased stability of FR over AR is contained within the intra-duplex interaction energy, which is favorable for 2′F-ANA (better in the duplex than in the single strand) but unfavorable for ANA (Table 4). A detailed analysis reveals that a pseudohydrogen bond between 2′-F and purine C8-H contributes substantially to the stabilization of the FR duplex (around 2 kcal/mol), while bad steric contributions (differential van der Waals energy + 0.5 kcal/mol) and the lack of favorable electrostatic contacts are responsible for the unfavorable intra-duplex binding energy of the AR derivative (Table 4).
DISCUSSION
Structural differences
Despite the general similarity between DR, AR and FR hybrids, subtle structural differences can be observed between the modified strands. Thus, while sugars in the DNA strand of a DR hybrid are in a dynamic equilibrium between north and south (or A and B-form) conformations, sugars in both ANA and 2′F-ANA strands are more rigid and sampled only the South and East regions. 2′F-ANA sugars appear slightly more displaced towards the East region, while the sugars of the ANA strand are closer to the canonical B-form (South) region. Excluding terminal residues, the average pseudorotational phase angle is 125° for the 2′F-ANA strand and 133° for the ANA strand. Particularly significant are the differences at nucleotides T5, A6 and T8, for which the ANA strand has a greater pseudorotational phase angle by 22–62° (Supplementary Tables S7 and S8). Consistent with this increase in B-form character for the ANA strand, the minor groove width of AR is smaller than that of FR or DR. A third line of evidence for the greater B-form character of the ANA strand is found in the strong sequential H2″-H6/8 NOEs observed in that strand. Our data are consistent with early expectations that ANA nucleotides are unlikely to adopt the A-conformation (47).
Since the RNA strand is maintained in a Northern conformation, a strand with a rigid Southern conformation is not easily tolerated, which might help justify the lower affinity of ANA to form hybrids with RNA. These subtle structural differences were not observed in the hairpin system (1,2), but a study on single inserts in A- and B-form DNA duplexes concluded that ANA was restricted to the southeast pucker range, while 2′F-ANA could adopt a much broader range of conformations including North-Eastern conformations (12). Our findings are also consistent with previous evidence from circular dichroism, which showed that the short wavelength (∼210 nm) negative band associated with the A-form helical structure is slightly reduced in 2′F-ANA•RNA hybrids and further reduced in ANA•RNA hybrids (8).
Pseudohydrogen bonding and 2′X–nucleobase interactions
The second clear difference that emerges between the 2′F-ANA- and ANA-based hybrids is a bad steric interaction involving the β-2′-OH group in AR which contrasts with a favorable 2′F–H8(purine) pseudohydrogen bond in the case of FR. Evidence for this comes directly from HOESY NMR spectra, MD/TI interaction energies and complementary energy analysis, as well as the fact that the duplex geometry appears to be adjusted to optimize 2′F–H8 interactions (Figure 7, also see Supplementary Table S11 for a listing of all intra and interresidual 2′F–H6/H8 distances).
Figure 7.

Details of X2′–H8 interactions in FR and AR. Left, the structure of the FR duplex, showing distances between F2′ and aromatic H6/H8 protons. These distances correlate well with cross-peak intensity of the 19F – 1H HOESY spectra shown in Figure 3. Right, the same region of the AR duplex showing unfavorable steric interactions with 2′-OH.
Structural results reported here strongly suggest that optimal 2′F-H8 pseudohydrogen bonding is achieved at pyrimidine–purine steps, where the base-stacking geometry can adjust to optimize the interaction without incurring a steric penalty (Figure 7 and Supplementary Table S11). In purine-only 2′F-ANA sequences, this optimization may be harder to achieve, causing either reduced pseudohydrogen bonding or other structural problems, and ultimately leading to less stabilization as predicted (48). All these findings agree with the experimental observations (8,49) that the highest increases in binding affinity upon replacement of a DNA strand by 2′F-ANA tend to occur for mixed base sequences.
