


Abstract
The mobility of restriction–modification (RM) gene complexes and their association with genome rearrangements is a subject of active investigation. Here we conducted systematic genome comparisons and genome context analysis on fully sequenced prokaryotic genomes to detect RM-linked genome rearrangements. RM genes were frequently found to be linked to mobility-related genes such as integrase and transposase homologs. They were flanked by direct and inverted repeats at a significantly high frequency. Insertion by long target duplication was observed for I, II, III and IV restriction types. We found several RM genes flanked by long inverted repeats, some of which had apparently inserted into a genome with a short target duplication. In some cases, only a portion of an apparently complete RM system was flanked by inverted repeats. We also found a unit composed of RM genes and an integrase homolog that integrated into a tRNA gene. An allelic substitution of a Type III system with a linked Type I and IV system pair, and allelic diversity in the putative target recognition domain of Type IIG systems were observed. This study revealed the possible mobility of all types of RM systems, and the diversity in their mobility-related organization.
INTRODUCTION
Restriction enzymes recognize and cut at specific DNA sequences, while their cognate modification enzymes methylate the same sequence to inhibit restriction enzyme cleavage. Restriction (R) and modification (M) enzyme genes are often tightly linked, forming a restriction–modification (RM) gene complex (1). When cells harboring an RM gene complex are invaded by foreign DNA, the R enzyme protects the cells by digesting the unmodified invading DNA, while the cellular DNA, which is protected by methylation from the M enzyme, is left intact. This benefit is the major reason RM systems are thought to be maintained in bacterial and archaeal genomes (2,3).
Four types of restriction systems (I–IV) are currently recognized (4). Type II R enzymes cleave DNA at definite positions within or near the recognition sequence (4,5). Fusion of R and M enzymes yields Type IIG (4,6). Type I systems consist of R and M genes, and sequence recognition (S) subunit genes, the products of which form multi-subunit enzymes for modification (SM) or restriction (SMR) (7). Type III systems consist of res and mod genes. The mod gene product has M activity on its own, while the complex of the two gene products has R enzyme activity (8). Type IV R enzymes, such as McrBC from Escherichia coli, cleave DNA near a methylated recognition sequence (9,10).
Some restriction systems are known to occasionally attack the host genome. If the RM gene complex is lost from a bacterial cell, the R and M enzymes gradually decrease in intracellular concentration as the cells grow and divide. Eventually, the M enzyme cannot methylate the chromosomal recognition sites sufficiently to protect against lethal attack by the remaining R enzyme molecules. This selfish post-segregational killing behavior forces host cells to maintain at least some Type II RM systems (11). Host cell killing also occurs with the Type IV enzyme McrBC when a particular DNA methylation system is introduced (10). Under some conditions of genome instability, Type I R enzymes attack the host chromosome at an arrested replication fork (12,13).
The mobility of RM genes has also been investigated. Phylogenetic trees of RM genes suggest horizontal transfer between distantly related prokaryotes (14–16). The average GC content and codon usage of RM genes often deviates from the rest of the genome (14,17–20). Genome context analysis has shown that some RM genes are on mobile elements such as plasmids and prophages (21–28), and some are linked to recombination-related genes such as integrases, invertases and transposases (29,30). RM systems and apparently solitary M genes flanked by insertion sequence (IS) elements have been observed (31–34). Genome comparisons have also shown that RM systems are involved in genome rearrangements such as insertion, deletion and transposition (14,17,35–37). Intragenomic comparisons of Helicobacter pylori demonstrated large inversion events next to RM genes (17). Allelic RM systems have also long been recognized. In E. coli, the hsd locus is occupied by either an EcoKI Type I system, an EcoB Type I system or other non-RM genes (38).
RM gene complexes are occasionally flanked by direct repeats (39,40). Genome context and genome comparison analysis led to the classification of the repeats into three groups: site-specific recombinations (Figure 1b), insertions with long target duplications (Figure 1c), and chance insertions between repeated sequences. The first class was observed for RM systems on prophages (21,23–28), or in the vicinity of integrase genes (30,41). We demonstrated the second class by genome comparison analysis revealing insertion of RM systems with long and variable target duplications, with no other mobile elements (37).
Figure 1.

Various modes of DNA recombination that result in target sequence duplication. (a) Insertion of a DNA transposon typically results in direct repeats of <10 bp, although the Mycoplasma transposon IS1630 forms long and variable target duplications of 19–26 bp. (b) Insertion by site-specific recombination. (c) Insertion with long and variable target duplication.
This study is the first report of a systematic, intraspecific genome comparison to explore the repertoire of genome rearrangements linked to RM genes within a given species. We also systematically analyzed RM gene linkage to flanking repeats. Our data strongly indicated putative mobility for all types of RM systems, and revealed organizational diversity related to mobility. Among the examples are novel, compact types of mobility units that are similar to DNA transposons, in which RM genes are flanked by long inverted repeats.
MATERIALS AND METHODS
Intraspecific pair-wise genome comparison
Sets of multiple complete genome sequences that were available for a single species were retrieved from NCBI (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov) on 1 April 2006, resulting in 760 pairs of syntenic regions that included RM genes in both or in one of the regions (Supplementary Table 2). The type, position and orientation of RM systems were obtained from REBASE (http://rebase.neb.com) (2). Sequence similarity between pairs of syntenic regions was visualized using the Artemis Comparison Tool (ACT, http://www.sanger.ac.uk/Software/ACT) (42) with default variables. Conserved domain was searched by NCBI Conserved Domain Search (CD-Search, http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). The 5-kb flanking sequences of RM systems were used for the classification.
Genomic variables
Relatedness between two intraspecific genome sequences was represented by two variables: identity and coverage (Supplementary Table 1). Identity was calculated by the equation:
where 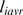 is the average nucleotide length of an ith orthologous region between two genomes as detected by the Comparative Genome Analysis Tool (CGAT) (43), and
is the average nucleotide length of an ith orthologous region between two genomes as detected by the Comparative Genome Analysis Tool (CGAT) (43), and 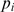 is the fraction of identity (percent identity/100) of the ith orthologous region. Coverage was calculated as the ratio of the sum length of the orthologous regions to the average whole-genome length.
is the fraction of identity (percent identity/100) of the ith orthologous region. Coverage was calculated as the ratio of the sum length of the orthologous regions to the average whole-genome length.
A phylogenetic tree was drawn using the neighbor-joining method of the MEGA4 program (44). Bootstrap values were from 1000 trials, and other variables were default. The GC content of third-codon nucleotides (GC3), and codon usage bias were calculated using CGAT (43). Codon usage bias of gene G against reference gene set R, B(G|R), was calculated using the equation (45):
where PG(a) is the ratio of amino acid a in a protein sequence of G, and fG(x,y,z) and fR(x,y,z) are the frequencies of the codon (x,y,z) in G and R, respectively. fG(a) and fR(a) are the frequencies of amino acid a in G and R, respectively. All genes in a genome were used as the reference gene set R, represented as B (G|all).
Detection of repeats flanking RM systems
The definition of RM genes from REBASE Genomes database (http://tools.neb.com/∼vincze/genomes/), accessed on 29 November 2008 was used. A line in the list of RM genes for a prokaryotic genome was assumed to represent an RM system. Systems including only M gene and lacking a gene labeled R were treated as solitary M systems and included in the analysis. RM systems that contained the same genes were removed manually, resulting in 4132 RM systems. One kb of flanking sequences was examined for each gene in an RM system. Within each RM system, all pairs of the left flanking sequence of a gene, and either the right flanking sequence of the same gene, or other genes to the right were compared. For example, if an RM system had three genes, six pairs of flanking sequences were analyzed (Figure 2). Totally 11 554 pairs were analyzed for RM systems in total. The longest bidirectional match detected by Blastn (46) in each pairs was assumed to represent repeat sequences if the match was longer than 20 bp, which was chosen over 30 bp as the threshold for intraspecific genome comparison analysis to increase sensitivity.
Figure 2.

Search design for finding repeats flanking RM genes. White boxes indicate an RM-related gene; thick black lines indicate 1 kb of flanking sequence. Each curved arrow indicates a pair of black line sequences. When three genes were included, six pairs result.