The existence of pseudohydrogen bonds involving 2′F-ANA is consistent with previous experimental data. For example, single 2′F-ANA residues in a DNA helix showed short inter-residue 2′F–C8 distances (<3 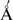 ) in an A-form helical environment (12). Interestingly, this was also for a TA step (5′-2′F-araT–dA-3′.) If these interactions were stabilizing, it would help explain the significant distortion observed in the local helical environment around the residues (12). Short 2′F-C6(pyrimidine) distances have also been observed in the crystal structure of a 2′F-ANA-modified B-form DNA sequence (2′F–C8 < 2.8 ) (48). Both inter- and intraresidual 2′F–H8/H6 distances are relatively short in our structure. However the geometry is more favorable for the interresidual 2′F-H8 in pyrimidine–purine steps, in which the C8-H8-F angle is higher than 145°. The NMR spectra of many 2′F-arabinonucleosides and oligonucleotides have often shown intraresidual 2′F–H8/H6 scalar coupling of 1–3 Hz (1,8,50,51). Empirical and theoretical data suggest that this is due to a ‘through-space’ electronic interaction (52,53), consistent with the pseudohydrogen bonding we propose. While still controversial, there is mounting evidence from crystal structures and binding affinities that fluorine-mediated pseudohydrogen bonding can be important in base pairing as well (54–56).
) in an A-form helical environment (12). Interestingly, this was also for a TA step (5′-2′F-araT–dA-3′.) If these interactions were stabilizing, it would help explain the significant distortion observed in the local helical environment around the residues (12). Short 2′F-C6(pyrimidine) distances have also been observed in the crystal structure of a 2′F-ANA-modified B-form DNA sequence (2′F–C8 < 2.8 ) (48). Both inter- and intraresidual 2′F–H8/H6 distances are relatively short in our structure. However the geometry is more favorable for the interresidual 2′F-H8 in pyrimidine–purine steps, in which the C8-H8-F angle is higher than 145°. The NMR spectra of many 2′F-arabinonucleosides and oligonucleotides have often shown intraresidual 2′F–H8/H6 scalar coupling of 1–3 Hz (1,8,50,51). Empirical and theoretical data suggest that this is due to a ‘through-space’ electronic interaction (52,53), consistent with the pseudohydrogen bonding we propose. While still controversial, there is mounting evidence from crystal structures and binding affinities that fluorine-mediated pseudohydrogen bonding can be important in base pairing as well (54–56).
In contrast to the 2′F-ANA situation, 2′-OH–H8(purine) interactions in AR are destabilizing by about 0.5 kcal/mol due to bad van der Waals contacts that are not compensated by favourable electrostatic contacts. This quite surprising difference in the behaviour of OH and F is justified by structural data which shows how the neighbouring phosphates impose conformational constraints to the 2′OH groups, preventing them from adopting a more favourable orientation for interacting with H8 or other acidic groups in their vicinities. Support for our claims on the importance of OH-mediated steric repulsion in ANA hybrids can be found in previous studies that found that a single ANA insert in a B-form DNA duplex showed steric hindrance between the 2′-OH and the Me5 and C6 groups of the neighboring thymine, and an associated computational study predicted that these steric effects would be more severe in the A-form (47). Thus while the concepts of 2′F–H8 mediated stabilization and 2′OH–C/H8 mediated destabilization are not totally unprecedented, we were surprised to find such strong evidence that they operate in the same sequence.
The preference for pseudohydrogen bonding at pyrimidine–purine steps can be confirmed by thermal stability studies of sequences containing identical nucleotide compositions and modifications but varying numbers of pyrimidine–purine steps. Indeed, preliminary results from our lab show a much higher degree of stabilization upon 2′F-ANA modification of an 11-mer sequence containing four TA steps than a sequence of identical base composition but without any TA steps (M. Yayahee, unpublished).
Flexibility and preorganization
Preorganization of oligonucleotide analogues into appropriate conformations leads to higher binding affinity, presumably by reducing the entropic penalty of duplex formation. Thus we chose to include an exploration of flexibility and preorganization in our study, through examining NMR relaxation data and molecular dynamics.
NMR-derived proton relaxation data indicates that the FR and AR duplexes have similar dynamics, which is supported by unrestricted MD simulations. Both ANA and 2′F-ANA strands have dynamics quite similar to the RNA strand, in contrast with the DR duplex in which the DNA strand is much more mobile (13,38).
The duplex rigidity of FR and AR appears to be similar; while AR is slightly more flexible by unrestricted MD simulations, this factor is probably not a major contributor to the stability difference. However, the ANA and 2′F-ANA strands might display a different preorganization for hybrid formation. Our studies suggest that the 2′F–H8 pseudohydrogen bond mentioned above in the case of 2′F-ANA would tend to preorganize the bases into the anti conformation, lowering the entropic penalty of duplex formation. In contrast, the unfavorable 2′-OH–base interactions in the case of ANA would tend to disfavor the glycosidic angles most suitable for duplex formation and thus increase the entropic penalty of duplex formation. Thus, besides stabilizing/destabilizing the duplexes once formed, the 2′-substituent–base interactions may contribute directly to the ease of duplex formation (Table 1 and Figure 7).
Hydration
Hydration plays a determining role in many of the properties of nucleic acids, including duplex stability (57,58). It has long been suspected that 2′F-ANA and ANA might differ in their hydration, contributing to their different binding affinities. Berger et al. observed an ordered water structure around the 2′-fluorines of FMAU residues incorporated into the Dickerson–Drew dodecamer. However, the F•water contacts in this crystal structure were relatively long and argued against strongly stabilizing hydrogen bonds to fluorine (48). More recently, Li et al. (12) observed that the groove regions around 2′F-ANA residues incorporated into DNA oligomers were dry, in contrast with ANA residues which were heavily hydrated.
By bringing together empirical and computational data, we conclude that hydration plays very little role in explaining the different thermal stabilities of 2′F-ANA- and ANA-based hybrids. The fluorine atoms of FR are not well hydrated as measured by comparisons of 19F NMR chemical shifts in D2O and H2O, while the 2′-OH of ANA remains relatively well hydrated in the AR duplex. However, the level of hydration in the duplex is not too different from that in the single strand, and accordingly the solvation free energies associated with hybrid formation are similar for the two hybrids.
The fact that hydration has little effect on the stability of these hybrids may help explain the contradictory results obtained in osmotic stressing and related experiments, since small changes in stability due to hydration can be masked by other effects, such as interactions with the osmolyte.
Ion uptake
As discussed above, melting studies in the presence of varying ion concentrations showed that the AR duplex had the lowest uptake of both monovalent and divalent counterions (Table 2 and Supplementary Figure S3). This would certainly be related to its lower stability, since counterion uptake compensates for the unfavorable increase in charge density associated with duplex formation. Thus, another difference between AR and FR is in their ion uptake: the latter appears to have a significantly higher number of counterions (especially magnesium) associated with the duplex relative to the single strands. Interestingly, however, DR and FR are very similar in their ion uptake behavior. Thus we might hypothesize that favorable ion uptake provides extra stability for FR over AR, especially in environments containing divalent cations, while the stability difference between FR and DR is achieved by other factors—mostly the increased rigidity of 2′F-ANA relative to DNA, preorganization of the single strands, and 2′-F–H8 pseudohydrogen bonding.
Conclusions
Several factors play a contributing role in the striking stability difference of DNA, ANA and 2′F-ANA-containing hybrid duplexes. The most significant differences appear to be on the level of structure, steric contacts and pseudohydrogen bonding.
Both ANA and 2′F-ANA-containing hybrids are structurally similar to DNA•RNA hybrid duplexes. The ANA strand is limited to more southern conformations, while the 2′F-ANA strand is somewhat closer to the east, a conformation which is more compatible with the fixed northern conformation of the RNA strand. This is reflected, among other features, in a narrower minor groove width for the ANA•RNA hybrid duplexes.
Interactions between the 2′-substituent and the nucleobase are crucial in determining the different stabilities of FR and AR. For FR, several independent pieces of evidence, both computational and empirical, point to a favorable 2′F–H8(purine) pseudohydrogen bond. On the other hand, the 2′–base interactions in the case of ANA destabilize the duplex due to unfavorable steric contacts.
Based on our structures and calculations, solvation is not expected to play a key role in explaining the differential stability of FR and AR. The FR duplex does feature a higher uptake of counterions, especially divalent counterions, than does AR, and this is expected to contribute to its greater stability in environments containing divalent cations.