The same analysis was carried out for other genes using all genes in 50 randomly selected bacterial genomes as controls, for 119 865 total genes. One-kb sequences flanking n genes (n = 1, 2, 3, 4, 5, 6, 7, 8, 9) were analyzed, resulting in 119 865 × 9 = 1 078 785 total pairs.
For RM system analysis, the numbers of the pair that flanked different number of genes were different. For example, the number of the pairs that flank one gene was 7049, while the number of the pairs that flank two genes was 2917. For control analysis, the numbers of pairs that flank different number of genes were the same. To compare results, the difference was corrected by calculating a weighted average for the control pairs to adopt the ratio of pairs in the RM systems analysis, using the following equation:
where Rw is the weighted average, rncon is the number of pairs for which the repeat sequences flanking n genes were detected in control genome analysis, Cncon is the total number of pairs flanking n genes in the control genome analysis, and CnRM is the total number of pairs flanking n genes in the RM system analysis. Nucleotide sequence alignment was carried out by ClustalW (47).
Genome sequences that corresponded to an ‘empty site’ allele of an RM system were searched using Blastn (46) against the nucleotide sequence database (nr, prokaryote, NCBI), with 1-kb flanking regions and the RM system region as queries. A genome sequence with >500 bp similarity in both flanking sequences, but with no similarity in the RM system, was selected as a subject sequence for genome comparison. Genome comparison results were visualized by ACT, and classified manually.
RESULTS
Intraspecific genome comparison of RM loci
We performed a comprehensive search for RM gene-linked genome rearrangements by comparing each RM locus to the syntenic regions of all other available complete genomes within the same species. Results of the pair-wise comparisons were classified according to nucleotide sequence similarity in the RM regions and 5 kb of flanking region (Table 1). Half of the pairs did not even have partial sequence similarity in the RM regions (Table 1), suggesting frequent insertions or deletions of RM genes in the history of that species. Pairs with no similarity in the RM region, but with similarity in the flanking sequences were classified into substitutions, or insertion/deletions (indel), based on the length of the unaligned region. Investigation of these cases allowed us to identify three types of potential rearrangements: (i) insertion with a long target duplication; (ii) substitution by other RM genes; and (iii) substitution of the target recognition domain in a Type IIG RM gene.
Table 1.
RM locus pairs classification
| Classification | RM loci pair |
|---|---|
| Homology detected in entire RM regions | |
| A. flanking 5-kb region ≥50% aligned | 244 |
| B. flanking 5-kb region <50% aligned | 24 |
| Homology partially detected in RM regions | |
| C. flanking 5-kb region ≥50% aligned | 99 |
| D. flanking 5-kb region <50% aligned | 18 |
| No homology detected in RM region | |
| flanking 5-kb region ≥50% aligned | |
| E. Substitutiona | 116 |
| F. Indela | |
| RM insertion with long target duplication | 9 |
| others | 149 |
| G. flanking 5-kb region <50% aligned | 101 |
| Total | 760 |
aCriteria for classifying substitution and indel is the length of the subject genome region which corresponds to the RM region in query genome. If it is longer than 1 kb, the case was classified as substitution. If shorter than 1 kb case, then classified as indel.
Insertion of RM with a long target duplication
We searched for cases where RM systems were inserted into the genome with long and variable target duplications, but with no other mobile elements (37). Cases classified as indels in Table 1 were assumed to be insertions with long target duplications if they satisfied the following criteria: (i) inclusion of both R gene and M gene homologs in the inserted sequence; (ii) inserted sequence length of less than 20 kb to exclude large mobile elements such as prophages (48); and (iii) target duplication length longer than 30 bp to exclude typical repeats formed by site-specific recombination events (49). Nine cases were found from H. pylori, Burkholderia pseudomallei, Haemophilus influenzae, Thermus thermophilus, Vibrio vulnificus and Xylella fastidiosa. Three cases in H. pylori were previously reported (37), and a case in B. pseudomallei was reported as a Type I RM on a prophage annotated as a genomic island 5 (50). The other cases are analyzed in detail here, with the length and identity of the repeat sequences summarized in Table 2.
Table 2.
Insertion with long target duplication
| Symbols (see Figure 3) | Species | Strain with/without insert | Inserted RM genes | Identity/repeat (bp/bp) |
||
|---|---|---|---|---|---|---|
| upstream- target | downstream- target | upstream- downstream | ||||
| a | Haemophilus influenzae | 86-028NP/Rd KW20 | NTHI0188 (I, M), NTHI0192 (I, S), NTHI0193 (I, R) | 45/46 | 45/46 | 44/46 |
| b | Haemophilus influenzae | 86-028NP/Rd KW20 | NTHI1460 (II, M), NTHI1459 (II, R) | 43/46 | 45/46 | 44/46 |
| c | Thermus thermophilus | HB27/HB8 | TTC1877 (IIGS, RM), TTC1880 (II, M) | 47/47 | 47/47 | 47/47 |
| d | Xylella fastidiosa | Temecula I/9a5c | PD1608 (II, R), PD1607 (II, M) | 45/45 | 45/45 | 45/45 |
| e | Vibrio vulnificus | CMCP6/YJ016 | VV1_2037 (I, R), VV1_2031 (I, M), VV1_2030 (I, S) | 49/49 | 49/49 | 49/49 |
If an inserted region contained only RM genes, it was classified as an RM insertion with a long and variable target duplication, as described previously (37). However, if the inserted region included an integrase gene homolog, and the flanking repeats had sequence similarity to a tRNA sequence, it might be the product of site-specific recombination, since tRNA sequences are known to be an integration target for bacteriophages (51–64) and integrative plasmids (65–68). Integration often involves a long sequence, such as the region from the anticodon loop through 3′ end (51,60,61,63–68), or the entire tRNA gene sequence (55,57), both of which are longer than our 30-bp threshold. Therefore, we examined each polymorphism in detail.
Two cases of RM insertion with a long target duplication were detected in a comparison of two H. influenzae genomes (Figure 3a and b). One case (Figure 3a1) showed an insertion of Type I M, S and R genes interrupted by a transcriptional regulator homolog and an ATP binding protein, with a duplication of a 46-bp long sequence that did not occur elsewhere in either genome. The repeated sequences showed high identity (Table 2). The GC content of the inserted region was 41%, slightly higher than the genome average of 38%. The other case (Figure 3b1) was a Type II RM system insertion with duplication of a 46-bp sequence. This sequence differed from the above repeat, and was also unique in the genome. The repeated sequences showed high identity (Table 2), and the GC content of the insert (32%) was much lower than the genome average (38%).
Figure 3.


RM systems inserted with long target duplications. White triangles indicate a repeated sequence. (a) Comparison within H. influenzae. The 46-bp repeat sequence in Rd KW20 overlaps with 15 bp at the 3′ end of the HI0105 gene. (b) Comparison within H. influenzae. The 46-bp repeat sequence in Rd KW20 overlaps with 2 bp at the 3′ end of HI1589 gene. (c) Comparison within T. thermophilus. Underlined sequences represent the entire tRNA coding region of the query genome in (c2), (d2) and (e2). (d) Comparison within V. vulnificus. The sequence in (d2) corresponds to the strand complementary to tRNA. (e) Comparison within X. fastidiosa. In the original annotation of X. fastidiosa Temecula 1, the C-terminus of an integrase family gene (PD1606) overlapped with 144 bp of the C-terminus of the M gene homolog (PD1607). No overlap of the two genes occurs in the annotation of the same sequence in X. fastidiosa M23, shown here.
Cases of site-specific recombination were observed in T. thermophilus, V. vulnificus and X. fastidiosa (Figure 3c–e, Table 2). The repeat sequences showed similarity to the 3′ terminus of a tRNA sequence (Figure 3c2, d2 and e2), and all had a gene in the insert with strong or weak sequence similarity to an integrase, which likely mediated the site-specific recombination.
Figure 3c shows a tyrosine-type phage integrase homolog next to the repeat sequence. A Type IIG RM gene and a Type II M gene are present in the insert, as well as a transposase homolog and genes for hypothetical proteins. Figure 3d shows another tyrosine-type integrase homolog adjacent to the repeat sequence. The insert carries Type I RMS genes, as well as multiple genes for DNA-binding proteins, a virulence-related gene (HipA-like protein), and a multidrug efflux pump gene involved in drug resistance. These two inserts may be considered genomic islands.