Both ANA and 2′F-ANA-based hybrids are more rigid than their DNA-containing counterparts. In principle this should benefit them both on an entropic level, but in practice any benefit conferred upon ANA is overwhelmed by the unfavorable structural characteristics discussed above. Furthermore, interactions between the 2′-substituent and the base may help preorganize the single strand into an appropriate conformation for duplex formation in the case of 2′F-ANA, while disfavoring that conformation in the case of ANA.
The dramatic stability difference between these two very similar hybrids is therefore caused by the cumulative influence of multiple effects. Indeed, thermal stability can often be a surprisingly complex property (59–62). While it is difficult to rationally design an oligonucleotide analogue to optimize so many factors at once, keeping these factors in mind can be informative as new analogues are developed. This study also beautifully illustrates the surprising effect of small changes—by mutating the 2′-substituent of ANA from –OH to –F, interactions with the nucleobases not only stopped being destabilizing, but started making a significant contribution to duplex stability.
Coordinates
Atomic coordinates have been deposited in the Protein Data Bank (accession numbers 2KP3 and 2KP4). The complete assignment list has been deposited at the BMRB.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministerio de Ciencia e Innovación (grants CTQ2007-68014-C02-02 to CG and BIO2009-10964 to MO); Fundacion Marcelino Botin (grant to MO); Canadian Institutes for Health Research (grant to M.J.D.); Natural Sciences and Engineering Research Council of Canada (postgraduate scholarship to J.K.W.). Funding for open access charge: Canadian Institutes for Health Research.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We greatly acknowledge Dr Eva de Alba for her help running 19F NMR experiments.
REFERENCES
- 1.Trempe JF, Wilds CJ, Denisov AY, Pon RT, Damha MJ, Gehring K. NMR solution structure of an oligonucleotide hairpin with a 2′F-ANA/RNA stem: implications for RNase H specificity toward DNA/RNA hybrid duplexes. J. Am. Chem. Soc. 2001;123:4896–4903. doi: 10.1021/ja003859p. [DOI] [PubMed] [Google Scholar]
- 2.Denisov AY, Noronha AM, Wilds CJ, Trempe J.-F, Pon RT, Gehring K, Damha MJ. Solution structure of an arabinonucleic acid (ANA)/RNA duplex in a chimeric hairpin: comparison with 2′-fluoro-ANA/RNA and DNA/RNA hybrids. Nucleic Acids Res. 2001;29:4284–4293. doi: 10.1093/nar/29.21.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damha MJ, Wilds CJ, Noronha A, Brukner I, Borkow G, Arion D, Parniak MA. Hybrids of RNA and arabinonucleic acids (ANA and 2′F-ANA) are substrates of Ribonuclease H. J. Am. Chem. Soc. 1998;120:12976–12977. [Google Scholar]
- 4.Noy A, Perez A, Marquez M, Luque FJ, Orozco M. Structure, recognition properties, and flexibility of the DNA-RNA hybrid. J. Am. Chem. Soc. 2005;127:4910–4920. doi: 10.1021/ja043293v. [DOI] [PubMed] [Google Scholar]
- 5.Noy A, Luque FJ, Orozco M. Theoretical analysis of antisense duplexes: determinants of the RNase H susceptibility. J. Am. Chem. Soc. 2008;130:3486–3496. doi: 10.1021/ja076734u. [DOI] [PubMed] [Google Scholar]
- 6.Wilds CJ. Synthesis, Physicochemical and Biological Properties of Oligonucleotides Containing 2-fluoro-2-deoxy-β-D-arabinose. PhD Thesis, McGill University, 1999, Chapter 6. [Google Scholar]
- 7.Noronha AM, Wilds CJ, Lok C.-N, Viazovkina K, Arion D, Parniak MA, Damha MJ. Synthesis and biophysical properties of arabinonucleic acids (ANA): circular dichroic spectra, melting temperatures, and ribonuclease H susceptibility of ANA.RNA hybrid duplexes. Biochemistry. 2000;39:7050–7062. doi: 10.1021/bi000280v. [DOI] [PubMed] [Google Scholar]
- 8.Wilds CJ, Damha MJ. 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides and oligonucleotides (2′F-ANA): synthesis and physicochemical studies. Nucleic Acids Res. 2000;28:3625–3635. doi: 10.1093/nar/28.18.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watts JK, Damha MJ. 2′F-Arabinonucleic acids (2′F-ANA) – History, properties, and new frontiers. Can. J. Chem. 2008;86:641–656. [Google Scholar]
- 10.Topigen Pharmaceuticals moves novel inhaled PDE inhibitor, TPI 1100, into clinical development for COPD. Press release (22 December 2008) available at http://www.topigen.com. [Google Scholar]
- 11.Venkateswarlu D, Ferguson DM. Effects of C2′-substitution on arabinonucleic acid structure and conformation. J. Am. Chem. Soc. 1999;121:5609–5610. [Google Scholar]
- 12.Li F, Sarkhel S, Wilds CJ, Wawrzak Z, Prakash TP, Manoharan M, Egli M. 2′-fluoroarabino- and arabinonucleic acid show different conformations, resulting in deviating RNA affinities and processing of their heteroduplexes with RNA by RNase H. Biochemistry. 2006;45:4141–4152. doi: 10.1021/bi052322r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.González C, Stec W, Reynolds MA, James TL. Structure and dynamics of a DNA-RNA hybrid duplex with a chiral phosphorothioate moiety - NMR and molecular dynamics with conventional and time-averaged restraints. Biochemistry. 1995;34:4969–4982. doi: 10.1021/bi00015a008. [DOI] [PubMed] [Google Scholar]
- 14.Marky LA, Breslauer KJ. Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers. 1987;26:1601–1620. doi: 10.1002/bip.360260911. [DOI] [PubMed] [Google Scholar]
- 15.Kaur H, Arora A, Wengel J, Maiti S. Thermodynamic, counterion, and hydration effects for the incorporation of locked nucleic acid nucleotides into DNA duplexes. Biochemistry. 2006;45:7347–7355. doi: 10.1021/bi060307w. [DOI] [PubMed] [Google Scholar]
- 16.Rozners E, Moulder J. Hydration of short DNA, RNA and 2′-OMe oligonucleotides determined by osmotic stressing. Nucleic Acids Res. 2004;32:248–254. doi: 10.1093/nar/gkh175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 18.Sklenar V, Miyashiro H, Zon G, Miles HT, Bax A. Assignment of the 31P and 1H resonances in oligonucleotides by two-dimensional NMR spectroscopy. FEBS Lett. 1986;208:94–98. doi: 10.1016/0014-5793(86)81539-3. [DOI] [PubMed] [Google Scholar]
- 19.Yu C, Levy GC. Two-dimensional heteronuclear NOE (HOESY) experiments: investigation of dipolar interactions between heteronuclei and nearby protons. J. Am. Chem. Soc. 2002;106:6533–6537. [Google Scholar]
- 20.Goddard DT, Kneller G. SPARKY 3. University of California, San Francisco. [Google Scholar]
- 21.Borgias BA, James TL. MARDIGRAS, a procedure for matrix analysis of relaxation for discerning geometry of an aqueous structure. J. Magn. Reson. 1990;87:475–487. [Google Scholar]
- 22.Case DA, Pearlman DA, Caldwell JW, III T.EC, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, et al. San Francisco: University of California; 1997. AMBER 5. [Google Scholar]
- 23.Soliva R, Monaco V, Gómez-Pinto I, Meeuwenoord NJ, Marel GA, Boom JH, González C, Orozco M. Solution structure of a DNA duplex with a chiral alkyl phosphonate moiety. Nucleic Acids Res. 2001;29:2973–2985. doi: 10.1093/nar/29.14.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darden TE, York D, Pedersen L. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 25.Pérez A, Marchán I, Svozil D, Sponer J, Cheatham T, Laughton C, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheatham TE, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 27.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz K, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 28.Jorgensen WL, Chandrasekhar J, Madura JD. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 29.Shields GC, Laughton CA, Orozco M. Molecular dynamics simulations of the d(T.A.T) triple helix. J. Am. Chem. Soc. 1997;119:7463–7469. [Google Scholar]