In Figure 3e, the insert in the X. fastidiosa Temecula 1 genome contains only Type II R and M gene homologs, and an integrase gene homolog. Perfect sequence identity between the repeats suggests that the integration is a relatively recent event. The last gene product shows very weak sequence similarity to an integrase family protein (e-value of 7e-4 in blastp) (69), so we could not determine if this gene in this putative mobile unit has decayed or has specialized.
Allelic RM systems of different types
Substitution-type allelic RM systems were found in Campylobacter jejuni, E. coli, X. campestris, Rhodopseudomonas palustris and T. thermophilus. The C. jejuni case was reported as a substitution in a region including an S subunit gene from a Type I RM system (70), and the E. coli example was also reported (38). The remaining cases are presented here.
Type III RM alleles at a locus in X. campestris pv. campestris str. ATCC33913 were substituted with Type I RM genes in two genomes of this species (Figure 4). When the same locus was analyzed in other Xanthomonas species genomes, a deletion was found in X. axonopodis pv. citri str. 306 that left only 125 bp of a short open reading frame (ORF), and a substitution of non-homologous Type I RM genes was found in X. oryzae KACC10331 and X. oryzae MAFF311018. These two genomes showed evidence of insertion of ISXo1 into the S subunit gene in the Type I RM genes.
Figure 4.

Allelic RM systems in a Xanthomonas locus. Homologous regions are indicated in gray.
Homologs for all RM genes at this locus were found in distantly related bacteria, suggesting the possibility of horizontal transfer (Supplementary Figure 4). The Type III system homolog genes in X. campestris ATCC 33913 were found in Bordetella pertussis Tohama I (Supplementary Figure 1). The two genera are distantly related according to the phylogenetic tree (Supplementary Figure 4), and the GC3 and codon usage of the homologs were different from the majority of genes in both species, suggesting different origins for these homologs. The Type I and Type IV system homolog genes in X. campestris 8004 were also found in X. campestris pv. vesicatoria 85–10, Methylobacillus flagellatus KT, and Alkalimnicola ehrlichei HLNE-1 (Supplementary Figure 2), and Type IV genes are frequently observed at the vicinity of Type I RM genes (10). These genera are phylogenetically very distant from each other (Supplementary Figure 4). The GC3 and codon usage of these homologs are different from the majority of X. campestris and A. ehrlichei genes, but most of the homologs in M. flagellatus did not show much bias, suggesting that M. flagellatus is the origin of these Type I and Type IV systems. Homologs of the Type I and Type IV systems in X. oryzae KACC 10331 were found in Nitrosomonas eutropha C71 and Methylococcus capsulatus Bath (Supplementary Figure 3), which are phylogenetically distant (Supplementary Figure 4). The bias in GC3 and the codon usage of these homologs suggests the possibility that N. eutropha C71 recently acquired the Type I RM genes.
Type II RM genes in T. thermophilus HB8 were substituted with a different Type II M gene in T. thermophilus HB27 (Figure 5a). These M genes were 68% identical in amino acid sequence, although the neighboring R gene was completely deleted in the latter. The R and M genes of the former genome showed biased codon use and GC content (Figure 5a2), suggesting horizontal transfer. The M gene of the HB27 genome did not show bias, which may indicate amelioration of a horizontally transferred gene after decay (71).
Figure 5.

Allelic diversity in Thermus and Rhodopseudomonas. (a) Allelic Type II systems in T. thermophilus HB8 and HB27. (a1) Alignment. Gray indicates sequence similarity. (a2) Codon usage and GC contents of the third nucleotides of codons (GC3) of relevant RM genes and all HB8 genes. (a3) Codon usage and GC3 of the relevant M gene and all HB27 genes. (b) Allelic Type II M gene and Type IIG RM genes in Rhodopseudomonas palustris HaA2 and CGA009. (b1) Alignment. (b2) Codon usage and GC3 of HaA2 genes. (b3) Codon usage and GC3 of genes of CGA009. Black dots indicate another RM gene.
A Type II M gene in R. palustris HaA2 was substituted with a Type IIG gene in R. palustris CGA009 (Figure 5b), and no sequence similarity was observed between the two genes. Codon usage and GC3 of these genes were biased from the majority of genes in the genome (Figure 5b2 and b3), suggesting separate horizontal transfer of these genes to form alleles of a locus.
Allelic diversity in the target recognition domains of Type IIG RM genes
Sequence similarity and diversity in the C-terminal region of a Type IIG enzyme compared to a Type IS subunit have been reported (72), and relationships of recognition sequences and the region were confirmed previously by in vitro analysis (73).
Allelic diversity in the putative sequence recognition domain of a Type IIG RM protein was found in six cases, by investigating RM loci that were classified as partially matched (Table 1). Omitting cases of frameshift mutation and cases in which the same extent of diversity covered the entire RM gene region and both flanking regions, left two cases to analyze in detail.
In Campylobacter, both sides of the Type IIG homolog showed sequence similarity in all strains, but divergence at the nucleotide sequence level was observed in the C-terminus of the Type IIG homolog (Figure 6a). Amino acid sequences of the genes aligned completely except for the two variable regions at the C-terminus (Supplementary Figure 5). An NCBI Conserved Domain Search (CD-Search) (74) showed that CJE1195, Type IIG enzyme of C. jejuni RM 1221, had modification subunit motif for a Type I RM (COG0286, e-value 4e-28), and the sequence recognition motif for a subunit of Type I RM systems (COG0732, e-value 2e-3). The regions of the sequence recognition domain matched the diverged region between the homologs. In the two unaligned regions, repeats of 20–40 amino acids were observed, which also supported the similarity to the Type I RM sequence recognition subunit. To our knowledge, this is the first reported example of allelic diversity in the target recognition domain of a Type IIG enzyme, in closely related genomes.
Figure 6.

Allelic diversity in the target recognition domain of Type IIG proteins. Homologous regions are in gray. (a) Alleles of Type IIG RM gene at a C. jejuni locus. (b) Alleles of Type II M and Type IIG RM genes at a S. thermophilus locus.
Type IIG genes (Sth1066ORF1376P) in S. thermophilus CNRZ1066, and (Sth18311ORF1376P) in S. thermophilus LMG18311 were compared and found to have unmatched regions at the C-terminus (Figure 6b).
Systematic detection of RM systems flanked by repeats and empty-site genome sequences
In the second part of this study, we systematically searched for repeat sequences flanking RM genes in completely sequenced genomes. Specifically, we wanted to determine the generality of RM system insertions with long and variable target duplications (37). We also wished to examine RM systems in a novel context that might suggest a mechanism for their insertion.
One kilobase pair of flanking sequence was analyzed for 4132 RM systems. The frequency of RM systems with repeat sequences longer than 20 bp was compared to the frequency for other genes (see ‘Materials and methods’ section). Both direct and inverted repeat sequences were observed at significantly higher frequencies in the flanking sequences of RM systems (Figure 7). The longest repeat sequence was chosen from each RM systems and used for further analysis.
Figure 7.

Frequency of genes flanked by (a) direct or inverted repeats, (b) direct repeats and (c) inverted repeats. The vertical axis indicates percentage of the 11 554 compared RM-system-flanking sequence pairs. See ‘Materials and Methods’ section for control gene calculations. Black and white bars represent frequencies of flanking repeats and control genes, respectively. White circles indicate the ratio of RM systems to control genes for repeat frequency.
Some cases appear to have been caused by insertion of repeat sequences such as ISs, independently of the action of RM genes or integration by site-specific recombination of an RM-carrying phage or other mobile element. Because we were primarily interested in RM-mediated rearrangements that did not involve other mobile elements, we excluded these cases based on mobility-related gene annotation, and genomic copy number of the repeats (Figure 8a).
Figure 8.

Screening and classification of RM rearrangements. (a) Screening procedure for RM systems flanked by repeats. The number of RM systems selected is boxed. See text for details. (b) Classification in the genome comparison analysis. (b1) Insertion with long target duplication or integration by site-specific recombination. Repeated sequences align with the sequence in the other genome. (b2) Substitution. The repeated sequences do not align with the other genome. (b3) Tranposon-like structure. The outer, shorter, and direct repeats (triangles) align with the other genome, but the inner, longer and inverted repeats (arrows) do not.