- 30.Hess B, Kutzner C, Van der Spoel D, Lindhal E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 31.Miyamoto S, Kollman PA. SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comp Chem. 1992;13:952–962. [Google Scholar]
- 32.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 33.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 34.Berendsen HJC. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 35.Lavery R, Sklenar H. 1990. CURVES, helical analysis of irregular nucleic acids. 3.0. Laboratory of Theoretical Biochemistry CNRS, Paris. [Google Scholar]
- 36.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:29–32, 51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 37.Orozco M, Luque FJ. Theoretical methods for the description of the solvent effect in biomolecular systems. Chem. Rev. 2000;100:4187–4226. doi: 10.1021/cr990052a. [DOI] [PubMed] [Google Scholar]
- 38.González C, Stec W, Kobylanska A, Hogrefe RI, Reynolds M, James TL. Structural study of a DNA-RNA hybrid duplex with a chiral phosphorothioate moiety by NMR - extraction of distance and torsion angle constraints and imino proton-exchange rates. Biochemistry. 1994;33:11062–11072. doi: 10.1021/bi00203a002. [DOI] [PubMed] [Google Scholar]
- 39.Fenna CP, Wilkinson VJ, Arnold JR, Cosstick R, Fisher J. The effect of 2′-fluorine substitutions on DNA i-motif conformation and stability. Chem. Commun. 2008:3567–3569. doi: 10.1039/b804833a. [DOI] [PubMed] [Google Scholar]
- 40.Spink CH, Chaires JB. Effects of hydration, ion release, and excluded volume on the melting of triplex and duplex DNA. Biochemistry. 1999;38:496–508. doi: 10.1021/bi9820154. [DOI] [PubMed] [Google Scholar]
- 41.Cupane A, Vitrano E, Sanbiago PL, Madonia F, Palma MU. Thermal stability of Poly(A) and Poly(U) complexes in H2O and D2O - isotopic effects on critical temperatures and transition widths. Nucleic Acids Res. 1980;8:4283–4303. doi: 10.1093/nar/8.18.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McFail-Isom L, Sines CC, Williams LD. DNA structure: cations in charge? Curr. Opin. Struct. Biol. 1999;9:298–304. doi: 10.1016/S0959-440X(99)80040-2. [DOI] [PubMed] [Google Scholar]
- 43.Tereshko V, Minasov G, Egli M. A "hydrat-ion" spine in a B-DNA minor groove. J. Am. Chem. Soc. 1999;121:3590–3595. [Google Scholar]
- 44.Brady GP, Sharp KA. Decomposition of interaction free energies in proteins and other complex systems. J. Mol. Biol. 1995;254:77–85. doi: 10.1006/jmbi.1995.0600. [DOI] [PubMed] [Google Scholar]
- 45.Boresch S, Karplus M. The meaning of component analysis: decomposition of the free energy in terms of specific interactions. J. Mol. Biol. 1995;254:801–807. doi: 10.1006/jmbi.1995.0656. [DOI] [PubMed] [Google Scholar]
- 46.Mark AE, van Gunsteren WF. Decomposition of the free energy of a system in terms of specific interactions. Implications for theoretical and experimental studies. J. Mol. Biol. 1994;240:167–176. doi: 10.1006/jmbi.1994.1430. [DOI] [PubMed] [Google Scholar]
- 47.Gao YG, Van der Marel GA, Van Boom JH, Wang A.HJ. Molecular structure of a DNA decamer containing an anticancer nucleoside arabinosylcytosine: conformational perturbation by arabinosylcytosine in B-DNA. Biochemistry. 1991;30:9922–9931. doi: 10.1021/bi00105a016. [DOI] [PubMed] [Google Scholar]