In 57 out of 179 cases, five protein-coding sequences that flanked the RM system, or that were within the RM system, included genes annotated as mobile elements such as transposons, integrases, resolvases, invertases, topoisomerases and phage-related sequences. Of these, 30 out of 57 included a gene annotated as a transposon, and 25 out of 57 included a gene annotated as an integrase. After omitting these, 122 cases remained for further analysis.
Each repeat sequence was analyzed by Blastn against the entire genome to find sequences that had a match longer than 90% of the length of the query repeat, and were assumed to be copies. Totally 24 out of 122 had more than 10 copies, in addition to the two flanking RM genes, and were excluded.
An empty site was searched in the other genome sequences for 98 out of 122 cases. More specifically, RM systems and 1 kb of flanking sequence on both sides were used as queries for Blastn homology searches against all sequenced genomes. An empty-site genome sequence, lacking the RM genes but with sequence similarity on both sides, was found for 29 out of 98 cases (Table 3), which were used for genome comparison. Although the pool before selection included archaea, no archaeal RM systems survived the selection.
Table 3.
RM systems flanked by repeat sequences aligned with subject genome sequence
| Query species | Sequene ID | class or phylum | Compared sequences | Sequence ID | Homology group | Left- right | Left- subject | Right- subject | Direction | RM type |
|---|---|---|---|---|---|---|---|---|---|---|
| (a) Insertion with long target duplication | ||||||||||
| Helicobacter pylori J99 | NC_000921 | ε-proteobacteria | Helicobacter pylori HPAG1 | NC_008086 | Figure S7(a) | 417/423 | 345/370 | 343/370 | D | Type IIP |
| Burkholderia sp. 383 chromosome 1 | NC_007510 | β-proteobacteria | Burkholderia cenocepacia J2315chromosome 1 | NC_011000 | Figure S7(b) | 384/412 | 233/263 | 234/270 | D | Type IV |
| Helicobacter pylori Shi470 | NC_010698 | ε-proteobacteria | Helicobacter pylori 26695 | NC_000915 | Figure S7(c) | 388/397 | 372/397 | 371/397 | D | Type IIP |
| Helicobacter pylori HPAG1 | NC_008086 | ε-proteobacteria | Helicobacter pylori G27 | NC_011333 | Figure S7(c) | 368/393 | 370/393 | 361/393 | D | Type IIP |
| Helicobacter pylori Shi470 | NC_010698 | ε-proteobacteria | Helicobacter acinonychis str. Sheeba | NC_008229 | Figure S7(d) | 224/238 | 140/153 | 201/223 | D | Type IIP |
| Campylobacter jejuni subsp. jejuni NCTC 11828 | NC_009839 | ε-proteobacteria | Campylobacter jejuni RM1221 | NC_003912 | Figure S7(e) | 195/208 | 195/208 | 208/208 | D | Type III |
| Campylobacter jejuni subsp. doylei 269.97 | NC_009707 | ε-proteobacteria | Campylobacter jejunisubsp. jejuni 81-176 | NC_008787 | Figure S7(e) | 106/110 | 105/107 | 98/107 | D | Type III |
| Neisseria gonorrhoeae FA 1090 | NC_002946 | β-proteobacteria | Neisseria cinerea tex gene | AM886294 | Figure S7(f) | 98/104 | 74/76 | 70/76 | D | Type IIS |
| Neisseria gonorrhoeae NCCP11945 | NC_011035 | β-proteobacteria | Neisseria cinerea tex gene | AM886294 | Figure S7(f) | 99/104 | 70/76 | 75/76 | D | Type IIS |
| Helicobacter pylori HPAG1 | NC_008086 | ε-proteobacteria | Helicobacter acinonychis str.Sheeba | NC_008229 | Figure S7(d) | 98/104 | 98/103 | 94/103 | D | Type IIP |
| Helicobacter pylori J99 | NC_000921 | ε-proteobacteria | Helicobacter acinonychis str.Sheeba | NC_008229 | Figure S7(d) | 99/103 | 84/90 | 82/90 | D | Type IIP |
| Helicobacter pylori 26695 | NC_000915 | ε-proteobacteria | Helicobacter acinonychis str.Sheeba | NC_008229 | Figure S7(d) | 100/103 | 98/103 | 95/103 | D | Type IIP |
| Helicobacter pylori 26695 | NC_000915 | ε-proteobacteria | Helicobacter pylori HPAG1 | NC_008086 | Figure S7(g) | 83/96 | 67/73 | 55/63 | D | Type IIP |
| Helicobacter pylori J99 | NC_000921 | ε-proteobacteria | Helicobacter pylori P12 | NC_011498 | Figure S7(g) | 89/95 | 81/93 | 82/93 | D | Type IIP |
| Microcystis aeruginosa NIES-843 | NC_010296 | cyanobacteria | Microcystis aeruginosa PCC 7806 | AM778953 | Figure S7(h) | 88/88 | 88/88 | 88/88 | D | Type II |
| Helicobacter pylori Shi470 | NC_010698 | ε-proteobacteria | Helicobacter pylori P12 | NC_011498 | Figure S7(i) | 59/64 | 60/62 | 57/62 | D | Type IIS,P |
| Acidovorax sp. JS42 | NC_008782 | β-proteobacteria | Diaphorobacter sp. TPSY | NC_011992 | Figure S7(j) | 57/64 | 36/38 | 37/41 | D | Type IIG |
| Helicobacter pylori Shi470 | NC_010698 | ε-proteobacteria | Helicobacter pylori HPAG1 | NC_008086 | Figure S7(g) | 48/51 | 24/25 | 19/20 | D | Type IIP |
| Helicobacter pylori 26695 | NC_000915 | ε-proteobacteria | Helicobacter pylori G27 | NC_011333 | Figure S7(k) | 41/41 | 37/37 | 37/37 | D | Type IIS |
| Haemophilus influenzae 86-028NP | NC_007146 | γ-proteobacteria | Haemophilus influenzae Rd KW20 | NC_000907 | Figure S7(l) | 38/39 | 38/39 | 39/39 | D | Type I |
| Haemophilus influenzae PittGG | NC_009567 | γ-proteobacteria | Haemophilus influenzae Rd KW20 | NC_000907 | Figure S7(l) | 38/39 | 38/39 | 39/39 | D | Type I |
| Clavibacter michiganensis subsp. michiganensis | NC_009480 | actinobacter | Clavibacter michiganensis subsp. sepedonicus | NC_010407 | Figure S7(m) | 29/29 | 23/25 | 23/25 | D | Type IIP |
| Haemophilus influenzae PittEE | NC_009566 | γ-proteobacteria | Haemophilus influenzae Rd KW20 | NC_000907 | Figure S7(l) | 24/25 | 25/25 | 24/25 | D | Type I |
| (b) Substitution | ||||||||||
| Desulfovibrio vulgaris subsp. vulgaris DP4 | NC_008751 | δ-proteobacteria | Desulfovibrio vulgarissubsp. Vulgaris str. Hildenborough | NC_002937 | Figure S6(a) | 37/43 | – | – | D | Type I |
| Neisseria meningitidis Z2491 Serotype A | NC_003116 | β-proteobacteria | Neisseria gonorrhoeae NCCP11945 | NC_011035 | Figure S6(b) | 23/24 | – | – | D | Type IIG |
| Helicobacter pylori J99 | NC_000921 | ε-proteobacteria | Helicobacter pylori Shi470 | NC_010698 | Figure S6(c) | 20/22 | – | – | D | Type IIP |
| (c) Transposon like RM system | ||||||||||
| Xanthomonas oryzae pv. oryzae KACC10331 | NC_006834 | γ-proteobacteria | Xanthomonas oryzaepv. oryzae PX099A | NC_010717 | Figure 9a | 60/65 | – | – | I | Type IIP |
| Xanthomonas oryzae pv. oryzae MAFF 311018 | NC_007705 | γ-proteobacteria | Xanthomonas oryzaepv. oryzae PX099A | NC_010717 | Figure 9a | 60/65 | – | – | I | Type IIP |
| Neisseria gonorrhoeae NCCP11945 | NC_011035 | β-proteobacteria | Neisseria meningitidis MC58 | NC_003112 | Figure 9b | 26/26 | – | – | I | Type IIG |
Genome comparison for RM system insertions with repeats
In the 29 cases described above, the RM region was compared to the subject genome to detect RM-related rearrangements. Cases were classified into (Figure 8b1–b3): (i) insertion with long target duplication; (ii) substitution; and (iii) transposon-like structure where RM genes are flanked by inverted repeats.
Insertion with long target duplication
Evidence of an RM system insertion with a long target duplication has been reported only for H. pylori (epsilon-proteobacteria), from a pairwise genome comparison (37). We found two other cases in H. influenzae (see above). We found more prominent examples of insertion with long target duplications in Campylobacter (ε-proteobacteria), Microcystis (cyanobacteria), Neisseria (β-proteobacteria), Acidovorax (β-proteobacteria), Haemophilus (γ-proteobacteria), Burkholderia (β-proteobacteria) and Clavibacter (actinobacter, gram-positive) (Table 3). With the exception of Burkholderia, Microcystis, Acidovorax and Clavibacter, most occur in naturally competent species (75).
Examples in all types of RM systems, Types I, II, III and IV, were found (Figure 9, Table 3 and Supplementary Figure 7). An mcrA gene (Type IV) appeared to have inserted without other genes through this mechanism (Figure 9b). The discovered mcrA gene is relatively short, and has sequence similarity to only the C-terminal half of mcrA gene homologs in other bacteria. However, CD search (74) detected an HNH nuclease domain (cd00085) that is common in other mcrA homologs, suggesting that this gene may be active. Orphan M was also found to be inserted by this mechanism (Figure 9c and Supplementary Figure 7h).
Figure 9.

RM systems flanked by direct repeats in one genome and with a corresponding empty site in another genome. White triangles indicate repeat sequences. (a)–(c) Subject genomes with an empty site are not depicted. (d) Insertion of partial M and R genes. Positions of motifs in M genes are depicted in squares.
In many cases, the repeat sequences spanned the translation start or stop site, which, unlike insertion into the coding region, may leave the target gene intact and confer a selective advantage. Insertion of a Type II RM into an operon-like structure was also observed (Supplementary Figure 7m), consistent with a previous example (76).
Several RM systems flanked by repeats have been detected in H. pylori. Two cases were previously reported (Supplementary Figure 7a and k) (48), and in four novel cases, the targets were a Type IIG RM gene (Supplementary Figure 7d), a Type II M gene (Figure 9d and Supplementary Figure 7g), a Type III M gene (Supplementary Figure 7i) and a hypothetical protein gene (Supplementary Figure 7c). In the third case, the M gene in the query genome was frameshifted, while its homolog in the subject genome was not. This suggested that the insertion led to decay of the target gene by frameshift mutation and gene fusion.
The second example (Figure 9d and Supplementary Figure 7g) may be a case of generation of a novel M gene by gene fusion, because the repeated sequence was within the Type II M gene. Insertion with duplication of this sequence was likely the initial event. The M gene in the subject (target) genome was short (480 bp), and carried only motifs I through VIII, in order, and lacked a target recognition domain and motifs IX and X. The insertion apparently fused this partial M gene to a target recognition domain and motifs IX and X, creating a typical m5C methyltransferase (77). In strain J99, this fusion is active (78). The short N-terminal M gene may have been generated by a rearrangement event.
Substitution
Several cases in which the subject genome did not align, or only partially aligned with the repeated sequence were observed (Figure 8b). These cases were observed in Desulfovibrio (δ-proteobacteria), Neisseria (β-proteobacteria) and Helicobacter (ε-proteobacteria) (Supplementary Figure 6).
In Desulfovibrio, the Type I RM system flanked by repeats was substituted in the subject genome by a prophage with unrelated Type II M and RM genes (Supplementary Figure 6a). The prophage was flanked by 45-bp attL/R sequences, which align with the tRNA-Gly sequence.
In Neisseria, the Type IIG RM gene flanked by repeats was substituted by a transposase homolog (Supplementary Figure 6b), whose homologs, annotated as IS1016, were frequent in both genomes, with eight in the query genome and 16 in the subject genome. Substitution of this RM might have occurred by the action of this transposase.
In Helicobacter, a Type II RM system flanked by direct repeats was substituted by a gene for a hypothetical protein with a transmembrane domain (Supplementary Figure 6c). Only one of the two repeat sequences of the query RM system aligned with the subject genome. Because we could not find a genome with a clean empty site, we could not determine if the Type II RM system inserted into a genome with duplication of the 22-bp target sequence.
Transposon-like structures with RM genes flanked by inverted repeats
Terminal inverted repeats are a feature of many DNA transposons (79). Of the 98 RM systems remaining after removal of linked mobile genes and high-copy number repeats, 24 cases contained inverted repeats (Supplementary Table 3b and Figure 10). These were found in all RM types and orphan M (Figure 10a–f). In some cases, one or more component genes, but not the entire RM system, were flanked by inverted repeats. For example, in one Type II system of M-M-R, one of the two M genes was flanked by inverted repeats (Figure 10i), possibly representing an intermediate status in replacement of M partner by an R gene. In a Type I RM system, the inverted repeats are embedded in two S subunit genes flanking R and M subunit genes (Figure 10g). In Ureaplasma species, inverted repeats within the S genes flank M gene and another S gene of an apparent Type I system, composed of one R, one M and three S genes (Figure 10h).
Figure 10.

RM system components flanked by inverted repeats. Black or white arrows indicate inverted repeats.
A genome with an empty site was found for comparison among these examples, which clearly revealed their insertion points. In X. oryzae (Figure 11a), the inverted repeat sequences had 60/65 sequence identity, and an incomplete match is a feature of the terminal inverted repeats of many DNA transposons (80). The repeats did not align with the subject genome sequence, and were not found elsewhere in the query genome, and therefore appear to be unique to the inserted unit (Figure 11a). The entire RM system unit with the terminal inverted repeats was flanked by 8-bp direct repeats, which perfectly matched the 8-bp sequence in the subject genome empty site. This strongly suggested that insertion took place with 8-bp target site duplication. The same relationship was detected for an RM system homolog in another X. oryzae strain (Table 3, panel c). In addition, the 8-bp target sequence is flanked by 5′CTGC and 5′CAG, which are contained in the recognition sequence, 5′CTGCAG (81). The significance, if any, of such target site organization in the life cycle of this element remains unclear.
Figure 11.

Transposon-like structure of RM systems flanked by repeat sequences. Triangles and arrows represent different sets of repeated sequences. (a) Type II RM genes in X. oryzae pv. oryzae KAC10331 are flanked by 65-bp inverted repeats (aligned below). The resulting unit is further flanked by 8-bp direct repeats (underlined), which are identical to the 8-bp sequence at the empty locus in X. oryzae pv. oryzae PX099A. The short direct repeat sequences are flanked by part of the predicted recognition sequence of the RM system (boxed) in the other genome. (b) Type II genes in N. gonorrhoeae NCCP 11 945 are flanked by 26-bp inverted repeats (aligned below). The resulting unit is further flanked by 8-bp direct repeats, which are identical to the 8-bp sequence at the empty locus in N. meningitides MC58.
In N. gonorrhoeae (Figure 11b), inverted repeats showed a 26/26 sequence identity and were not found the subject genome. A unit with these terminal inverted repeats appeared to have inserted with direct duplication of 8-bp target sequence in this case. The inserted unit contained three ORFs, a Type IIG RM gene homolog, a Type I system S subunit homolog, and a hypothetical protein gene. RM systems with a Type IIG RM and a Type I S subunit homolog, such as BcgI (82) or Sau42I (28), are already known. Compared to these examples, the Type IIG RM homolog in this unit appears truncated, lacking its N terminal half. The third gene in this case had a transposase motif, and a likely inactivated derivative (COG3677, e-value 4e-12) by CD search (74). Whether this unit inserted through the activity of the transposase-like gene or through the activity of the RM genes is unknown.
DISCUSSION
By genome comparison and genome context analysis, we detected genome rearrangements linked to RM systems and their putative mobility forms. Intraspecific genome comparison revealed new examples of RM-linked genome rearrangements, such as insertion with a long target duplication, allelic substitution by different RM types, and allelic diversity of the target recognition domain in a Type IIG gene.