- 48.Berger I, Tereshko V, Ikeda H, Marquez VE, Egli M. Crystal structures of B-DNA with incorporated 2′-deoxy-2′-fluoro-arabino-furanosyl thymines: implications of conformational preorganization for duplex stability. Nucleic Acids Res. 1998;26:2473–2480. doi: 10.1093/nar/26.10.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tennila T, Azhayeva E, Vepsalainen J, Laatikainen R, Azhayev A, Mikhailopulo IA. Oligonucleotides containing 9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-adenine and -guanine: synthesis, hybridization and antisense properties. Nucleosides Nucleotides Nucleic Acids. 2000;19:1861–1884. doi: 10.1080/15257770008045466. [DOI] [PubMed] [Google Scholar]
- 50.Wright JA, Taylor NF, Fox JJ. Nucleosides. LX. Fluorocarbohydrates. 22. Synthesis of 2-deoxy-2-fluoro-D-arabinose and 9-(2-deoxy-2-fluoro-α and β-D-arabinofuranosyl)adenines. J. Org. Chem. 1969;34:2632–2636. doi: 10.1021/jo01261a031. [DOI] [PubMed] [Google Scholar]
- 51.Barchi JJ, Jr, Marquez VE, Driscoll JS, Ford H, Jr, Mitsuya H, Shirasaka T, Aoki S, Kelley JA. Potential anti-AIDS drugs. Lipophilic, adenosine deaminase-activated prodrugs. J. Med. Chem. 1991;34:1647–1655. doi: 10.1021/jm00109a018. [DOI] [PubMed] [Google Scholar]
- 52.Herdewijn P, Van Aerschot A, Kerremans L. Synthesis of nucleosides fluorinated in the sugar moiety. The application of diethylaminosulfur trifluoride to the synthesis of fluorinated nucleosides. Nucleosides Nucleotides. 1989;8:65–96. [Google Scholar]
- 53.Bergstrom DE, Swartling DJ, Wisor A, Hoffmann MR. Evaluation of thymidine, dideoxythymidine and fluorine substituted deoxyribonucleoside geometry by the Mindo/3 technique: the effect of fluorine substitution on nucleoside geometry and biological activity. Nucleosides Nucleotides. 1991;10:693–697. [Google Scholar]
- 54.Pallan PS, Egli M. Pairing geometry of the hydrophobic thymine analogue 2,4-difluorotoluene in duplex DNA as analyzed by X-ray crystallography. J. Am. Chem. Soc. 2009;131:12548–12549. doi: 10.1021/ja905739j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parsch J, Engels JW. C-F … H-C hydrogen bonds in ribonucleic acids. J. Am. Chem. Soc. 2002;124:5664–5672. doi: 10.1021/ja012116g. [DOI] [PubMed] [Google Scholar]
- 56.Sun Z, McLaughlin LW. Probing the nature of three-centered hydrogen bonds in minor-groove ligand-DNA interactions: the contribution of fluorine hydrogen bonds to complex stability. J. Am. Chem. Soc. 2007;129:12531–12536. doi: 10.1021/ja073981p. [DOI] [PubMed] [Google Scholar]
- 57.Mrevlishvili GM, Carvalho A.P.S.MC, da Silva M.A.VR, Mdzinarashvili TD, Razmadze GZ, Tarielashvili TZ. The role of bound water on the energetics of DNA duplex melting. J. Therm. Anal. Calorim. 2001;66:133–144. [Google Scholar]
- 58.Egli M, Portmann S, Usman N. RNA hydration: a detailed look. Biochemistry. 1996;35:8489–8494. doi: 10.1021/bi9607214. [DOI] [PubMed] [Google Scholar]
- 59.Freier SM, Altmann KH. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Egli M, Minasov G, Tereshko V, Pallan PS, Teplova M, Inamati GB, Lesnik EA, Owens SR, Ross BS, Prakash TP, et al. Probing the influence of stereoelectronic effects on the biophysical properties of oligonucleotides: comprehensive analysis of the RNA affinity, nuclease resistance, and crystal structure of ten 2′-O-ribonucleic acid modifications. Biochemistry. 2005;44:9045–9057. doi: 10.1021/bi050574m. [DOI] [PubMed] [Google Scholar]
- 61.Kolarovic A, Schweizer E, Greene E, Gironda M, Pallan PS, Egli M, Rozners E. interplay of structure, hydration and thermal stability in formacetal modified oligonucleotides: RNA may tolerate nonionic modifications better than DNA. J. Am. Chem. Soc. 2009;131:14932–14937. doi: 10.1021/ja904926e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schlegel MK, Xie X, Zhang L, Meggers E. Insight into the high duplex stability of the simplified nucleic acid GNA. Angew. Chem. Int. Ed. 2009;48:960–963. doi: 10.1002/anie.200803472. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.