Our group previously discovered the insertion of genes with long target duplication in H. pylori, using whole genome comparison analysis (37). All examples in that study involved Type IIS RM systems, a subtype that cuts DNA outside of the recognition sequence. Examples of this type of insertion have been found in various species of bacteria, and for Type I, II, III and IV RM systems. The lengths of the repeated sequences vary, but are much longer than the range reported for the transposon of Mycoplasma (83). The ubiquitous occurrence of this type of insertion suggests that it involves a property common to all RM types, such as DNA double-strand breakage. RM systems flanked by long direct repeats are known to amplify themselves (84). This led us to hypothesize a virus-like life cycle for RM systems (84), which invade a genome by insertion with a long direct duplication, amplify themselves using the repeats, then release to invade other host cells in subsequent cycles (85). Although there is direct evidence for the amplification step (84), there is yet no experimental evidence for release or subsequent infection.
Genome context analysis revealed flanking repeats at a significantly higher frequency for RM systems than for average genes. The insertion of RM systems with long target duplication was found in several bacterial types, and for all RM system types. We discovered a novel mobile form of RM system, similar to classical DNA transposons, in that RM genes are flanked by imperfect inverted repeats (Figure 10 and Supplementary Table 2). Although mobility-related genes such as transposase and integrase genes are often linked to RM genes (3,10), to our knowledge, this is the first report for these structures (Figures 10 and 11), which were found for all RM types. Some inserted into a genome with a short target duplication (Figure 11) similar to classical DNA transposons. We do not know if these RM gene products act as a transposase in this unit. We cannot exclude the possibility that this unit was inserted by a transposase acting in trans, as a non-autonomous transposon. In some cases, only part of an RM gene cluster was flanked by inverted repeats, which might contribute to the diversification of RM systems by component replacement. The presence of the inverted repeats within an S gene of Type I systems might be related to the phase variation of S genes known in a Mycoplasma species (86).
Another interesting form of RM mobility is composed of RM genes and an integrase homolog inserted into a tRNA gene, resulting in flanking long direct repeats (Figure 3d). Restriction modification genes with a similar form are active (87).
In addition, we detected gene fusion during insertion with a long target duplication to generate a novel modification methyltransferase gene (Figure 9d and Supplementary Figure 7g). This might be an intermediate form in evolution of modification methyltransferases (88), explaining the circular permutation of their sequences (89,90). The formation of new specificity in M genes through gene fusion and duplication is an interesting prospect.
Our systematic genome comparison analysis revealed both the generality and variety of RM system mobility, including putative mobile forms of RM systems. This approach will reveal more RM system diversity, as prokaryote sequence data accumulates with metagenomics and innovations in sequencing technology.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The 21st century COE project of ‘Elucidation of Language Structure and Semantic behind Genome and Life System’; the global COE project of ‘Genome Information Big Bang’ from Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (to I.K.); ‘Grants-in–Aid for Scientific Research’ from Japan Society for the Promotion of Science (JSPS) (21370001, 19657002) (to I.K.); Medical Genome Science Program in Support Program for Improving Graduate School Education of JSPS (to I.K.) Funding for open access charge: The global COE project of ‘Genome Information Big Bang’ from Ministry of Education, Culture, Sports, Science, and Technology (MEXT).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Mikihiko Kawai, Ikuo Uchiyama, and Iwona Mruk for helpful discussions and suggestions. The authors thank an anonymous reviewer for explanation of Figure 6S (b).
REFERENCES
- 1.Wilson GG. Organization of restriction-modification systems. Nucleic Acids Res. 1991;19:2539–2566. doi: 10.1093/nar/19.10.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2009;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001;29:3742–3756. doi: 10.1093/nar/29.18.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden D.TF, Dybvig K, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sistla S, Rao DN. S-Adenosyl-L-methionine-dependent restriction enzymes. Crit. Rev. Biochem. Mol. Biol. 2004;39:1–19. doi: 10.1080/10409230490440532. [DOI] [PubMed] [Google Scholar]
- 7.Murray NE. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle) Microbiol. Mol. Biol. Rev. 2000;64:412–434. doi: 10.1128/mmbr.64.2.412-434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dryden DTF, Murray NE, Rao DN. Nucleoside triphosphate-dependent restriction enzymes. Nucleic Acids Res. 2001;29:3728–3741. doi: 10.1093/nar/29.18.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart FJ, Panne D, Bickle TA, Raleigh EA. Methyl-specific DNA binding by McrBC, a modification-dependent restriction enzyme. J. Mol. Biol. 2000;298:611–622. doi: 10.1006/jmbi.2000.3697. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda E, Kaminska KH, Bujnicki JM, Kobayashi I. Cell death upon epigenetic genome methylation: a novel function of methyl-specific deoxyribonucleases. Genome Biol. 2008;9:R163. doi: 10.1186/gb-2008-9-11-r163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mochizuki A, Yahara K, Kobayashi I, Iwasa Y. Genetic addiction: selfish gene's; strategy for symbiosis in the genome. Genetics. 2006;172:1309–1323. doi: 10.1534/genetics.105.042895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishikawa K, Handa N, Kobayashi I. Cleavage of a model DNA replication fork by a Type I restriction endonuclease. Nucleic Acids Res. 2009;37:3531–3544. doi: 10.1093/nar/gkp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blakely GW, Murray NE. Control of the endonuclease activity of type I restriction-modification systems is required to maintain chromosome integrity following homologous recombination. Mol. Microbiol. 2006;60:883–893. doi: 10.1111/j.1365-2958.2006.05144.x. [DOI] [PubMed] [Google Scholar]
- 14.Nobusato A, Uchiyama I, Kobayashi I. Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene. 2000;259:89–98. doi: 10.1016/s0378-1119(00)00455-8. [DOI] [PubMed] [Google Scholar]
- 15.Jeltsch A, Kroger M, Pingoud A. Evidence for an evolutionary relationship among type-II restriction endonucleases. Gene. 1995;160:7–16. doi: 10.1016/0378-1119(95)00181-5. [DOI] [PubMed] [Google Scholar]
- 16.Bujnicki JM, Radlinska M. Molecular phylogenetics of DNA 5mC-methyltransferases. Acta Microbiol. Pol. 1999;48:19–30. [PubMed] [Google Scholar]
- 17.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 18.Jeltsch A, Pingoud A. Horizontal gene transfer contributes to the wide distribution and evolution of type II restriction-modification systems. J. Mol. Evol. 1996;42:91–96. doi: 10.1007/BF02198833. [DOI] [PubMed] [Google Scholar]
- 19.Mrazek J, Karlin S. Detecting alien genes in bacterial genomes. Ann. NY Acad. Sci. 1999;870:314–329. doi: 10.1111/j.1749-6632.1999.tb08893.x. [DOI] [PubMed] [Google Scholar]
- 20.Parkhill J, Achtman M, James KD, Bentley SD, Churcher C, Klee SR, Morelli G, Basham D, Brown D, Chillingworth T, et al. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature. 2000;404:502–506. doi: 10.1038/35006655. [DOI] [PubMed] [Google Scholar]
- 21.Kita K, Kawakami H, Tanaka H. Evidence for horizontal transfer of the EcoT38I restriction-modification gene to chromosomal DNA by the P2 phage and diversity of defective P2 prophages in Escherichia coli TH38 strains. J. Bacteriol. 2003;185:2296–2305. doi: 10.1128/JB.185.7.2296-2305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Betlach M, Hershfield V, Chow L, Brown W, Goodman H, Boyer HW. A restriction endonuclease analysis of the bacterial plasmid controlling the ecoRI restriction and modification of DNA. Fed. Proc. 1976;35:2037–2043. [PubMed] [Google Scholar]
- 23.Humbelin M, Suri B, Rao DN, Hornby DP, Eberle H, Pripfl T, Kenel S, Bickle TA. Type III DNA restriction and modification systems EcoP1 and EcoP15. Nucleotide sequence of the EcoP1 operon, the EcoP15 mod gene and some EcoP1 mod mutants. J. Mol. Biol. 1988;200:23–29. doi: 10.1016/0022-2836(88)90330-0. [DOI] [PubMed] [Google Scholar]
- 24.Tyndall C, Lehnherr H, Sandmeier U, Kulik E, Bickle TA. The type IC hsd loci of the enterobacteria are flanked by DNA with high homology to the phage P1 genome: implications for the evolution and spread of DNA restriction systems. Mol. Microbiol. 1997;23:729–736. doi: 10.1046/j.1365-2958.1997.2531622.x. [DOI] [PubMed] [Google Scholar]
- 25.Kita K, Tsuda J, Kato T, Okamoto K, Yanase H, Tanaka M. Evidence of horizontal transfer of the EcoO109I restriction-modification gene to Escherichia coli chromosomal DNA. J. Bacteriol. 1999;181:6822–6827. doi: 10.1128/jb.181.21.6822-6827.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohshima H, Matsuoka S, Asai K, Sadaie Y. Molecular organization of intrinsic restriction and modification genes BsuM of Bacillus subtilis Marburg. J. Bacteriol. 2002;184:381–389. doi: 10.1128/JB.184.2.381-389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. Evolutionary relationships among diverse bacteriophages and prophages: all the world's; a phage. Proc. Natl Acad. Sci. USA. 1999;96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dempsey RM, Carroll D, Kong H, Higgins L, Keane CT, Coleman DC. Sau42I, a BcgI-like restriction-modification system encoded by the Staphylococcus aureus quadruple-converting phage Phi42. Microbiol. 2005;151:1301–1311. doi: 10.1099/mic.0.27646-0. [DOI] [PubMed] [Google Scholar]
- 29.Anton BP, Heiter DF, Benner JS, Hess EJ, Greenough L, Moran LS, Slatko BE, Brooks JE. Cloning and characterization of the BglII restriction-modification system reveals a possible evolutionary footprint. Gene. 1997;187:19–27. doi: 10.1016/s0378-1119(96)00638-5. [DOI] [PubMed] [Google Scholar]
- 30.Brassard S, Paquet H, Roy PH. A transposon-like sequence adjacent to the AccI restriction-modification operon. Gene. 1995;157:69–72. doi: 10.1016/0378-1119(94)00734-a. [DOI] [PubMed] [Google Scholar]
- 31.Stiens M, Becker A, Bekel T, Godde V, Goesmann A, Niehaus K, Schneiker-Bekel S, Selbitschka W, Weidner S, Schluter A, et al. Comparative genomic hybridisation and ultrafast pyrosequencing revealed remarkable differences between the Sinorhizobium meliloti genomes of the model strain Rm1021 and the field isolate SM11. J. Biotechnol. 2008;136:31–37. doi: 10.1016/j.jbiotec.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 32.van Zyl LJ, Deane SM, Louw LA, Rawlings DE. Presence of a family of plasmids (29 to 65 kilobases) with a 26-kilobase common region in different strains of the sulfur-oxidizing bacterium Acidithiobacillus caldus. Appl. Environ. Microbiol. 2008;74:4300–4308. doi: 10.1128/AEM.00864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rochepeau P, Selinger LB, Hynes MF. Transposon-like structure of a new plasmid-encoded restriction-modification system in Rhizobium leguminosarum VF39SM. Mol. Gen. Genet. 1997;256:387–396. doi: 10.1007/s004380050582. [DOI] [PubMed] [Google Scholar]
- 34.Siguier P, Gagnevin L, Chandler M. The new IS1595 family, its relation to IS1 and the frontier between insertion sequences and transposons. Res. Microbiol. 2009;160:232–241. doi: 10.1016/j.resmic.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Stein DC, Gunn JS, Piekarowicz A. Sequence similarities between the genes encoding the S.NgoI and HaeII restriction/modification systems. Biol. Chem. 1998;379:575–578. [PubMed] [Google Scholar]
- 36.Chinen A, Uchiyama I, Kobayashi I. Comparison between Pyrococcus horikoshii and Pyrococcus abyssi genome sequences reveals linkage of restriction-modification genes with large genome polymorphisms. Gene. 2000;259:109–121. doi: 10.1016/s0378-1119(00)00459-5. [DOI] [PubMed] [Google Scholar]
- 37.Nobusato A, Uchiyama I, Ohashi S, Kobayashi I. Insertion with long target duplication: a mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene. 2000;259:99–108. doi: 10.1016/s0378-1119(00)00456-x. [DOI] [PubMed] [Google Scholar]
- 38.Sibley MH, Raleigh EA. Cassette-like variation of restriction enzyme genes in Escherichia coli C and relatives. Nucleic Acids Res. 2004;32:522–534. doi: 10.1093/nar/gkh194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lubys A, Lubiene J, Kulakauskas S, Stankevicius K, Timinskas A, Janulaitis A. Cloning and analysis of the genes encoding the type IIS restriction-modification system HphI from Haemophilus parahaemolyticus. Nucleic Acids Res. 1996;24:2760–2766. doi: 10.1093/nar/24.14.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunn JS, Stein DC. The Neisseria gonorrhoeae S.NgoVIII restriction/modification system: a type IIs system homologous to the Haemophilus parahaemolyticus HphI restriction/modification system. Nucleic Acids Res. 1997;25:4147–4152. doi: 10.1093/nar/25.20.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naderer M, Brust JR, Knowle D, Blumenthal RM. Mobility of a restriction-modification system revealed by its genetic contexts in three hosts. J. Bacteriol. 2002;184:2411–2419. doi: 10.1128/JB.184.9.2411-2419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 43.Uchiyama I, Higuchi T, Kobayashi I. CGAT: a comparative genome analysis tool for visualizing alignments in the analysis of complex evolutionary changes between closely related genomes. BMC Bioinformatics. 2006;7:472. doi: 10.1186/1471-2105-7-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 45.Karlin S, zek J, Campbell AM. Codon usages in different gene classes of the Escherichia coli genome. Mol. Microbiol. 1998;29:1341–1355. doi: 10.1046/j.1365-2958.1998.01008.x. [DOI] [PubMed] [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 47.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. Prophage genomics. Microbiol. Mol. Biol. Rev. 2003;67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campbell AM. Chromosomal insertion sites for phages and plasmids. J. Bacteriol. 1992;174:7495–7499. doi: 10.1128/jb.174.23.7495-7499.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holden MT, Titball RW, Peacock SJ, Cerdeno-Tarraga AM, Atkins T, Crossman LC, Pitt T, Churcher C, Mungall K, Bentley SD, et al. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl Acad. Sci. USA. 2004;101:14240–14245. doi: 10.1073/pnas.0403302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Mellaert L, Mei L, Lammertyn E, Schacht S, Anne J. Site-specific integration of bacteriophage VWB genome into Streptomyces venezuelae and construction of a VWB-based integrative vector. Microbiology. 1998;144(Pt 12):3351–3358. doi: 10.1099/00221287-144-12-3351. [DOI] [PubMed] [Google Scholar]
- 52.Freitas-Vieira A, Anes E, Moniz-Pereira J. The site-specific recombination locus of mycobacteriophage Ms6 determines DNA integration at the tRNA(Ala) gene of Mycobacterium spp. Microbiology. 1998;144(Pt 12):3397–3406. doi: 10.1099/00221287-144-12-3397. [DOI] [PubMed] [Google Scholar]
- 53.Dupont L, Boizet-Bonhoure B, Coddeville M, Auvray F, Ritzenthaler P. Characterization of genetic elements required for site-specific integration of Lactobacillus delbrueckii subsp. bulgaricus bacteriophage mv4 and construction of an integration-proficient vector for Lactobacillus plantarum. J. Bacteriol. 1995;177:586–595. doi: 10.1128/jb.177.3.586-595.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruttin A, Foley S, Brussow H. The site-specific integration system of the temperate Streptococcus thermophilus bacteriophage phiSfi21. Virology. 1997;237:148–158. doi: 10.1006/viro.1997.8769. [DOI] [PubMed] [Google Scholar]
- 55.Waldman AS, Goodman SD, Scocca JJ. Nucleotide sequences and properties of the sites involved in lysogenic insertion of the bacteriophage HP1c1 genome into the Haemophilus influenzae chromosome. J. Bacteriol. 1987;169:238–246. doi: 10.1128/jb.169.1.238-246.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez MA, Herrero M, Suarez JE. The site-specific recombination system of the Lactobacillus species bacteriophage A2 integrates in gram-positive and gram-negative bacteria. Virology. 1998;250:185–193. doi: 10.1006/viro.1998.9353. [DOI] [PubMed] [Google Scholar]
- 57.McShan WM, Tang YF, Ferretti JJ. Bacteriophage T12 of Streptococcus pyogenes integrates into the gene encoding a serine tRNA. Mol. Microbiol. 1997;23:719–728. doi: 10.1046/j.1365-2958.1997.2591616.x. [DOI] [PubMed] [Google Scholar]
- 58.Gindreau E, Torlois S, Lonvaud-Funel A. Identification and sequence analysis of the region encoding the site-specific integration system from Leuconostoc oenos (OEnococcus oeni) temperate bacteriophage phi 10MC. FEMS Microbiol. Lett. 1997;147:279–285. doi: 10.1016/s0378-1097(96)00540-x. [DOI] [PubMed] [Google Scholar]
- 59.Magrini V, Creighton C, Youderian P. Site-specific recombination of temperate Myxococcus xanthus phage Mx8: genetic elements required for integration. J. Bacteriol. 1999;181:4050–4061. doi: 10.1128/jb.181.13.4050-4061.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pierson LS, 3rd, Kahn ML. Integration of satellite bacteriophage P4 in Escherichia coli. DNA sequences of the phage and host regions involved in site-specific recombination. J. Mol. Biol. 1987;196:487–496. doi: 10.1016/0022-2836(87)90026-x. [DOI] [PubMed] [Google Scholar]
- 61.Uchiumi T, Abe M, Higashi S. Integration of the temperate phage phiU into the putative tRNA gene on the chromosome of its host Rhizobium leguminosarum biovar trifolii. J. Gen. Appl. Microbiol. 1998;44:93–99. doi: 10.2323/jgam.44.93. [DOI] [PubMed] [Google Scholar]
- 62.Gabriel K, Schmid H, Schmidt U, Rausch H. The actinophage RP3 DNA integrates site-specifically into the putative tRNA(Arg)(AGG) gene of Streptomyces rimosus. Nucleic Acids Res. 1995;23:58–63. doi: 10.1093/nar/23.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pena CE, Stoner JE, Hatfull GF. Positions of strand exchange in mycobacteriophage L5 integration and characterization of the attB site. J. Bacteriol. 1996;178:5533–5536. doi: 10.1128/jb.178.18.5533-5536.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Semsey S, Blaha B, Koles K, Orosz L, Papp PP. Site-specific integrative elements of rhizobiophage 16-3 can integrate into proline tRNA (CGG) genes in different bacterial genera. J. Bacteriol. 2002;184:177–182. doi: 10.1128/JB.184.1.177-182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vogtli M, Cohen SN. The chromosomal integration site for the Streptomyces plasmid SLP1 is a functional tRNA(Tyr) gene essential for cell viability. Mol. Microbiol. 1992;6:3041–3050. doi: 10.1111/j.1365-2958.1992.tb01762.x. [DOI] [PubMed] [Google Scholar]
- 66.Brown DP, Idler KB, Katz L. Characterization of the genetic elements required for site-specific integration of plasmid pSE211 in Saccharopolyspora erythraea. J. Bacteriol. 1990;172:1877–1888. doi: 10.1128/jb.172.4.1877-1888.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown DP, Idler KB, Backer DM, Donadio S, Katz L. Characterization of the genes and attachment sites for site-specific integration of plasmid pSE101 in Saccharopolyspora erythraea and Streptomyces lividans. Mol. Gen. Genet. 1994;242:185–193. doi: 10.1007/BF00391012. [DOI] [PubMed] [Google Scholar]
- 68.Mazodier P, Thompson C, Boccard F. The chromosomal integration site of the Streptomyces element pSAM2 overlaps a putative tRNA gene conserved among actinomycetes. Mol. Gen. Genet. 1990;222:431–434. doi: 10.1007/BF00633850. [DOI] [PubMed] [Google Scholar]
- 69.Esposito D, Scocca JJ. The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res. 1997;25:3605–3614. doi: 10.1093/nar/25.18.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller WG, Pearson BM, Wells JM, Parker CT, Kapitonov VV, Mandrell RE. Diversity within the Campylobacter jejuni type I restriction-modification loci. Microbiology. 2005;151:337–351. doi: 10.1099/mic.0.27327-0. [DOI] [PubMed] [Google Scholar]
- 71.Lawrence JG, Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- 72.Cesnaviciene E, Petrusyte M, Kazlauskiene R, Maneliene Z, Timinskas A, Lubys A, Janulaitis A. Characterization of AloI, a restriction-modification system of a new type. J. Mol. Biol. 2001;314:205–216. doi: 10.1006/jmbi.2001.5049. [DOI] [PubMed] [Google Scholar]
- 73.Jurenaite-Urbanaviciene S, Serksnaite J, Kriukiene E, Giedriene J, Venclovas C, Lubys A. Generation of DNA cleavage specificities of type II restriction endonucleases by reassortment of target recognition domains. Proc. Natl Acad. Sci. USA. 2007;104:10358–10363. doi: 10.1073/pnas.0610365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lorenz MG, Wackernagel W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 1994;58:563–602. doi: 10.1128/mr.58.3.563-602.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sekizaki T, Otani Y, Osaki M, Takamatsu D, Shimoji Y. Evidence for horizontal transfer of SsuDAT1I restriction-modification genes to the Streptococcus suis genome. J. Bacteriol. 2001;183:500–511. doi: 10.1128/JB.183.2.500-511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Posfai J, Bhagwat AS, Posfai G, Roberts RJ. Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res. 1989;17:2421–2435. doi: 10.1093/nar/17.7.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kong H, Lin LF, Porter N, Stickel S, Byrd D, Posfai J, Roberts RJ. Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 2000;28:3216–3223. doi: 10.1093/nar/28.17.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mahillon J, Chandler M. Insertion sequences. Microbiol. Mol. Biol. Rev. 1998;62:725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chandler M, Mahillon J. Insertion sequences revisited. In: Berg DE, Howe MM, editors. Mobile DNA II. American Society for Microbiology Press, Vol. 1. 2002. pp. 305–366. [Google Scholar]
- 81.Moon WJ, Cho JY, Chae YK. Recombinant expression, purification, and characterization of XorKII: a restriction endonuclease from Xanthomonas oryzae pv. oryzae. Protein Expr. Purif. 2008;62:230–234. doi: 10.1016/j.pep.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 82.Kong H. Analyzing the functional organization of a novel restriction modification system, the BcgI system. J. Mol. Biol. 1998;279:823–832. doi: 10.1006/jmbi.1998.1821. [DOI] [PubMed] [Google Scholar]
- 83.Calcutt MJ, Lavrrar JL, Wise KS. IS1630 of Mycoplasma fermentans, a novel IS30-type insertion element that targets and duplicates inverted repeats of variable length and sequence during insertion. J. Bacteriol. 1999;181:7597–7607. doi: 10.1128/jb.181.24.7597-7607.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sadykov M, Asami Y, Niki H, Handa N, Itaya M, Tanokura M, Kobayashi I. Multiplication of a restriction-modification gene complex. Mol. Microbiol. 2003;48:417–427. doi: 10.1046/j.1365-2958.2003.03464.x. [DOI] [PubMed] [Google Scholar]
- 85.Kobayashi I. Life cycle of restriction-modification gene complexes, powers in genome evolution. International Congress Series. 2002;1246:191. [Google Scholar]
- 86.Dybvig K, Sitaraman R, French CT. A family of phase-variable restriction enzymes with differing specificities generated by high-frequency gene rearrangements. Proc. Natl Acad. Sci. USA. 1998;95:13923–13928. doi: 10.1073/pnas.95.23.13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khan F, Furuta Y, Kawai M, Kaminska KH, Ishikawa K, Bujnicki JM, Kobayashi I. 2010. A putative mobile genetic element carrying a novel Type IIF restriction-modification system (PluTI) Nucleic Acids Res., doi:10.1093/nar/gkp1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995;253:618–632. doi: 10.1006/jmbi.1995.0577. [DOI] [PubMed] [Google Scholar]
- 89.Bujnicki JM. Sequence permutations in the molecular evolution of DNA methyltransferases. BMC Evol. Biol. 2002;2:3. doi: 10.1186/1471-2148-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jeltsch A. Circular permutations in the molecular evolution of DNA methyltransferases. J. Mol. Evol. 1999;49:161–164. doi: 10.1007/pl00006529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.